

Abstract
Background
Oxidative stress during reperfusion of ischemia is associated with a phenotypic change in circulating monocytes from CD14++CD16- to a pro-inflammatory CD14+CD16+ subpopulation resulting in altered immunity and development of organ failure. However, the mechanism responsible remains unknown. We hypothesize that this phenotypic change, modeled by hydrogen peroxide exposure in vitro, is due to oxidative-induced intracellular calcium flux, and distinct cytoskeletal and lipid raft changes.
Methods
Peripheral blood monocytes obtained from healthy volunteers underwent 100 mM H2O2 exposure for 0-24 hours. Selected cells were pretreated with 2 μM cytochalasin D (CD), 1 μM lactrunculin A (LA) or 30 μM 1,2-Bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA) for 30 minutes. Cells underwent FACS for CD14, CD16 and cytokine expression. Cellular and lipid raft CD16 expression was determined by immunoblot and confocal microscopy.
Results
H2O2 exposed monocytes underwent a rapid time dependent increase in the surface expression of CD16 from 12.81 ± 3.53% to 37.12 ± 7.61% at 24 hours (p=0.001). Total cellular CD16 was not changed by H2O2, but an increase in lipid raft and decrease in intracellular CD16 expression was seen following H2O2 exposure. This increase in CD16 expression was associated with a 27% increase in intracellular TNF-α, an alteration in actin polymerization, and the formation of raft macrodomains. These changes induced by H2O2 were inhibited by inhibition of actin polymerization (CD and LA) and intracellular calcium flux (BAPTA).
Conclusion
This study provides the first evidence that phenotypic alterations induced by oxidative stress during reperfusion may occur as a result of changes in cytoskeletal architecture due to calcium flux that result in lipid raft alterations rather than solely from demargination and/or production of bone marrow derived CD16+ monocytes.
INTRODUCTION
Phenotypic alterations within immune cells, a condition that alters subsequent responsiveness to inflammatory stimuli, appears to be responsible for the development of acute respiratory distress syndrome (ARDS) and multiple organ dysfunction syndrome (MODS) following trauma(1-3). Development of these conditions results in significant morbidity and mortality(4-6). Although the mechanisms involved in these immune cell alterations remain ill-defined, recent work has suggested oxidative stress following ischemia/reperfusion during trauma may be directly responsible(7-9).
Central toward the alterations in cellular immunity by oxidant stress during reperfusion is the peripheral blood monocyte. This cell is critical to the regulation of innate immunity given its capability to differentiate into macrophages and dendritic cells. Although differentiation is tightly regulated, cellular differentiation is initially regulated on the phenotypic characteristics of each individual monocyte. As a result, the peripheral blood monocyte has been divided into several distinct subsets with overlapping but distinct functions. Among these subpopulations, making up about 10% of the circulating monocyte population, is the rare subset CD14+CD16+(10). The existence of this subpopulation was first described by Ziegler-Heitbrock and colleagues. They found a population distinct from the classical population of monocytes that expresses low levels of CD14 antigens and high levels of CD16 (CD14+CD16+) that was distinct from the commonly circulating strongly positive CD14 subpopulation (CD14++CD16-).
Although the CD14+CD16+ makes up only a small percentage of the circulating phenotype, recent work by several different investigators have demonstrated marked alterations in this circulating phenotype following ischemia/reperfusion induced by trauma hemorrhage(11-13). In fact, distinct expansion of this relatively rare phenotype occurs following trauma that appears to correlate with the degree of underlying injury. Additionally, enhanced expression of this population has been associated with increased morbidity during sepsis and may be associated with the development of organ dysfunction, in particular ARDS(14).
However, the mechanism in which the CD14+CD16+ population expression is altered following an inflammatory or ischemic insult has recently come into question. Originally, it was thought that disease related alteration in CD14+CD16+ expression was due increased circulating levels of this subpopulation from the marginal pool due to demargination. But recent work has suggested that exposure to a number of different inflammatory mediators, such as TGF-β and IL-10, could potential alter phenotypic expression in other cell types(15). As a result, we hypothesized that oxidant stress which characterizes ischemia/reperfusion following trauma, similar to TGF-β and IL-10, would result in a lipid raft and cytoskeletal alterations due to calcium flux leading to expansion in the CD14+CD16+ subpopulation.
MATERIALS AND METHODS
Reagents
Cytochalasin D (Sigma) was dissolved in sterile DMSO at a concentration of 2 mM. Lactrunculin A (Calbiochem, San Diego, CA) was dissolved in sterile DMSO at a concentration of 2 mM. Endotoxin contamination of cytochalasin D and lactrunculin A was tested by the Limulus Amebocyte Lysate assay (E-TOXATE Kit, Sigma) and found to be less than 0.05 ng/ml.
PBMC preparation
Venous blood (60 ml) was collected from healthy human volunteers in syringes containing 5 ml of 3.6& sodium citrate. Each aliquot of 15 ml whole blood was diluted with 20 ml of Dulbecco phosphate-buffered saline (DBPS) without calcium or magnesium (BioWhittaker, Walkersville, MD) and underlay with 15 ml of Ficoll-Paque Plus (Amersham Bioscience, Piscataway, NJ). Cells were then centrifuged at room temperature for 30 minutes at 1200 rpm, and the buffy coat resuspended in RPMI supplemented with 0.1 mg/ml of gentamicin (BioWhittaker). Cells then underwent further monocyte isolation using the MACS CD14 Microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Cells were then resuspended in RPMI-gentamicin. Following isolation, cells were then treated with various inhibitors 2 μM cytochalasin D (CD), 1 μM lactrunculin A (LA), 30 μM 1,2-Bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA) and/or hydrogen peroxide as indicated in the figure legends. Dimethyl sulfoxide (DMSO) was used as the vector control for all experiments.
For cells undergoing confocal evaluation the PBMC were allowed to adhere on tissue culture plates by incubation at 37°C for 2 hours. The cells were then rinsed 2 times with RPMI-gentamycin and incubated for 30 minutes at 37°C with RPMI-gentamicin + adult bovine serum (Hyclone, Logan, UT) prior to further treatment as indicated in the figure legends.
Fluorescence-activated cell sorter (FACS)
Peripheral blood monocytes were prepared as described earlier. Adherent cells were rinsed 2 times with 10 ml of DPBS without Ca++ or Mg++ and then with 10 ml of Versene (0.2 mg/ml ethylenediaminetetraactic acid; BioWhittaker) and incubated with 3 ml of TypLE Express (Gibco, Grand Island, NY). As soon as the cells detached, the monocytes were resuspended in 10 ml of 0.1% bovine serum albumin (BSA)-DPBS. Cells were then transferred onto ice and washed with ice cold FACS buffer (10 mM phosphate buffered saline (PBS) without Ca2+ and Mg2+, 10 mM HEPES and 0.25% BSA) before fluorescent staining with directly labeled antibodies. Additionally, cells that were to be analyzed for intracellular cytokines were permeabilized with FACS Permeablizing Solution (BD Biosciences). Cells were blocked with mouse IgG (50 mg/ml) for 10 min at room temperature then stained with a 1/10 dilution of PE-conjugated anti-mouse IgG monoclonal antibody against CD14 (BD Biosciences, Franklin Lake, NJ), FITC-conjugated anti-mouse IgG monoclonal antibody against CD16 (BD Biosciences), APC-conjugated anti-mouse IgG monoclonal antibody against TNF-α (BD Biosciences), or isotype control antibody for 45 minutes at 4°C. Cell were then washed with FACS buffer and fixed using 1 × Cellfix (BD Biosciences). Flow cytometry was performed on a dual-laser FACS Caliber (BD Biosciences) using CellQuest software (BD Biosciences). Analysis gates and quadrant markers were set to define positive and negative populations according to the nonspecific staining of isotype-matched negative controls.
Lipid raft and non-raft protein extraction
Following H2O2 stimulation, cells were lysed at 4°C in 2 ml of 1% Triton X-100 and TNE/P (25 mM Tris, 150 mM NaCl, 5mM EDTA, μM sodium orthovanadate, 100 μM DTT, 200 μM PMSF, 10 μg/ml leupeptin, 0.15 U/ml aprotinin, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 2.5 μg/ml pepstatin A, 1 mM benzamidine) for 20 minutes. Lysates were then mixed with 2.5 ml of 80% sucrose in TNE/P. Samples were then overlaid with 7 ml 35% sucrose in TNE/P and then 3 ml 5% sucrose in TNE/P. Lysates were then spun for 24 hours at 100,000 g at 4°C. The gradient was then divided into 10 fractions, with fractions 2-4 representing the lipid raft fraction and fraction 6-9 representing the non-raft fraction. Protein within the combined fractions were isolated and resuspened in 200 μl of TNE/P. Protein concentration was determined using the Pierce BCA protein assay (Pierce, Rockford, IL).
Cellular protein extraction
Following H2O2 stimulation, total cellular protein was extracted at 4°C in 500 μl of lysis buffer (20 mM Tris, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Triton X-100, μM sodium orthovanadate, 100 μM DTT, 200 μM PMSF, 10 μg/ml leupeptin, 0.15 U/ml aprotinin, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 2.5 μg/ml pepstatin A, 1 mM benzamidine, 40 mM α-glycerophosphate). Protein concentration was determined using the Pierce BCA protein assay (Pierce, Rockford, IL).
Western blots
Lipid raft, non-raft, and total cellular proteintwere electrophoresed in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to Hybond-ECL nitrocellulose membrane (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). The membrane was blocked for one hour at room temperature with 5% milk and incubated with either anti-CD14 (BD Biosciences), or anti-CD16 (BD Biosciences) for 12 hours at 4°C. Blots were then incubated in a horseradish peroxidase-conjugated secondary antibody against the primary at room temperature for 1 hour. The blot was developed using the SuperSignal chemiluminescent substrate (Pierce) and exposed on Kodak KAR-5 film (Eastman Kodak, Rochester, NY). Densitometry was performed by the NIH.image program (National Institutes of Health, Bethesda, MD) to quantitate optical density.
Immunohistochemistry
Following oxidant exposure, cells adherent to cover slips were washed with PBS. Cells were then fixed for 30 minutes with 3% paraformaldehyde in PBS with calcium and magnesium. Cells were once again washed with PBS and allowed to dry, and quenched with NH4Cl/PBS for 10 minutes at room temperature. Following this, cells were washed with PBS without Ca2+ and Mg2+, and blocked with 10% milk and 5% goat serum in PBS. Cells were then stained with rhodamine-labeled cholera toxin (1:300) for 30 minutes in order to stain lipid rafts. Cells were then washed, and incubated with rabbit anti-human CD16 antibody (BD Biosciences) for 30 minutes, washed again, and blocked with 10%milk and 5% goat serum in PBS for 10 minutes. Cells were subsequently stained with anti-rabbit FITC-conjugated antibody (Molecular Probes, Eugene, OR) at 1:500 dilution for 30 minutes that was followed by washing, fixing and examining the cells under an Axiovert 40 microscope (Carl Zeiss, Inc., Thornwood, NY).
Additionally, following oxidant exposure adherent and fixed cells as described above were permabilized with 0.1% Triton X-100. Cells were then blocked with 1% BSA, and stained with FITC-conjugated phalloidin (Molecular Probes, Eugene, OR) for 30 minutes. Cells were then washed and examined.
Cell viability and morphologic features
Representative cell populations from each condition were examined under light microscopy. No significant change was noted under any condition. Cell viability was also confirmed by trypan blue exclusion.
Statistic analysis
Values are expressed as means ± SEM. Group means are compared by unpaired Student t-tests. A probability value of 0.05 or less was considered significant.
RESULTS
CD16 surface expression is increased in response to H2O2, a condition dependent on cytoskeletal integrity
To investigate the effect of cytoskeletal stability on oxidant induced CD16 surface expression, we pretreated selected human monocytes obtained by negative selection from healthy human volunteers (Figure 1A) with either 2 μM CD, 1 μM LA or vector. Surface expression of CD16 was determined by FACS with an appropriate isotype control used to determine relative changes induced by H2O2 with/without CD or LA pretreatment (Figure 1B). A time dependent increase in CD16 surface expression was demonstrated following H2O2 exposure from a baseline of 12.81 ± 3.53% to 37.12 ± 7.61% following 24 hours of H2O2 exposure (Figure 2). This time dependent increase in CD16 expression was inhibited by both CD and LA, and maintained near baseline levels at all time points. Neither CD or LA alone had any effect on CD16 surface expression at any time (data not shown).
Figure 1. Oxidant exposure results in a time dependent surface expression of CD16 that requires maintenance of cytoskeletal integrity.

Human monocytes were stimulated with 100 mM H2O2 for 24 hours. Selected cells were pretreated with either 2 μM CD or 1 μM LA. (A) Surface expression of both CD16 was then examined under all conditions with appropriate isotype control antibody (demonstrated in grey) by FACscan.
Figure 2.
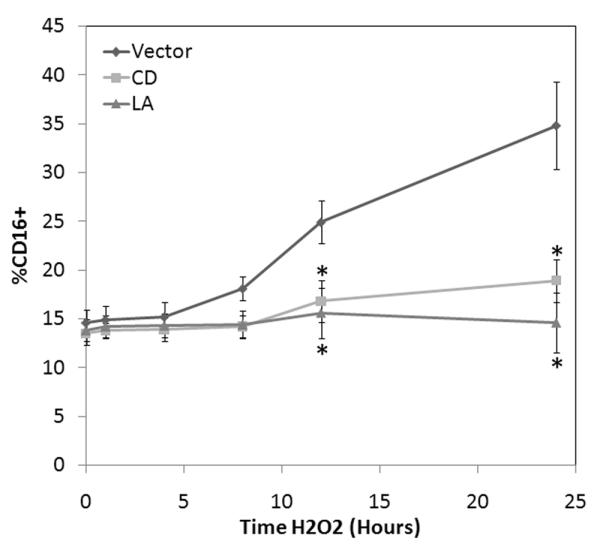
Oxidant induced CD16 surface expression is exposure dependent. To determine the rate of CD16 surface expression changes, human monocytes were stimulated with 100 mM 100 mM H2O2 for up to 24 hours. Selected cells were pretreated with either 2 μM CD or 1 μM LA. Time dependent surface expression of CD16 was determined under the various conditions by FACscan. * P <0.05 vs. time-matched control values based on 5 separately performed experiments.
In order to determine if the corresponding surface expression of CD16 was due to mobilization from an intracellular store, raft, non-raft and total protein were extracted under the same conditions (Figure 3). CD16 was found within the non-raft fraction in control cells, and was mobilized to the surface raft following H2O2 exposure. Pretreatment with either CD or LA inhibited this apparent cellular mobilization of CD16 from the non-raft fraction to the surface raft. This mobilization and association of CD16 with lipid rafts was further verified by confocal microscopy (Figure 4). As demonstrated by confocal imaging, oxidant exposure lead CD16 and lipid raft co-localization, and the coalescence of lipid rafts forming lipid raft macrodomains. The expression of CD14 within the lipid raft was not affected under any of the conditions (data not shown).
Figure 3. Oxidant induced CD16 surface expression originates from non-raft intracellular CD16.

Human monocytes were stimulated with 100 mM H2O2 for 24 hours. Selected cells were pretreated with either 2 μM CD or 1 μM LA. Lipid raft, non-raft and total protein were harvested, and analyzed by Western blot for CD16 . Constitutively found lipid raft SRC, non-raft cytoskeletal actin and cytoplasmic ERK 1 under the various extractions served as controls to access equal loading (data not shown). Representative blots from one of three separately performed experiments are shown.
Figure 4. Lipid raft mobilization of CD16.

Human monocyte colocalization of CD16 and lipid rafts was determined by immunohistochemistry. Both control and H2O2 exposed cellular expression of lipid rafts (green) and CD16 (red) were determined. Colocalization was determined by overlaying images resulting in yellow fluorescence. Data based on 5 separately performed experiments
CD16 surface expression is associated with pro-inflammatory phenotype
To determine the inflammatory nature of the CD14+CD16+ subpopulation, we examined the intracellular cytokine expression for TNF-α following H2O2 by FACS that underwent pretreatment with either 2 μM CD, 1 μM LA or vector (Figure 5). Oxidant exposure resulted in increased intracellular expression of TNF-α in both subpopulations, but to a higher degree in the CD14+CD16+ subpopulation increasing the level of TNF-α by nearly 30% over baseline. However, this increased expression was attenuated following pretreatment with either CD or LA.
Figure 5. Oxidant exposure results in enhanced intracellular expression of TNF-α within monocyte subpopulations.

Human monocytes were stimulated with 100 mM H2O2 for up to 24 hours. Selected cells were pretreated with either 2 μM CD or 1 μM LA. Intracellular TNF-a expression was then examined with appropriate isotype control antibody by FACscan. Data are expressed as percentage change in mean fluorescence intensity (ΔMFI) compared to baseline conditions (set at 100%). Significant differences: * P <0.05 vs. time-matched control values based on 6 separately performed experiments.
Oxidant induced cytoskeletal alterations are calcium induced
As a result of previous observations demonstrating oxidant induced reprogramming being calcium dependent, we set out to determine if the cytoskeletal changes induced by oxidant exposure were calcium induced(16, 17). In order to study this, we subjected cells to pretreatment with 30 μM BAPTA or vector. Stress fiber polymerization was then determined by confocal microscopy following H2O2 exposure for 30 minutes. Oxidant exposure resulted in stress fiber polymerization following H2O2 exposure, but this polymerization was inhibited by BAPTA pretreatment (Figure 6).
Figure 6. Oxidant induced cytoskeletal changes are calcium dependent.

Human monocytes were stimulated with 100 mM H2O2 for 30 minutes. Selected cells were pretreated with 30 μM BAPTA. Cells were fixed and stained for actin, and then examined by confocal microscopy. Representative images of three separately performed experiments are shown.
Oxidant induced CD16 surface expression is calcium dependent
To determine if calcium mobilization is critical to CD16 surface expression, we pretreated selected human monocytes with 30 μM BAPTA. Surface expression of both CD14 and CD16 were determined by FACS following H2O2 exposure. A time dependent increase in CD16 surface expression was demonstrated following H2O2 exposure (Figure 7). This time dependent increase in CD16 expression was inhibited by BAPTA, similar to CD and LA.
Figure 7. Oxidant induced surface expression of CD16 requires intracellular calcium flux.
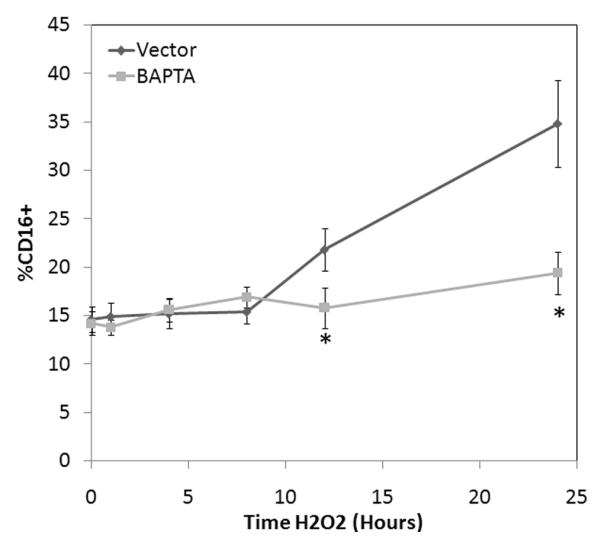
Human monocytes were stimulated with 100 mM H2O2 for 24 hours. Selected cells were pretreated with 30 μM BAPTA. Surface expression of both CD14 and CD16 was then examined under all conditions with appropriate isotype control antibody by FACscan. * P <0.05 vs. time-matched control values based on 5 separately performed experiments.
DISCUSSION
Oxidative stress generated following ischemia/reperfusion is, in part, responsible for the subsequent development of ARDS and MODS(18). Although the underlying mechanism is complex, it appears that altered monocytic phenotypes are central to this process. These altered phenotypes are associated with dysregulated responses to subsequent stimuli leading to unregulated production of various inflammatory mediators(17, 19). This dysregulated response has been modeled in vitro by a number of investigators, simulating oxidant stress by hydrogen peroxide in a similar fashion to our experiments(20, 21). In fact these previous investigations, have demonstrated enhanced liberation of various pro-inflammatory products including TNF by endotoxin if cells were exposed to as little as 100 mM of H2O2.
These studies, however, do not clearly lead to an understanding of the underlying membrane changes that occur that may be responsible for the altered phenotype. This lead us to investigate potential alterations in the surface expression of CD16 based on recent evidence has that the subpopulation expressing CD16 undergoes significant expansion under a number of inflammatory states including ischemia/reperfusion(22). The current prevailing mechanism in the expansion of this subpopulation during ischemia/reperfusion is due to a cathecolamine-dependent mobilization of these cells from the marginal pool(10). However, recent work questions this theory by demonstrating alterations in the phenotypic expression of monocytes under similar inflammatory states due to the liberation of various inflammatory mediators, such as TGF-β and IL-10(15, 23).
As a result, we set out to determine if oxidative stress a condition that occurs during reperfusion would directly result in phenotypic alterations in the peripheral blood monocytes. In a series of experiments we were able to clearly demonstrate that an increase in the CD16+ subpopulation occurs following oxidant exposure. The mechanism of the increased surface expression of CD16 did not appear to be dependent on initiation of transcription and translation of CD16, since total cellular levels of CD16 were not altered. Rather, this increase in surface expression of CD16 appeared to result from mobilization of intracellular CD16 to the surface. This data is consistent with a previous observation demonstrating similar changes in CD16 in response TGF-β in patients with rheumatoid arthritis(24). Novel to our observations is that CD16 is found within the cytosolic component, and it is this fraction which is mobilized within lipid rafts following oxidant exposure. Furthermore, we demonstrate that the integrity of the actin cytoskeleton is essential for this phenotypic alteration and this occurs with concomitant formation of lipid raft macrodomains. This was demonstrated following both CD and LA pretreatment. As a result of this pretreatment, oxidant exposure was associated with attenuated mobilization of CD16 to lipid rafts. Interestingly, this cytoskeletal redistribution of CD16 to lipid rafts is consistent with previous observation in human neutrophils treated with jasplakinolide demonstrating alteration in CD16 surface expression following jasplakinolide exposure(25).
Although this data clearly demonstrates an increase in the surface expression of CD16 on lipid rafts on the plasma membrane, it provides little insight into the potential phenotypic effects of this altered surface expression. Previously, we and others have demonstrated that following oxidant exposure cytokine expression is modified and altered following various inflammatory stimuli(9, 17). Based on a series of experiments we were able to verify these findings (data not shown), and additionally demonstrate that the CD14+CD16+ subpopulation expresses the higher levels of TNF-α following oxidant exposure. This data is consistent with the initial observations suggesting the pro-inflammatory phenotype that the CD14+CD16+ subpopulation represents(10, 26).
Demonstrating these alterations in cytoskeletal structure, lipid raft structure and CD16 surface expression, we set out to determine if our previous finding implicating calcium flux as a dependent factor in oxidant induced mononuclear cell reprogramming. In order to study this, we pretreated cells with BAPTA. This inhibition of intracellular calcium flux was associated with an inhibition of stress fiber polymerization and CD16 mobilization by oxidant exposure. Although the mechanisms in which this calcium flux is responsible for the mobilization of CD16 to lipid rafts remains unknown, it appears that cytoskeletal dependent mobilization is required(16). This requirement is consistent with previous observations in adhesion induced CD16 receptor assembly in neutrophils(25).
Although mobilization of the marginal pool in vivo may be a critical mechanism involved in ischemia/reperfusion induced CD14+CD16+ expression, our data provides insight into a potentially novel mechanism by which oxidative stress during reperfusion directly alters phenotypic expression within monocytes. The mobilization of CD16 to the membrane raft appears to require initial mobilization of intracellular calcium leading to alterations in the actin cytoskeleton which in turn leads to CD16 mobilization. This mobilization, however, does not immediately follow the initial cellular alterations, therefore suggesting other unknown mechanisms such as protein modification as a component in membrane expression. Taken together, these findings suggest potential new and novel therapeutic targets that may serve to regulate monocyte subpopulations during reperfusion.
Footnotes
Supported by National Institutes of Health Grant KO8 GM68816-05 and RO1 GM078054-01
REFERENCES
- 1.Birkenmaier C, Hong YS, Horn JK. Modulation of the endotoxin receptor (CD14) in septic patients. J Trauma. 1992;32:473–478. discussion 478-479. [PubMed] [Google Scholar]
- 2.Laudanski K, De A, Brouxhon S, Kyrkanides S, Miller-Graziano C. Abnormal PGE(2) regulation of monocyte TNF-alpha levels in trauma patients parallels development of a more macrophage-like phenotype. Shock. 2004;22:204–212. doi: 10.1097/01.shk.0000135289.62159.ad. [DOI] [PubMed] [Google Scholar]
- 3.Rosseau S, Hammerl P, Maus U, Walmrath HD, Schutte H, Grimminger F, Seeger W, Lohmeyer J. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2000;279:L25–35. doi: 10.1152/ajplung.2000.279.1.L25. [DOI] [PubMed] [Google Scholar]
- 4.Afessa B, Gajic O, Keegan MT. Severity of illness and organ failure assessment in adult intensive care units. Crit Care Clin. 2007;23:639–658. doi: 10.1016/j.ccc.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Butt I, Shrestha BM. Two-hit hypothesis and multiple organ dysfunction syndrome. JNMA J Nepal Med Assoc. 2008;47:82–85. [PubMed] [Google Scholar]
- 6.de Hemptinne Q, Remmelink M, Brimioulle S, Salmon I, Vincent JL. ARDS: A Clinicopathologic Confrontation. Chest. 2008 doi: 10.1378/chest.08-1741. [DOI] [PubMed] [Google Scholar]
- 7.Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36:691–709. doi: 10.1016/j.injury.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 8.Laplace C, Huet O, Vicaut E, Ract C, Martin L, Benhamou D, Duranteau J. Endothelial oxidative stress induced by serum from patients with severe trauma hemorrhage. Intensive Care Med. 2005;31:1174–1180. doi: 10.1007/s00134-005-2737-7. [DOI] [PubMed] [Google Scholar]
- 9.Powers KA, Szaszi K, Khadaroo RG, Tawadros PS, Marshall JC, Kapus A, Rotstein OD. Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J Exp Med. 2006;203:1951–1961. doi: 10.1084/jem.20060943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81:584–592. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 11.Frankenberger M, Passlick B, Hofer T, Siebeck M, Maier KL, Ziegler-Heitbrock LH. Immunologic characterization of normal human pleural macrophages. Am J Respir Cell Mol Biol. 2000;23:419–426. doi: 10.1165/ajrcmb.23.3.4182. [DOI] [PubMed] [Google Scholar]
- 12.Kampalath B, Cleveland RP, Chang CC, Kass L. Monocytes with altered phenotypes in posttrauma patients. Arch Pathol Lab Med. 2003;127:1580–1585. doi: 10.5858/2003-127-1580-MWAPIP. [DOI] [PubMed] [Google Scholar]
- 13.Rizoli SB, Rhind SG, Shek PN, Inaba K, Filips D, Tien H, Brenneman F, Rotstein O. The immunomodulatory effects of hypertonic saline resuscitation in patients sustaining traumatic hemorrhagic shock: a randomized, controlled, double-blinded trial. Ann Surg. 2006;243:47–57. doi: 10.1097/01.sla.0000193608.93127.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aloulou M, da Silva F. Pinheiro, Skurnik D, Benhamou M, Monteiro RC. [Deleterious role of CD16 in sepsis] Med Sci (Paris) 2008;24:231–233. doi: 10.1051/medsci/2008243231. [DOI] [PubMed] [Google Scholar]
- 15.Stec M, Weglarczyk K, Baran J, Zuba E, Mytar B, Pryjma J, Zembala M. Expansion and differentiation of CD14+CD16(-) and CD14+ +CD16+ human monocyte subsets from cord blood CD34+ hematopoietic progenitors. J Leukoc Biol. 2007;82:594–602. doi: 10.1189/jlb.0207117. [DOI] [PubMed] [Google Scholar]
- 16.Cuschieri J, Bulger E, Garcia I, Maier RV. Oxidative-induced calcium mobilization is dependent on annexin VI release from lipid rafts. Surgery. 2005;138:158–164. doi: 10.1016/j.surg.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Cuschieri J, Maier RV. Oxidative stress, lipid rafts, and macrophage reprogramming. Antioxid Redox Signal. 2007;9:1485–1497. doi: 10.1089/ars.2007.1670. [DOI] [PubMed] [Google Scholar]
- 18.Motoyama T, Okamoto K, Kukita I, Hamaguchi M, Kinoshita Y, Ogawa H. Possible role of increased oxidant stress in multiple organ failure after systemic inflammatory response syndrome. Crit Care Med. 2003;31:1048–1052. doi: 10.1097/01.CCM.0000055371.27268.36. [DOI] [PubMed] [Google Scholar]
- 19.Mendez C, Garcia I, Maier RV. Oxidants augment endotoxin-induced activation of alveolar macrophages. Shock. 1996;6:157–163. [PubMed] [Google Scholar]
- 20.Nakao N, Kurokawa T, Nonami T, Tumurkhuu G, Koide N, Yokochi T. Hydrogen peroxide induces the production of tumor necrosis factor-alpha in RAW 264. 7 macrophage cells via activation of p38 and stress-activated protein kinase. Innate Immun. 2008;14:190–196. doi: 10.1177/1753425908093932. [DOI] [PubMed] [Google Scholar]
- 21.Park SY, Ji GE, Ko YT, Jung HK, Ustunol Z, Pestka JJ. Potentiation of hydrogen peroxide, nitric oxide, and cytokine production in RAW 264.7 macrophage cells exposed to human and commercial isolates of Bifidobacterium. Int J Food Microbiol. 1999;46:231–241. doi: 10.1016/s0168-1605(98)00197-4. [DOI] [PubMed] [Google Scholar]
- 22.Sbrana S, Bevilacqua S, Buffa M, Spiller D, Parri MS, Gianetti J, De Filippis R, Clerico A. Post-reperfusion changes of monocyte function in coronary blood after extracorporeal circulation. Cytometry B Clin Cytom. 2005;65:14–21. doi: 10.1002/cyto.b.20049. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Hangoc G, Broxmeyer HE. Interleukin-10 in combination with M-CSF and IL-4 contributes to development of the rare population of CD14+CD16++ cells derived from human monocytes. Biochem Biophys Res Commun. 2004;322:637–643. doi: 10.1016/j.bbrc.2004.07.172. [DOI] [PubMed] [Google Scholar]
- 24.Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, Iwahashi M, Ueno A, Ohmoto Y, Makino H. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–2586. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- 25.Middelhoven PJ, van Buul JD, Kleijer M, Roos D, Hordijk PL. Actin polymerization induces shedding of FcgammaRIIIb (CD16) from human neutrophils. Biochem Biophys Res Commun. 1999;255:568–574. doi: 10.1006/bbrc.1999.0244. [DOI] [PubMed] [Google Scholar]
- 26.Tsujimoto H, Ono S, Hiraki S, Majima T, Kawarabayashi N, Sugasawa H, Kinoshita M, Hiraide H, Mochizuki H. Hemoperfusion with polymyxin B-immobilized fibers reduced the number of CD16+ CD14+ monocytes in patients with septic shock. J Endotoxin Res. 2004;10:229–237. doi: 10.1179/096805104225005814. [DOI] [PubMed] [Google Scholar]