

Abstract
Glial activation participates in the mediation of pain including neuropathic pain, due to release of neuroexcitatory, proinflammatory products. Glial activation is now known to occur in response to opioids as well. Opioid-induced glial activation opposes opioid analgesia and enhances opioid tolerance, dependence, reward and respiratory depression. Such effects can occur, not via classical opioid receptors, but rather via non-stereoselective activation of toll-like receptor 4 (TLR4), a recently recognized key glial receptor participating in neuropathic pain as well. This discovery identifies a means for separating the beneficial actions of opioids (opioid receptor mediated) from the unwanted side-effects (TLR4/glial mediated) by pharmacologically targeting TLR4. Such a drug should be a stand-alone therapeutic for treating neuropathic pain as well. Excitingly, with newly-established clinical trials of two glial modulators for treating neuropathic pain and improving the utility of opioids, translation from rats-to-humans now begins with the promise of improved clinical pain control.
Introduction
Normally, a painful stimulus is perceived via a chain of events beginning with the activation of “pain”-receptive sensory nerve fibers. The resultant action potentials relay information of potential or actual tissue injury to pain transmission neurons in the spinal cord dorsal horn. These, in turn, send the information to multiple sites within the brain where various aspects of the pain experience (sensation, analysis of meaning, emotional reactions, etc) are analyzed and responded to. However, pain processing is not a passive process but rather is under powerful modulatory control. Pain messages can be suppressed by drugs like morphine, relayed unaltered, or amplified under conditions such as chronic pain. When chronic pain develops as a result of peripheral nerve injury, for example, these conditions have typically been attributed to a variety of neuronal changes, including altered excitability of sensory neurons, alterations in which neurotransmitters are synthesized and released by various sensory neurons, alterations in pain transmission neuron excitability via multiple changes in receptor and ion channel functions, and so on1.
Intriguingly, powerful modulatory control exists not only for pain, but also for an organism’s responses to opioids, such as morphine. Opioids not only suppress pain, they also activate endogenous counter-regulatory mechanisms that, for example, actively oppose opioid-induced pain suppression, enhance analgesic tolerance wherein repeated opioids lose their ability to suppress pain, and enhance dependence wherein organisms require continued opioid exposure to stave off drug withdrawal. These modulatory controls have again been attributed to a variety if neuronal mechanisms, including release of endogenous anti-opioid peptides such as cholecystokinin, internalization and/or desensitization of opioid receptors, alterations in opioid receptor signaling cascades, and so on2,3.
While modulatory control systems regulating pain and opioid actions have been thought to involve separate mechanisms, the present paper will review recent evidence that suggests that these two phenomena are closely related and involve overlapping mechanisms. This development has been predicted from prior studies focused on the neuronal bases of chronic pain and opioid tolerance, where striking commonalities such as upregulation of NMDA function transcend what initially appeared to be quite different phenomena4. The present review will extend the commonalities between chronic pain and various opioid effects to include a non-neuronal component (glial cells, especially microglia and astrocytes) and a distinctly non-traditional mechanism, that being activation of the innate immune receptor expressed by glia, called toll like receptor 4 (TLR4).
Concepts of chronic pain and opioid actions have evolved in recent years with the realization that alterations in neuronal functions fail to capture all of the critical mechanisms involved. Recognition of a role for microglia and astrocytes in these processes first occurred for pain in the early 1990s, but the involvement of glia in modulating opioid actions was not discovered until a decade later. Indeed, the growing recognition of striking similarities in mechanisms underlying chronic pain and opioid tolerance directly led to the discovery of glial involvement in modulating opioid actions. Since glia were so convincingly important in chronic pain, it became a natural question whether they regulated the actions of opioids as well.
The goals of this article are to explore how glial activation impacts both pain and opioid actions. Pain will be considered first, including how glial activation increases neuronal excitability and how glial activation occurs under conditions leading to chronic pain. Included in this latter topic will be a discussion of TLR4 as a key glial activation receptor for the initiation and maintenance of chronic pain via TLR4-induced release of neuroexcitatory products such as proinflammatory cytokines. TLR4 will also be discussed as having a unique role in regulating the actions of opioids, as opioids have now been found to activate TLR4 on glia, in addition to their classical actions occurring through neuronal opioid receptors. Glial activation by opioids is an important phenomenon to understand, as glial activation opposes opioid analgesia and enhances opioid tolerance, dependence, reward and other negative side effects such as respiratory depression. Lastly, the implications of such glial activation will be considered for drug development aimed at improving both clinical pain control and clinical efficacy of opioids.
Role of glial activation in pain enhancement
The conclusion that the activation of microglia and astrocytes are critical to pain enhancement arose from 3 independent lines of study5: (a) cell culture studies showing that spinal cord is one of the rare CNS sites where astrocytes are activated in response to substance P, providing the first evidence that spinal cord glia are responsive to “pain” neurotransmitters; (b) anatomy studies that recognized that microglia and astrocytes each upregulate their expression of so-called activation markers in response to peripheral nerve injury that causes neuropathic pain, and that drugs known to block neuropathic pain block glial activation as well; and (c) in vivo studies of sickness-induced changes in physiology and behavior (increased sleep, fever, suppressed food and water intake, enhanced pain, etc.) that recognized that sickness responses, including enhanced pain, are all created as a result of glial activation and proinflammatory cytokine release within the brain and/or spinal cord. These three diverse lines of evidence predicted that glial activation would prove to be intimately important in the creation and maintenance of chronic pain.
The idea that glia contribute to pain has gained widespread support in the past decade6. Spinal microglia and astrocytes are activated in every clinically relevant animal model of pain enhancement, including pain arising from trauma-, inflammation- or chemotherapy-induced peripheral nerve damage, bone cancer, spinal cord injury, spinal nerve injury, multiple sclerosis, migraine, radiculopathy, and among others7 (Textbox 1). Comparable results have now been reported for trigeminal pain models as well8,9. Drugs that inhibit proinflammatory glial activation or their proinflammatory products block and/or reverse such pain states (Table 1)7. Although microglia and astrocytes release diverse neuroexcitatory substances, key among these for pain sensitivity are proinflammatory cytokines: interleukin-1β (IL-1), tumor necrosis factor-α (TNF), and IL-6, and much of the pain literature has focused on these classically glially derived modulators of neuronal excitability.
TABLE 1.
Pain facilitation in animal models is prevented (p) or reversed (r) by inhibition of spinal glial activation or proinflammatory cytokine actions:
| Model | Intervention [prevention (p) vs. reversal (r) noted for each] |
|---|---|
| Bone cancer | IL-1ra (r)61 |
| Carrageenan, subcutaneous | minocycline (p), IL-1 knockout (p) |
| Chemotherapy induced neuropathy | propentofylline (SLC022) (p)62 IL-10 (r)63, IL-1ra (r)63, ibudilast (AV411) (r)64, CB2 agonist (p)65, minocycline (p)66 |
| Colon irritation | minocycline (r)67 |
| Complete Freund’s adjuvant | IL-1ra (p,r)68,69, IL-1 KO (p), fluorocitrate (p)69, CB2 agonist (r)70 |
| Dorsal root compression/inflammation | IL-1ra, sTNFR minocycline (p)71 |
| Dynorphin, intrathecal | IL-1ra (p), IL-10 (p) |
| EAE model of multiple sclerosis pain | IL-10 (r)72 |
| Facial nerve chronic constriction injury | IL-1ra (r)73 |
| Formalin, subcutaneous | fluorocitrate (p), IL-1ra (p), minocycline (p), IL-1 KO (p) |
| Fractalkine, intrathecal | minocycline (p), IL-1ra (p), anti-IL-6 (p) |
| HIV-1 gp120, intrathecal | fluorocitrate (p), IL-1ra (p), sTNFR (p), anti-IL6 (p), minocycline (p)74, IL-10 (p)75 |
| Inferior alveolar and mental nerve transection | minocycline (p) |
| Infraorbital nerve injury | propentofylline (SLC022) (r)8, fluorocitrate (r)8, minocycline (r)8, sTNFR (r)8, IL-1ra (r)8 |
| Monoarthritis | Fluorocitrate (r)6, minocycline (p)6 |
| Myositis | minocycline (p)76, anti-TNF (p)76 |
| Mustard oil, topical | fluorocitrate (p) |
| Phospholipase A2, subcutaneous | fluorocitrate (p), IL-1ra (p), sTNFR (p) |
| Postoperative incisional pain | fluorocitrate (r), IL1ra overexpressing transgenetics & IL1 receptor KO (p)77, IL-1ra (r)77, pentoxifylline (p)78, CB2 agonist (r)79 |
| Sciatic nerve injury | IL-1ra (r), IL-10 (r), IL-1 knockout (p), ibudilast (AV411) (r)58, fluorocitrate (r)80, minocycline (p)81, 82, pentoxifylline (p)81, CB2 agonist (r)70 |
| Sciatic nerve inflammation | fluorocitrate (p), minocycline (p), IL-1ra (p), sTNFR (p), anti-IL-6 (p), IL-10 (p)83 |
| Sciatic nerve tetanic stimulation | fluorocitrate (p) |
| Spinal cord injury | IL-10 (p), sTNFR (p)84, minocycline (p,r) 84,85, ibudilast (AV411) (r), propentofylline (p)86 |
| Spinal nerve root injury | methotrexate (p,r) |
| Spinal nerve transection or ligation | propentofylline (SLC022) (p,r), minocycline (p)87, ibudilast (AV411) (r)58, pentoxifylline (p)88, IL-1 KO (p), CB2 agonist (r)89, IL-1ra + sTNFR (p,r), IL-1 receptor KO (p), IL1ra overexpresssing transgenic (p) |
| Supradural “inflammatory soup” | ibudilast (AV411) (r) |
| Tempormandibular joint region inflammation | anti-IL6 (p)90 |
| Tibia fracture model of complex regional pain syndrome | pentoxifylline (p)91 |
| Zymosan, subcutaneous | fluorocitrate (r)80 |
Abbreviations: anti-IL6: neutralizing antibody against interleukin-6, anti-TNF: neutralizing antibody against tumor necrosis factor-α, CB2: cannabinoid type 2, KO: knockout mice, IL-1ra: interleukin-1 receptor antagonist, sTNFR: soluble TNF receptor, IL-10: interleukin-10.
Modified and updated from Watkins et al.7. Citations are found in this 2007 review unless otherwise indicated.
Impact of glial activation on neuronal excitability
Under conditions of normal pain responsivity, microglia actively survey the extracellular space in search of potential danger, but are quiescent in terms of releasing neuroexcitatory substances10. By contrast, astrocytes are active players in synaptic signaling even under basal conditions because they both receive signals from and signal to synapses11. Astrocytes also maintain “housekeeping” functions such as providing energy sources and neurotransmitter precursors to neurons, clearing debris, maintaining homeostasis of extracellular ions, and taking up released neurotransmitters to terminate their actions. On appropriate stimulation (see below), microglia and astrocytes can each shift from their basal-but-active state to an activated state, characterized by a reactive, proinflammatory response profile. In this state, glia release substances that increase neuronal excitability, leading to pain enhancement. These include proinflammatory cytokines (IL-1, TNF, IL-6), chemokines, arachidonic acid and prostaglandins, ATP, excitatory amino acids and D-serine, nerve growth factors, reactive oxygen species, nitric oxide, and enkephalinases. These glial products can directly enhance neuronal excitability12,13, increase pain-associated neurotransmitter release from sensory afferents7, upregulate the number and conductance of calcium-permeable AMPA and NMDA receptors14, potentiate inward currents in tetrodotoxin-resistant sodium channels15, downregulate GABA receptors, downregulate outward potassium currents16, downregulate expression of glial glutamate transporters, and downregulate GPCR kinase 2, thereby removing a “brake” on excitability17. Thus, both microglia and astrocytes can be triggered by special circumstances to enter a state in which they release a variety of neuroexcitatory substances. How that can occur in response to cues for pathological pain states, such as neuropathic pain, and by exposure to opioids, are explored below.
Triggering glia to enter an activated, proinflammatory state
Several glial activating factors have been identified (Figure 1), including many neuron-to-glia signals. The basic concept is that the peripheral tissue or nerve injury must generate a signal that in turn causes glia in the spinal cord to become activated and to release neuroexcitatory, proinflammatory signals. Neurons can release fractalkine (CX3CL1), monocyte chemotactic protein-1 (CCL2), nitric oxide, substance P, calcitonin gene related peptide, ATP, glutamate and prostaglandins, each of which can activate glia to release pain-enhancing products7.
Figure 1. Glial activation by peripheral neuropathy.
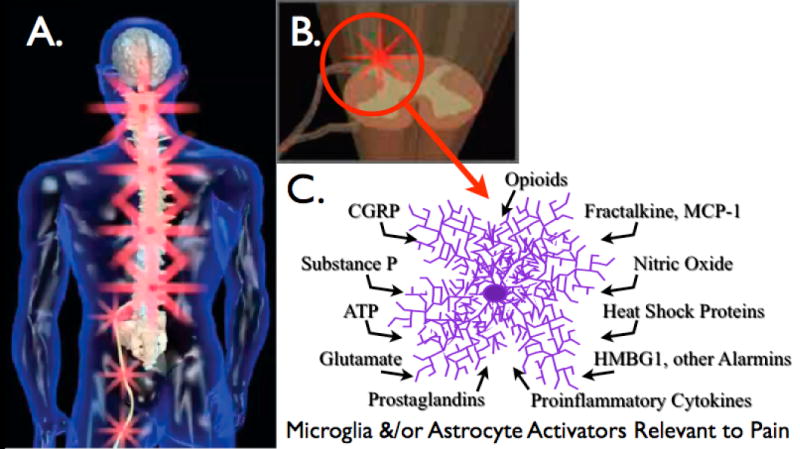
(A) Peripheral nerve injury leads to spinal amplification of incoming pain messages in addition to spontaneous pain signaling. This is indicated by the magnification of red “stars” symbolizing pain signals moving from the periphery into spinal cord, with amplification of pain messages being relayed to higher brain centers. (B) Spinal amplification of pain occurs within the spinal cord dorsal horn, a key site for the dynamic regulation of pain processing. This is the site where incoming sensory fibers synapse with neurons that relay pain messages up to brain via the spinothalamic and other pathways. This is also the site where glia and other immunocompetent cells can amplify pain via the release of neuroexcitatory substances such as proinflammatory cytokines. (C) The release of neuroexcitory, proinflammatory products by glia occurs in response to microglial and astrocyte activation. This activated state can occur in response to a variety of neuron-to-glia signals including neuronal chemokines (fractalkine, MCP-1), neurotransmitters (glutamate, ATP, substance P, CGRP), neuromodulators (nitric oxide, prostaglandins), endogenous danger signals (also called “alarmins”; e.g., heat shock proteins, the nuclear protein HMGB1), in addition to xenobiotics including opioids.
Intriguingly, conditions such as peripheral nerve injury can also elicit the release of what have recently come to be called “endogenous danger signals” or “alarmins”18. These are signals that something is wrong; that is, there is cellular stress/damage, independent of the release of classical neuronal neurotransmitters or neuromodulators. Such signals include degradation products of the extracellular matrix, components of circulating blood not normally having access to the extracellular space such as fibrinogen, and substances released by stressed, damaged and dying cells, such as nuclear protein HMBG1, heat shock proteins, DNA, and related “self” substances normally hidden from immunological surveillance. On release of these signals, the innate immune pattern recognition receptors TLR4 and TLR2 detect the presence of the danger signal resulting in the activation of TLR-expressing cells.
TLR4 as a glial activation receptor: role in neuropathic pain
Within the CNS, TLR4 is predominantly expressed by microglia, but its expression may be upregulated by astrocytes under neuroinflammatory conditions19. Given that TLR4 signaling is activated by messengers of cellular stress, damage and death, TLR4 and hence microglia are well positioned to be key for enhancing pain resulting from injury/inflammation of peripheral or central tissues that release endogenous danger signals (Textbox 2). Indeed, TLR4 has been shown to be a key glial activation receptor for the initiation and maintenance of neuropathic pain19,20. Although TLR2 and TLR3 have recently been implicated in neuropathic pain21,22, most studies have been done on TLR4, and so this receptor is our focus here.
TLR4 signaling occurs via a cascade of events (Figure 2)23. Using Gram-negative bacterial lipopolysaccharide (LPS) as the prototypic TLR4 agonist, LPS is transported to the cell by LPS-binding protein, which transfers LPS to the co-receptor cluster determinent-14 (CD14) on the cell membrane. This leads to intracellular activation of acid sphingomyelinase that generates ceramide, which in turn induces generation of a lipid raft containing the co-receptor myeloid differentiation factor 2 (MD2), TLR4, heat shock proteins 70 and 90, among others. CD14 transfers LPS to MD2, leading to first MD2-TLR4 heterodimerization and then homodimerization of MD2-TLR4 pairs. Ensuing intracellular signaling occurs through at least 3 parallel pathways: cell motility and cell survival/apoptosis occurs through the PI3K/Akt pathway; and proinflammatory products such as cytokines result from activation of the NFκB and MAPK pathways. The details of how various endogenous danger signals interact with, bind to, and subsequently activate the TLR4 signaling pathway, and the specific identity of the endogenous danger signals that are involved in neuropathic pain are unknown. What evidence does exist suggests that a similar cascade is involved because neuropathic pain is suppressed in CD14 knockout mice24, by TLR4 competitive antagonists, by novel TLR4 signaling inhibitors such as (+)-naloxone19 (see below), by inhibitors of MD2-TLR4 docking25, and by inhibitors of heat shock protein 9026.
Figure 2. TLR4 signaling cascade and evidence for modulation by opioids.
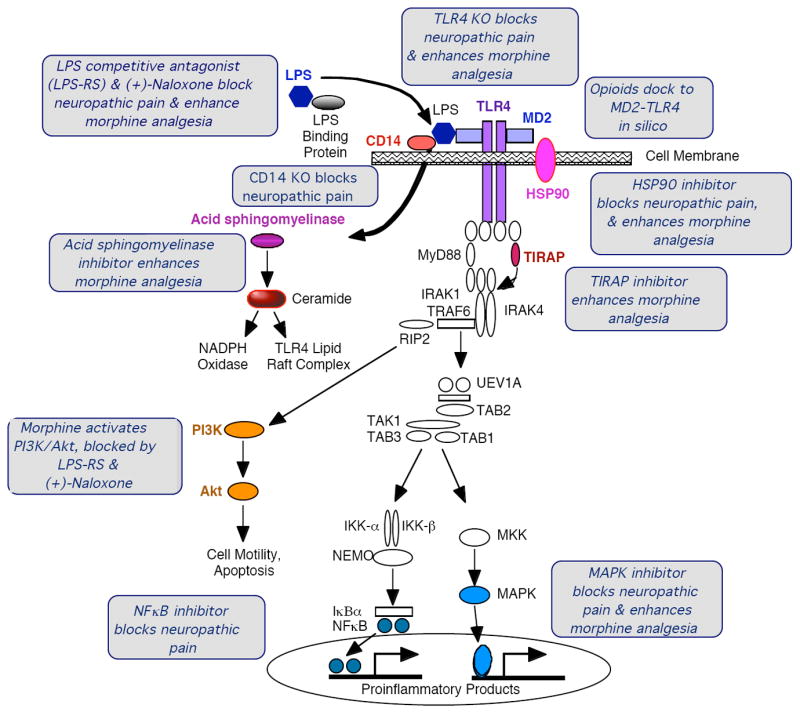
TLR4 signaling occurs via a cascade of events. Gram-negative bacterial lipopolysaccharide (LPS; dark blue hexagon) is the prototypic TLR4 agonist that is transported to the cell via LPS-binding protein (gray oval), which transfers LPS to CD14 on the cell membrane (red oval). This leads to intracellular activation of acid sphingomyelinase (purple oval), which generates ceramide (red oval). Ceramide induces the generation of a lipid raft containing the co-receptor myeloid differentiation factor 2 (MD2) (light blue rectangle), TLR4 (purple rectangles), and heat shock protein (HSP) 70 and HSP90 (pink oval), among other elements. Ceramide also activates NADPH oxidase, that leads to peroxynitrite formation. CD14 transfers LPS to MD2 (light blue rectangle), leading to both MD2-TLR4 heterodimerization and then homodimerization of MD2-TLR4 pairs. Ensuing intracellular signaling occurs through toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) to at least 3 parallel pathways: cell motility and cell survival/apoptosis occur through the IP3K/Akt pathway (yellow ovals), and proinflammatory products such as cytokines result from activation of the NFκB (dark blue circles) and MAPK (medium blue oval) pathways. The gray boxes provide summaries of the converging evidence that opioids interact with the TLR4 signaling cascade. The TLR4 signaling pathway schema is a modified from illustrative figure of Dauphinee and Karsan http://www.nature.com/labinvest/journal/v86/n1/fig_tab/3700366f1.html#figure-title
In summary, using peripheral nerve injury-induced neuropathic pain as the exemplar, nerve damage leads to the activation of microglia and astrocytes within the spinal cord. This occurs as a consequence of signals released by stressed and damaged neurons, including factors that activate the “endogenous danger signal” receptor, TLR4. Upon activation, glia release a variety of neuroexcitatory, pain-enhancing substances, key amongst these being proinflammatory cytokines.
Beyond pathological pain: glial modulation of opioid actions
In the past decade, a series of discoveries have revised our views of the pharmacological actions of opioids. Since 2001, several laboratories have reported that glia become activated in response to opioids and this glial activation leads to the release of proinflammatory products, including proinflammatory cytokines27-29. In vivo, opioid-induced glial activation has been inferred from: (a) morphine-induced upregulation of microglial and astrocytic activation markers30,31, (b) morphine-induced upregulation and/or release of proinflammatory cytokines and chemokines28,31-33, (c) enhanced morphine analgesia produced by the glial activation inhibitors fluorocitrate, minocycline or ibudilast (AV411)31,34,35, (d) enhanced morphine analgesia produced by blocking proinflammatory cytokine actions28,36, and (e) opioid-induced selective activation of microglial p38 MAPK and enhanced morphine analgesia by p38 MAPK inhibitors30. In vitro studies also document direct actions of opioids on glia34,37-39.
Until recently, it was assumed that opioids affect glia through opioid receptors. However, opioids can exert nonstereoselective effects. In contrast to classical opioid receptors, where only (-)-opioid isomers possess function, (+)-opioid agonists suppress (-)-opioid analgesia40, an effect attributed to glial activation based on propentofylline blockade41 and independent of classical μ-opioid receptors in knockout mice studies42. Opioid hyperalgesia is still observed in mu, delta, and kappa opioid receptor triple knockout mice43, which again is suggestive of the existence of a non-classical opioid receptor whose nonstereoselective activation opposes analgesia.
Role of TLR4 in counteracting the beneficial actions of opioids
The non-classical, non-stereoselective effects of opioids remained a puzzle until a link was made in 2007 to TLR444. In vivo, in vitro, and in silico approaches provided converging lines of evidence that members of each structural class of opioids activate TLR4 (some nonstereoselectively) and that opioid antagonists such as naloxone and naltrexone nonstereoselectively block TLR4 signaling45,46. The consequences of opioid-induced TLR4 signaling are extensive (Textbox 2). Acute blockade of TLR4, genetic knockout of TLR4, or blockade of TLR4 downstream signaling each lead to a marked potentiation of the magnitude and duration of opioid analgesia, with TLR4 modulation of opioid actions in wildtype animals occurring within minutes. Both spinal and supraspinal sites of opioid-TLR4 interactions are implicated45. Given the breadth of opioids now documented to interact with TLR4, many off-target opioid effects previously attributed to unilateral opioid action at classical neuronal opioid receptors might in fact result, at least in part, from the duality of opioid actions at TLR4. This dual opioid action hypothesis (glial TLR4 and neuronal opioid receptor) raises the benchmark for establishing unilateral neuronal opioid receptor involvement in a response, over simple naloxone blockade or the use of “opioid-selective” agonists or antagonists, unless they are proven not to be TLR4 signaling activators.
Mechanism of TLR4 activation by opioids
Intriguingly, TLR4 is activated by morphine-3-glucoronide (M3G), a morphine metabolite that is inactive at classical opioid receptors, but not by morphine-6-glucoronide (M6G), the opioid receptor active metabolite45,46. Along with nonstereoselectivity, the TLR4 signaling activation by M3G, but not M6G, points to a major difference from the structure activity relationship of the opioid receptor with the reliance of 4,5-epoxymorphinans on the 3’OH for classical opioid receptor activity but not for TLR4 activity. M3G action at TLR4 predicts that intrathecal M3G would induce pain enhancement mediated by TLR4, microglia (given their predominance in TLR4 expression), and proinflammatory cytokines (a downstream product of TLR4 activation). Indeed, this is the case46. TLR4 activation by opioids is now implicated in opposing acute and chronic opioid analgesia, and contributing to opioid-induced hyperalgesia, dependence and reward45,46.
Nonstereoselectivity sets TLR4 apart from classical opioid receptors that bind (-)-opioid isomers but not (+)-isomers. Additionally, this nonstereoselectivity provides a means of specifically blocking opioid-induced glial activation by using drugs such as (+)-naloxone. This would allow (-)-opioids to act neuronally to suppress pain via their actions on classical opioid receptors, while preventing (-)-opioids from simultaneously activating glial TLR4, which causes the release of pain-enhancing proinflammatory cytokines that oppose or counter-regulate the pain suppressive effects of opioids. The conclusion that (+)- and (-)-naloxone and naltrexone are TLR4 signaling inhibitors derives from their dose-dependent blockade of TLR4 activation by LPS, a classical TLR4 agonist, as indicated by suppression of LPS-induced reporter protein in a TLR4 expressing cell line19. Further, using a microglial cell line, (+)- and (-)-naloxone each blocked TLR4 activation-induced gene expression of both proinflammatory cytokines and a microglial activation marker19.
Although it is clear that TLR4 signaling is activated and inhibited by opioid agonists and opioid antagonists, respectively, where along the signaling cascade this non-stereoselective interaction is occurring remains under investigation (Figure 2). This is because other xenobiotics, for example thalidomide and tricyclic antidepressants, have been shown to modify TLR4 signaling at least in part by exerting intracellular effects downstream of TLR4. Their effects include suppressing MD2 expression and inhibiting acid sphingomyelinse47,48. For opioids, the interaction with TLR4 is probably occurring at or very close to MD2/TLR4. In silico modeling of opioid interactions with MD2, TLR4, and the MD2-TLR4 complex point to docking of opioids in the LPS binding pocket of MD2, in a manner predictive of altering the MD2-TLR4 complex45 (Figure 3). Opioid-induced activation of TLR4 signaling is blocked by a competitive inhibitor of LPS, again supportive of an action at the TLR4 complex. Also, morphine analgesia is potentiated in TLR4 knockout mice, supportive of opioid actions at TLR4 rather than downstream sites. Additionally, in vitro studies have revealed a requirement for soluble factors in media conditioned by macrophages cultures for allowing opioids to signal effectively through TLR4, suggestive of the requirement of known soluble member(s) of the TLR4 lipid raft complex, such as MD2. From in vitro and in vivo studies, opioids activate toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) and all three of its downstream signaling pathways (PI3K/Akt, NFkB, MAPK) leading to both membrane ruffling and motility (PI3K/Akt) (Figure 4) and proinflammatory cytokines (NFkB, MAPK). While such data support a role of TLR4, they do not preclude opioid interactions with other TLRs, because opioid effects on TLR2 and TLR9 have been reported49.
Figure 3. Computer modeling of opioid interaction with the TLR4-MD2 complex.

In silico docking of (-)-morphine to the 3D crystalline structure of the human MD2 (blue) and TLR4 (green) complex demonstrates that the preferred binding conformation of (-)-morphine is to the LPS binding domain of MD2.
Figure 4. Live cell imaging of opioid-induced TLR4 signaling.

Opioid interactions with TLR4 can be imaged in real time using a RAW264.7 mouse macrophage cell line that stably expresses green fluorescent protein labeled Akt1 (GFP-Akt1), as Akt is a component of one of the 3 parallel signaling pathways activated in response to TLR4 agonists. (A) Under basal conditions, Akt1 is diffusely distributed in the cytosol. (B) On activation, Akt1 rapidly moves to the membrane site where an Akt1 activating event is occurring. Membrane ruffling is also reflective of activation of the PI3K/Akt pathway downstream of TLR4. (C) Mobilization of GFP-Akt1 from the cytosol is easily quantified. At the first (downward) arrow, a TLR4 antagonist [Gray Triangles; either the competitive TLR4 antagonist LPS-RS (an LPS variant expressed by the bacterium Rhodobacter sphaeroides which helps the pathogen evade the host’s immune system by blocking TLR4), (+)-naloxone, or (-)-naloxone] or vehicle [Black Squares] is added to the live cell cultures. Cytoplasmic Akt1 fluorescence remains unchanged until TLR4 agonists (the classic TLR4 agonist LPS, (+)-morphine, or (-)-morphine) are added at the second (upward) arrow. Cells that are exposed only to TLR4 agonists (Black Squares) show a rapid loss of fluorescence from the cytosol as GFP-Akt1 mobilizes to the cell membrane. In the presence of TLR4 antagonists (Grey Triangles), TLR4 agonists now fail to induce GFP-Akt1 activation, so cytosolic fluorescence remains stable. As a positive control that these antagonist-blocked cells are in fact responsive, complement 5a (C5a) is then added at the third arrow (White Triangles) as a positive control since C5a activates PI3K/Akt1 but via a pathway independent of TLR4. Panel 3 is modified from Hutchinson et al.45.
TLR4 and glial activation: implications for drug development
Gaining insights into glial modulation of pain and opioid effects in humans has been constrained by the challenges of assaying glial activation and glial products within the CNS. In rodents, positron emission tomography allows analyses of glial activation through visualizing uptake of labeled fluoroacetate (metabolic inhibitor specific to the Krebs cycle in glia)50 or labeled ligands of the translocator protein (TSPO; formerly known as the peripheral benzodiazepine receptor), the upregulation of which is a hallmark of activation in microglia51. No published studies have used such approaches for studying glial modulation of pain or opioid actions. The only human study of imaging of glial activation in patients reported thalamic microglial activation in amputees with long-duration phantom limb pain52. Protein or gene activation analyses of pain- or opioid-related glial activation in humans is in its infancy, with reports supportive of glial activation, enhanced proinflammatory cytokines and suppressed anti-inflammatory cytokines in CSF and/or spinal cord of chronic pain53-55 and chronic opioid using patients56. Genomic analyses of polymorphisms also support alterations in the cytokine system of patients with chronic pain7 and more recently predisposition to opioid dependence57. Given the strength of the animal studies, human research using such approaches is needed.
On the basis of the overwhelming evidence from animal studies supportive of a role of glia in pathological pain states, the U.S. Food and Drug Administration (FDA) has now approved two glia-targeting drugs for Phase 2 clinical trials for the treatment of neuropathic pain: ibudilast (Avigen’s AV411) and propentofylline (Solace’s SLC022). In addition, ibudilast has FDA approval for testing its ability to increase the clinical efficacy of opioids. While having distinct mechanisms of action58,59, both ibudilast and propentofylline are orally available, blood-brain barrier permeable, glial activation inhibitors (based on microglial and astrocyte activation marker suppression) that have strong support treating pain from various rodent models58,59. Ibudilast has a long history of safety in humans for the treatment of asthma and post-stroke dizziness. Propentofylline has previously been tested in humans as far as Phase 3 trials for treating Alzheimer’s disease, with the trial discontinued for lack of efficacy. In addition to their well-characterized effects in vitro and in vivo, a recent novel finding is that ibudilast, but not pentoxyfyline (close relative of propentofylline), is a TLR4 signaling inhibitor45. Furthermore, given the structural diversity of exogenous pharmacotherapies and endogenous small molecule “alarmins” that have TLR4 activity, a far wider range of existing xenobiotics (as well as those in development for a myriad of TLR4 indications) may have TLR4 activity. This presents the possibility of drug-drug and/or drug/-TLR4 interactions at the previously unrecognized xenobiotics-TLR4-glial interface. Further, both AV411 and SLC022 possess the ability to enhance the production of anti-inflammatory cytokines, in addition to suppressing proinflammatory ones. This is important as it would replace the important glially-derived neuroprotective, negative feedback factors that would be missing if only proinflammatory products were to be suppressed.
This action of ibudilast as a TLR4 signaling inhibitor raises the more general issue of what is desirable in a glial inhibitor. Pervasive glial inhibitors such as fluorocitrate or fluoroacetate are not feasible, as these metabolic poisons shut down glial uptake of extracellular excitatory amino acids, leading to seizures59 and have mostly been tested in inflammatory models (Table 1). Drugs such as ibudilast and propentofylline have both neuronal and glial effects58,59, and it is clear that they have multiple effects45,58,60. This suggests that (+)-naloxone analogs should be considered that specifically target TLR4 rather than having broad effects as do propentofylline and ibudilast. While one could argue that blocking TLR4, classically known as the endotoxin receptor, could have detrimental effects, the immune system is redundant in its safeguards and has multiple avenues for recognizing and eliminating bacteria, such as opsonization and internalization by pathways independent of TLR4. In addition, the clinical experience of chronic high dose (-)-naltrexone use is well tolerated in other indications, and studies of TLR4 knockout and TLR4 mutant mice by-and-large reveal no health concerns in the absence of a functional TLR4 system. Such evidence, in addition to the data reviewed above, advocates serious consideration of orally available, blood-brain barrier permeable, TLR4 (and probably TLR2) inhibitors for clinical trials as stand alone treatments of chronic pain and as adjunct therapeutics for improving the clinical utility of opioids.
Concluding remarks
As reviewed above, while glia in their basal state play important roles in maintaining the health and normal functioning of the nervous system, their inappropriate proinflammatory activation concurrent with chronic pain pathologies and opioid administration can dramatically amplify pain and detrimentally alter the actions of opioids. While proinflammatory responses by glia can be important for inducing resolution of CNS immune challenges, neuroprotection, and repair, under conditions of chronic pain and opioid exposure, glial activation is not advantageous and instead detrimental to health and CNS functioning. Importantly, the glial actions described here do not stand and act alone to produce the behavioral consequences, since previously established neuronal pathways will unquestionably have to signal, precipitate, and propogate the response. Instead, the data reviewed here suggests that glia activation, in some cases resulting from TLR4 signaling, contributes significantly to the dysregulation of neuronal functioning under these altered physiological states. While TLR4 is by no means the only receptor-mediated activation pathway possessed by glia, TLR4 activation facilitates glial activation and neuroexcitability under conditions of chronic pain where endogenous danger signals are elevated and in response to opioids. The results from animal studies to date predict that controlling endogenous danger signal- and opioid-induced glial activation and the resultant proinflammation using pharmacotherapies will: be stand-alone treatments for diverse pathological pain states, enhance the ability of opioids to suppress pain, suppress the development of opioid tolerance, suppress the development of opioid dependence, suppress opioid reward linked to drug abuse, and suppress other negative side-effects of opioids such as respiratory depression. The broader implications of glial attenuation by novel pharmacotherapies beyond their intended beneficial actions will need to be examined and carefully monitored, but the opportune clinical experience based on existing previously uncharacterized glial modulators designed and prescribed for other purposes, such as naltrexone and minocycline, is promising as no negative consequences of glial attenuation have been identified to date with these agents. With two glial modulatory drugs now entering clinical trials, the exciting translation to humans is finally beginning.
TABLE 2.
Clinically relevant efficacy of opioids is improved in animal models by inhibition of glial activation or proinflammatory cytokine actions:
| Model | Direction of effect | Intervention |
|---|---|---|
| Opioid induced acute analgesia | enhanced | minocycline28, 34, ibudilast (AV411)31, IL-1032, IL-1ra28, 32, 36, IL-1 signaling KOs36, IL-ra over-expressing transgenics36, classic TLR4 antagonists45, (+)-naloxone45, (-)- naloxone45, ultra-low (-)-opioid antagonists 92, sTNFR28, anti-IL-628, p38 MAPK inhibitor28 |
| Opioid induced analgesia for neuropathic pain | enhanced | propentofylline (SLC022)93, pentoxifylline93 IL-1ra + TNF soluble receptors + anti-IL694 |
| Morphine analgesic tolerance | suppressed | ibudilast (AV411)64, IL-1032, IL-1ra32, fluorocitrate95, minocycline35, pentoxifylline81, (+)-naloxone45, propentofylline (SLC022)33, IL-1 signaling KOs36, IL-ra overexpressing transgenics36, IL-10 + IL-1ra28, IL-1 converting enzyme inhibitor + IL1ra28 |
| Morphine withdrawal-induced pain enhancement | suppressed | IL-1032, IL-1ra32, propentofylline (SLC022)33, (+)-naloxone45, IL-10 + IL-1ra28, IL-1 converting enzyme inhibitor + IL1ra28 |
| Morphine withdrawal-induced pain enhancement in neuropathic rats | suppressed | IL-1ra + TNF soluble receptors + anti-IL694 |
| Precipitated opioid withdrawal behaviors | suppressed | ibudilast (AV411)31, (+)-naloxone45, minocycline31 |
| Spontaneous opioid withdrawal behaviors | suppressed | ibudilast (AV411)31 |
| Morphine induced respiratory depression | suppressed | minocycline34 |
| Morphine induced dopamine release from brain “reward” area (nuc. Accumbens) | suppressed | (+)-naloxone, ibudilast (AV411)96 |
| Morphine induced conditioned place preference | suppressed | minocycline34, propentofylline (SLC022)97 |
| Morphine induced glial activation (IHC) | suppressed | ibudilast (AV411)31, pentoxifylline98, propentofylline (SLC022)99, fluorocitrate95 minocycline74 |
| Morphine induced proinflammatory cytokines & chemokines | suppressed | propentofylline (SLC022)99, (+)-naloxone19, ibudilast (AV411)31 |
Abbreviations: anti-IL6: neutralizing antibodies against interleukin-6, IL-1ra: interleukin-1 receptor antagonist, KOs: knock out mice, TLR4: toll like receptor 4, sTNFR: soluble TNF receptor, IL-10: interleukin-10.
Acknowledgments
International Association for the Study of Pain International Collaborative grant, American Australian Association Merck Company Foundation Fellowship, National Health and Medical Research Council CJ Martin Fellowship (ID 465423;M.R.H.) and NIH Grants DA015642, DA017670, DA024044, and DE017782. This work was partially supported by the by the NIH Intramural Research Programs of the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. We thank Avigen for permission to use their images in Figure 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ueda H, Ueda M. Mechanisms underlying morphine analgesic tolerance and dependence. Front Biosci. 2009;14:5260–5272. doi: 10.2741/3596. [DOI] [PubMed] [Google Scholar]
- 3.Watkins LR, et al. Potentiation of opiate analgesia and apparent reversal of morphine tolerance by proglumide. Science. 1984;224:395–396. doi: 10.1126/science.6546809. [DOI] [PubMed] [Google Scholar]
- 4.Chang G, et al. Opioid tolerance and hyperalgesia. Med Clin North Am. 2007;91:199–211. doi: 10.1016/j.mcna.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Watkins LR, Maier SF. Glia and pain: past, present and future. In: Merskey H, et al., editors. The Paths of Pain 1975-2005. 2005. pp. 165–176. IASP Press. [Google Scholar]
- 6.Cao H, Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev. 2008;32:972–983. doi: 10.1016/j.neubiorev.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Watkins LR, et al. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56:148–169. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei F, et al. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28:10482–10495. doi: 10.1523/JNEUROSCI.3593-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terayama R, et al. Activation of microglia and p38 mitogen-activated protein kinase in the dorsal column nucleus contributes to tactile allodynia following peripheral nerve injury. Neuroscience. 2008;153:1245–1255. doi: 10.1016/j.neuroscience.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 10.Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 11.Haydon PG, et al. Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology. 2009;56(Suppl 1):83–90. doi: 10.1016/j.neuropharm.2008.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu YL, et al. Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52:708–715. doi: 10.1016/j.neuropharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Binshtok AM, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang RX, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135:232–239. doi: 10.1016/j.pain.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin X, Gereau RWt. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeda M, et al. Activation of interleukin-1beta receptor suppresses the voltage-gated potassium currents in the small-diameter trigeminal ganglion neurons following peripheral inflammation. Pain. 2008 doi: 10.1016/j.pain.2008.06.015. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 17.Kleibeuker W, et al. IL-1 beta signaling is required for mechanical allodynia induced by nerve injury and for the ensuing reduction in spinal cord neuronal GRK2. Brain Behav Immun. 2008;22:200–208. doi: 10.1016/j.bbi.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 19.Hutchinson MR, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanga FY, et al. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282:14975–14983. doi: 10.1074/jbc.M607277200. [DOI] [PubMed] [Google Scholar]
- 22.Obata K, et al. Toll-like receptor 3 contributes to spinal glial activation and tactile allodynia after nerve injury. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05353.x. [DOI] [PubMed] [Google Scholar]
- 23.Laird MH, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. 2009 doi: 10.1189/jlb.1208763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao L, et al. The contributing role of CD14 in toll-like receptor 4 dependent neuropathic pain. Neuroscience. 2009;158:896–903. doi: 10.1016/j.neuroscience.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slivka PF, et al. A Peptide Antagonist of the TLR4-MD2 Interaction. Chembiochem. 2009 doi: 10.1002/cbic.200800769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchinson MR, Sholar PW, Kearney JJ, Zhang Y, Maier SF, Rice KC, Watkins LR. Evidence for a role of heat shock protein-90 (HSP90) in TLR4 mediated pain enhancement. Neuroscience. 2009 doi: 10.1016/j.neuroscience.2009.09.046. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watkins LR, et al. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Hutchinson MR, et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008 doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tawfik VL, et al. Transcriptional and translational regulation of glial activation by morphine in a rodent model of neuropathic pain. J Pharmacol Exp Ther. 2005;313:1239–1247. doi: 10.1124/jpet.104.082420. [DOI] [PubMed] [Google Scholar]
- 30.Cui Y, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 31.Hutchinson MR, et al. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast) Brain Behav Immun. 2009;23:240–250. doi: 10.1016/j.bbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston IN, et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghavendra V, et al. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–334. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- 34.Hutchinson MR, et al. Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun. 2008 doi: 10.1016/j.bbi.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui Y, et al. A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun. 2008;22:114–123. doi: 10.1016/j.bbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 36.Shavit Y, et al. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–59. doi: 10.1016/j.pain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Narita M, et al. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology. 2006;31:2476–2488. doi: 10.1038/sj.npp.1301007. [DOI] [PubMed] [Google Scholar]
- 38.Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci. 2009;29:998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takayama N, Ueda H. Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci. 2005;25:430–435. doi: 10.1523/JNEUROSCI.3170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu HE, et al. Stereoselective action of (+)-morphine over (-)-morphine in attenuating the (-)-morphine-produced antinociception via the naloxone-sensitive sigma receptor in the mouse. Eur J Pharmacol. 2007;571:145–151. doi: 10.1016/j.ejphar.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu HE, et al. Antianalgesia: stereoselective action of dextro-morphine over levo-morphine on glia in the mouse spinal cord. J Pharmacol Exp Ther. 2005;314:1101–1108. doi: 10.1124/jpet.105.087130. [DOI] [PubMed] [Google Scholar]
- 42.Wu HE, et al. dextro- and levo-morphine attenuate opioid delta and kappa receptor agonist produced analgesia in mu-opioid receptor knockout mice. Eur J Pharmacol. 2006;531:103–107. doi: 10.1016/j.ejphar.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 43.Juni A, et al. Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience. 2007;147:439–444. doi: 10.1016/j.neuroscience.2007.04.030. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson MR, et al. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. ScientificWorldJournal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hutchinson MR, et al. Novel toll-like receptor 4 activity of opioids: implications for opioid analgesia and dependence. Journal of Neuroscience. 2009 revision in review. [Google Scholar]
- 46.Lewis SS, et al. Morphine-3-glucuronide cuases enhance pain through proinflammatory glial mechanisms. Neuroscience. 2009 in review. [Google Scholar]
- 47.Noman AS, et al. Thalidomide inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production via down-regulation of MyD88 expression. Innate Immun. 2009;15:33–41. doi: 10.1177/1753425908099317. [DOI] [PubMed] [Google Scholar]
- 48.Cuschieri J, et al. Acid sphingomyelinase is required for lipid Raft TLR4 complex formation. Surg Infect (Larchmt) 2007;8:91–106. doi: 10.1089/sur.2006.050. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, et al. Morphine promotes apoptosis via TLR2, and this is negatively regulated by beta-arrestin 2. Biochem Biophys Res Commun. 2009;378:857–861. doi: 10.1016/j.bbrc.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Marik J, et al. PET of Glial Metabolism Using 2-18F-Fluoroacetate. J Nucl Med. 2009 doi: 10.2967/jnumed.108.057356. [DOI] [PubMed] [Google Scholar]
- 51.Chauveau F, et al. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009;50:468–476. doi: 10.2967/jnumed.108.058669. [DOI] [PubMed] [Google Scholar]
- 52.Banati RB, et al. Long-term trans-synaptic glial responses in the human thalamus after peripheral nerve injury. Neuroreport. 2001;12:3439–3442. doi: 10.1097/00001756-200111160-00012. [DOI] [PubMed] [Google Scholar]
- 53.Backonja MM, et al. Altered cytokine levels in the blood and cerebrospinal fluid of chronic pain patients. J Neuroimmunol. 2008;195:157–163. doi: 10.1016/j.jneuroim.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 54.Del Valle L, et al. Spinal cord histopathological alterations in a patient with longstanding complex regional pain syndrome. Brain Behav Immun. 2009;23:85–91. doi: 10.1016/j.bbi.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Alexander GM, et al. Changes in immune and glial markers in the CSF of patients with Complex Regional Pain Syndrome. Brain Behav Immun. 2007;21:668–676. doi: 10.1016/j.bbi.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 56.Weber M, et al. Increased polysialic acid neural cell adhesion molecule expression in human hippocampus of heroin addicts. Neuroscience. 2006;138:1215–1223. doi: 10.1016/j.neuroscience.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 57.Liu L, et al. Association of IL-1beta genetic polymorphisms with an increased risk of opioid and alcohol dependence. Pharmacogenetics and Genomics. 2009 doi: 10.1097/FPC.0b013e328331e68f. in review. [DOI] [PubMed] [Google Scholar]
- 58.Ledeboer A, et al. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs. 2007;16:935–950. doi: 10.1517/13543784.16.7.935. [DOI] [PubMed] [Google Scholar]
- 59.Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- 60.Si Q, et al. Differential regulation of microglial activation by propentofylline via cAMP signaling. Brain Res. 1998;812:97–104. doi: 10.1016/s0006-8993(98)00954-8. [DOI] [PubMed] [Google Scholar]
- 61.Zhang RX, et al. Interleukin 1beta facilitates bone cancer pain in rats by enhancing NMDA receptor NR-1 subunit phosphorylation. Neuroscience. 2008;154:1533–1538. doi: 10.1016/j.neuroscience.2008.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sweitzer SM, et al. Propentofylline attenuates vincristine-induced peripheral neuropathy in the rat. Neurosci Lett. 2006;400:258–261. doi: 10.1016/j.neulet.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 63.Ledeboer A, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21:686–698. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ledeboer A, et al. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol. 2006;2:279–291. doi: 10.1017/S1740925X0700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Naguib M, et al. MDA7: a novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br J Pharmacol. 2008;155:1104–1116. doi: 10.1038/bjp.2008.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cata JP, et al. The effects of thalidomide and minocycline on taxol-induced hyperalgesia in rats. Brain Res. 2008;1229:100–110. doi: 10.1016/j.brainres.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saab CY, et al. Microglia: a newly discovered role in visceral hypersensitivity? Neuron Glia Biol. 2007;2:271–277. doi: 10.1017/S1740925X07000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li A, et al. Interleukin-1ra inhibits Fos expression and hyperalgesia in rats. Neuroreport. 2007;18:495–498. doi: 10.1097/WNR.0b013e3280586839. [DOI] [PubMed] [Google Scholar]
- 69.Guo W, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Whiteside GT, et al. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur J Pharmacol. 2005;528:65–72. doi: 10.1016/j.ejphar.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 71.Rothman SM, et al. Spinal microglial proliferation is evident in a rat model of painful disc herniation both in the presence of behavioral hypersensitivity and following minocycline treatment sufficient to attenuate allodynia. J Neurosci Res. 2009;87:2709–2717. doi: 10.1002/jnr.22090. [DOI] [PubMed] [Google Scholar]
- 72.Sloane E, et al. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental Multiple Sclerosis: MOG-EAE behavioral and anatomical symptom treatment with cytokine gene therapy. Brain Behav Immun. 2009;23:92–100. doi: 10.1016/j.bbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lewis SS, et al. Constriction of the facial nerve leads to increased sensitivity to somatosensory and auditory stimuli: characterization of an inflammatory and glial component. Proc Soc Neurosci. 2009 in press. [Google Scholar]
- 74.Ledeboer A, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 75.Milligan ED, et al. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci. 2005;21:2136–2148. doi: 10.1111/j.1460-9568.2005.04057.x. [DOI] [PubMed] [Google Scholar]
- 76.Chacur M, et al. Role of spinal microglia in myositis-induced central sensitisation: An immunohistochemical and behavioural study in rats. Eur J Pain. 2008 doi: 10.1016/j.ejpain.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 77.Wolf G, et al. Interleukin-1 signaling is required for induction and maintenance of postoperative incisional pain: genetic and pharmacological studies in mice. Brain Behav Immun. 2008;22:1072–1077. doi: 10.1016/j.bbi.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 78.Lu CH, et al. Preincisional intravenous pentoxifylline attenuating perioperative cytokine response, reducing morphine consumption, and improving recovery of bowel function in patients undergoing colorectal cancer surgery. Anesth Analg. 2004;99:1465–1471. doi: 10.1213/01.ANE.0000132974.32249.C8. table of contents. [DOI] [PubMed] [Google Scholar]
- 79.Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- 80.Clark AK, et al. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. 2007;11:223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 81.Mika J, et al. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol. 2007;560:142–149. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 82.Padi SS, Kulkarni SK. Minocycline prevents the development of neuropathic pain, but not acute pain: possible anti-inflammatory and antioxidant mechanisms. Eur J Pharmacol. 2008;601:79–87. doi: 10.1016/j.ejphar.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 83.Milligan ED, et al. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol Pain. 2005;1:9. doi: 10.1186/1744-8069-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marchand F, et al. Effects of Etanercept and Minocycline in a rat model of spinal cord injury. Eur J Pain. 2009;13:673–681. doi: 10.1016/j.ejpain.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 85.Tan AM, et al. Early microglial inhibition preemptively mitigates chronic pain development after experimental spinal cord injury. J Rehabil Res Dev. 2009;46:123–133. [PubMed] [Google Scholar]
- 86.Gwak YS, et al. Propentofylline attenuates allodynia, glial activation and modulates GABAergic tone after spinal cord injury in the rat. Pain. 2008;138:410–422. doi: 10.1016/j.pain.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin CS, et al. Chronic intrathecal infusion of minocycline prevents the development of spinal-nerve ligation-induced pain in rats. Reg Anesth Pain Med. 2007;32:209–216. doi: 10.1016/j.rapm.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 88.Liu J, et al. Pentoxifylline attenuates the development of hyperalgesia in a rat model of neuropathic pain. Neurosci Lett. 2007;412:268–272. doi: 10.1016/j.neulet.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 89.Romero-Sandoval A, et al. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008;108:722–734. doi: 10.1097/ALN.0b013e318167af74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang S, et al. Regulation of the trigeminal NR1 subunit expression induced by inflammation of the temporomandibular joint region in rats. Pain. 2009;141:97–103. doi: 10.1016/j.pain.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wei T, et al. Pentoxifylline attenuates nociceptive sensitization and cytokine expression in a tibia fracture rat model of complex regional pain syndrome. Eur J Pain. 2009;13:253–262. doi: 10.1016/j.ejpain.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsai RY, et al. Ultra-low-dose naloxone restores the antinociceptive effect of morphine and suppresses spinal neuroinflammation in PTX-treated rats. Neuropsychopharmacology. 2008;33:2772–2782. doi: 10.1038/sj.npp.1301672. [DOI] [PubMed] [Google Scholar]
- 93.Mika J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol Rep. 2008;60:297–307. [PubMed] [Google Scholar]
- 94.Raghavendra V, et al. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 96.Bland ST, et al. The Glial Activation Inhibitor AV411 Reduces Morphine-Induced Nucleus Accumbens Dopamine Release. Brain Behav Immun. 2009 doi: 10.1016/j.bbi.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Narita M, et al. Implication of activated astrocytes in the development of drug dependence: differences between methamphetamine and morphine. Ann N Y Acad Sci. 2008;1141:96–104. doi: 10.1196/annals.1441.032. [DOI] [PubMed] [Google Scholar]
- 98.Mika J, et al. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav Immun. 2009;23:75–84. doi: 10.1016/j.bbi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 99.Romero-Sandoval EA, et al. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs. 2008;9:726–734. [PMC free article] [PubMed] [Google Scholar]
- 100.Inoue K, Tsuda M. Microglia and neuropathic pain. Glia. 2009 doi: 10.1002/glia.20871. in press. [DOI] [PubMed] [Google Scholar]
- 101.Raghavendra V, et al. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- 102.Wieseler-Frank J, et al. A novel immune-to-CNS communication pathway: cells of the meninges surrounding the spinal cord CSF space produce proinflammatory cytokines in response to an inflammatory stimulus. Brain Behav Immun. 2007;21:711–718. doi: 10.1016/j.bbi.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 103.Takeda M, et al. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci Biobehav Rev. 2009 doi: 10.1016/j.neubiorev.2008.12.005. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 104.Roberts J, et al. Glial activation in the rostroventromedial medulla promotes descending facilitation to mediate inflammatory hypersensitivity. Eur J Neurosci. 2009 doi: 10.1111/j.1460-9568.2009.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wieseler J, et al. A novel method for modeling facial allodynia associated with migraine in awake and freely moving rats. Journal of Neuroscience Methods. 2009 doi: 10.1016/j.jneumeth.2009.10.006. invited revision in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sloane EM, et al. Long-term control of neuropathic pain in a non-viral gene therapy paradigm. Gene Ther. 2009;16:470–475. doi: 10.1038/gt.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tawfik VL, et al. Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav Immun. 2007;21:238–246. doi: 10.1016/j.bbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 108.Shen W, et al. Inhibition of TLR activation and up-regulation of IL-1R-associated kinase-M expression by exogenous gangliosides. J Immunol. 2008;180:4425–4432. doi: 10.4049/jimmunol.180.7.4425. [DOI] [PubMed] [Google Scholar]
- 109.Yoon HJ, et al. Contribution of TLR2 to the initiation of ganglioside-triggered inflammatory signaling. Mol Cells. 2008;25:99–104. [PubMed] [Google Scholar]
- 110.Bhat RS, et al. Morphine-induced macrophage apoptosis: oxidative stress and strategies for modulation. J Leukoc Biol. 2004;75:1131–1138. doi: 10.1189/jlb.1203639. [DOI] [PubMed] [Google Scholar]
- 111.Qin L, et al. Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. Faseb J. 2005;19:550–557. doi: 10.1096/fj.04-2857com. [DOI] [PubMed] [Google Scholar]
- 112.Salvemini D, Neumann WL. Peroxynitrite: a strategic linchpin of opioid analgesic tolerance. Trends Pharmacol Sci. 2009;30:194–202. doi: 10.1016/j.tips.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 113.Ndengele MM, et al. Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther. 2009;329:64–75. doi: 10.1124/jpet.108.146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang DX, et al. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am J Physiol Heart Circ Physiol. 2003;284:H605–612. doi: 10.1152/ajpheart.00697.2002. [DOI] [PubMed] [Google Scholar]