

Abstract
The Meckel syndrome (MKS) is a lethal fetal disorder characterized by diffuse renal cystic dysplasia, polydactyly, a brain malformation that is usually occipital encephalocele and/or vermian agenesis, with intrahepatic biliary duct proliferation. Joubert syndrome (JBS) is a viable neurological disorder with a characteristic “molar tooth sign” (MTS) on axial images reflecting cerebellar vermian hypoplasia/dysplasia. Both conditions are classified as ciliopathies with an autosomal recessive mode of inheritance. Allelism of MS and JBS has been reported for TMEM67/MKS3, CEP290/MKS4, and RPGRIP1L/MKS5. Recently, one homozygous splice mutation with a founder effect was reported in the CC2D2A gene in Finnish fetuses with MKS, defining the 6th locus for MKS. Shortly thereafter, CC2D2A mutations were reported in JBS also. The analysis of the CC2D2A gene in our series of MKS fetuses, identified 14 novel truncating mutations in 11 cases. These results confirm the involvement of CC2D2A in MKS and reveal a major contribution of CC2D2A to the disease. We also identified three missense CC2D2A mutations in two JBS cases. Therefore and in accordance with the data reported regarding RPGRIP1L, our results indicate phenotype-genotype correlations, as missense and presumably hypomorphic mutations lead to JBS while all null alleles lead to MKS.
Keywords: Meckel-Gruber syndrome, MKS, Joubert syndrome, JBS, CC2D2A, ciliopathy
INTRODUCTION
Meckel-Gruber syndrome (MKS; MIM# 249000), is a rare autosomal recessive lethal ciliopathy characterized by central nervous system malformations (typically occipital meningoencephalocele), postaxial polydactyly, diffuse renal cystic dysplasia, intrahepatic biliary duct proliferation, and other malformations such as situs inversus, or bone anomalies. MKS is genetically heterogeneous with 6 loci and 5 genes yet identified, respectively on 17q23 (MKS1; MIM# 609883) (Kyttala et al., 2006), 8q24 (TMEM67 or MKS3; MIM# 609884) (Smith et al., 2006), 12q21(CEP290 or MKS4; MIM# 610142) (Baala et al., 2007a) 16q12 (RPGRIP1L or MKS5; MIM# 611561) (Delous et al., 2007) and most recently on 4p15 (CC2D2 or MKS6; MIM# 612013) (Tallila et al., 2008). The disease causing gene at the MKS2 locus remains unknown (Roume et al., 1998).
Joubert syndrome (JBS; MIM# 213300) is an autosomal recessive multisystem ciliopathy also, characterized by developmental delay, hypotonia, irregular breathing pattern, eye movement abnormalities (Joubert et al., 1968) and cerebellar vermis hypoplasia/dysplasia with accompanying brainstem abnormalities resulting in the radiological “molar tooth sign” (MTS) (Patel and Barkovich, 2002). Other variable features include retinal dystrophy, renal anomalies, polydactyly, liver fibrosis and occipital encephalocele, which define a group of Joubert Syndrome Related Disorders (JSRD).
JBS and MKS were shown to be allelic at 4 loci: TMEM67/MKS3/JBTS6 (Baala et al., 2007b), CEP290/MKS4/JBTS5 (Baala et al., 2007a), RPGRIP1L/MKS5/JBTS7 (Delous et al., 2007) and CC2D2A/MKS6/JBTS9 (Gorden et al., 2008; Tallila et al., 2008). In addition, mutations in 3 other genes have also been associated with JBS: AHI1 (JBTS3, 6q23.2) (Ferland et al., 2004), NPHP1 (JBTS4, 2q13) (Parisi et al., 2004), and ARL13B (JBTS8, 3q11.2) (Cantagrel et al., 2008). Two more loci JBTS1/CORS1 (Saar et al., 1999) and JBTS2/CORS2 (Keeler et al., 2003; Valente et al., 2003) map to chromosome 9q34.3 and 11p12-11q13.3 respectively.
Recently, a homozygous CC2D2A splice mutation was reported in Finnish MKS fetuses (Tallila et al., 2008) and CC2D2A mutations were also reported in patients with JBS (Gorden et al., 2008). This confirms further the allelism between these 2 disorders. As only one CC2D2A splice mutation was reported in six Finnish MKS fetuses, clearly indicating a founder effect, we decided to screen a distinct cohort of MKS fetuses and JBS cases in order to flesh out the spectrum of CC2D2A mutations in MKS and JS. In our cohort, while all 14 MKS mutations predicted null alleles (11 cases), JBS mutations were missense (3 mutations in 2 patients). Our data suggest that CC2D2A mutations are a major cause of MKS, contributing to 10 % of our cohort of 120 MKS fetuses. In addition, the data indicate phenotype-genotype correlations. Finally we investigated the expression pattern of this gene using in situ hybridization at early stages of human development. We show that CC2D2A, like other MKS and JBS genes examined to date (Kyttälä et al., 2006; Smith et al.; 2006, Baala et al., 2007b; Delous et al., 2007; Dawe et al., 2007 Talila et al., 2008; Gorden et al., 2008) is ubiquitously expressed during early human development.
MATERIALS and METHODS
Patients
Our MKS cohort is a large multiethnic series of 120 fetuses. Inclusion criteria for MKS were based on characteristic: (1) diffuse renal cystic dysplasia of the kidneys, (2) intrahepatic biliary duct proliferation and/or fibrotic changes of the liver (3) malformation of the CNS, and (4) normal karyotype based on either blood, cultured amniocytes or chorionic villi samples. In familial cases, these criteria were present in at least one sibling. Pregnancies were terminated after genetic counselling in accordance with national legislation. We also analysed the CC2D2A gene in 10 Joubert cases ascertained by a brain imaging showing a molar tooth sign, and excluding all other JBS loci. For all, chromosome analysis and clinicopathological examination were performed for at least one sibling in all cases. Informed consent was obtained for all participating families.
Genome-wide linkage screening and linkage analysis
Genome-wide homozygosity mapping was performed using 10K Affymetrix SNP arrays in 19 consanguineous MKS families, excluding all 4 known MKS genes. Data were evaluated by calculating multipoint lod scores across the whole genome using MERLIN software, assuming recessive inheritance with complete penetrance. Six were found homozygous at the CC2D2A locus. Nine other familial or consanguineous cases were tested for linkage with two intragenic microsatellite markers that were generated respectively in intron 1 and intron 3 of the gene. Primers are: CC2D2A i1-Forward: TTGTTTGTTTCCCTTCATTGC, CC2D2A-i1-Reverse: CCCAGCAAATTCTGAGCTTC; CC2D2A-i3-Forward: AGCCTAACAAATGCAGTCAT, CC2D2A-i3-Reverse: TGGAGCATATGTAGAGATCTGA.
CC2D2A mutation analysis
Genomic DNA was extracted from frozen tissue or cultured amniotic fluid cells in fetal cases and from peripheral blood samples for parents and unaffected siblings. Primers were designed in introns flanking the 36 exons (3–38 coding exons) using the “Primer 3” program (http://fokker.wi.mit.edu/primer3/input.htm) and are listed in Supp. Table S1. All PCR were all performed in the same conditions, with a touchdown protocol consisting of denaturation for 30s at 96°C, annealing for 30s at a temperature ranging from 64°C to 50°C (decreasing 1° during 14 cycles, then 20 cycles at 50°) and extension at 72°C for 30s. PCR products were treated with Exo-SAP IT (AP Biotech). Both strands were sequenced with the appropriate primer and the “BigDye” terminator cycle sequencing kit (Applied Biosystems Inc.) and analyzed on ABI3130 automated sequencers. In silico analyses were carried out using SIFT (http://blocks.fhcrc.org/sift/SIFT.html), Polyphen (http://genetics.bwh.harvard.edu/pph/), NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html) and HSF (http://www.umd.be/HSF/) online software. Segregation of the identified mutations was investigated in all family members available. Mutation numbering is based on cDNA sequence with a ‘c.’ symbol before the number, where +1 corresponds to the A of ATG translation codon (codon 1) of the cDNA reference sequences (EU450799). Mutation names were checked by the Mutalyzer programm (Wildeman et al., 2008).
Gene expression analyses using in situ hybridization
Normal human embryos and fetal tissues were obtained after elective pregnancy termination in agreement with French legislation (law no. 2004-800), National Ethics Committee recommendations (report no. 1 of May 22, 1984), and the Necker Hospital ethics committee. Embryonic stages were established according to Carnegie staging (CS) classification. Tissues were fixed in 4% phosphate buffered paraformaldehyde, dehydrated, and embedded in paraffin blocks. Five micron-thick serial sections were cut. Four different embryonic stages CS13 (gestational day 30), CS14 (d33), CS17 (d42), and CS22 (d54) were studied. Exon 17 primers were selected for PCR amplification (17F: TGACCTGCCTGATTACACACTT; 17R: CCTGATGCTCGTGTAGGTCA). A T7 promotor sequence extension (TAATACGACTCACTATAGGGAGA) was added at the end of each primer. T7F/R and F/T7R primers allowed the amplification of sense and antisense templates specific to the CC2D2A gene. Riboprobe labelling, tissue fixation, hybridisation, and developing were carried out according to standard protocols as described previously. Sections were hybridized with a digoxygenin labelled probe at 70°C overnight and digoxygenin was revealed with an anti-DIG-Fab’ antibody (Roche) at 1:1000.
RESULTS
In our cohort of 120 MKS fetuses, 28 consanguineous or multiplex MKS families excluded all 4 known genes. Linkage to CC2D2A was tested either by Affymetrix SNP 10K microarray analysis (19 cases), or by microsatellite-based linkage analysis using 2 intragenic markers (9 families). Among the consanguineous families, 7 fetuses were found homozygous at the CC2D2A locus, while in multiplex non consanguineous families, 2 showed potential linkage. CC2D2A mutational screening was performed in these 9 fetuses and in 12 additional sporadic cases in which all 4 known MKS genes were excluded by directed sequencing. This study identified a total of 11 cases with various CC2D2A mutations: 6 were homozygous mutations, 3 were compound heterozygous. In two cases, only one mutation could be identified. The molecular screening of the other sporadic cases of our cohort is ongoing, but these data already indicate that CC2D2A mutations are responsible for at least 10% of MKS cases in a multiethnic cohort. Clinical and molecular data are summarized on Figure 1 and Table 1.
Figure 1. Phenotypic features of cases with CC2D2A mutations.

Histology pattern (hematoxylin-eosin stain) of kidney (a–e), liver (f–j) and eye (k) in CC2D2A-mutated cases. Kidney histology shows conserved corticomedullary organisation with little generation of mature glomerules. Cysts are found in the deep cortex and medulla, and are smaller at the periphery than in the center (a–e). Liver histology shows portal fibrosis with important and diffuse bile duct proliferation in all cases (arrows f–j). Sagittal section of the eye in case MKS-54 (k) shows an optic nerve cystic coloboma. X-rays show femoral bowing in case MKS-143 (l) and a bell-shaped thorax in case MKS-10 (m).
Table 1.
Clinical data of Joubert patients and Meckel fetuses with CC2D2A mutations
| Fam | Case | Age * | Origin | C** | Kidney | Liver | Ocular | Central Nervous System | PD | Other | Nucleotide alterations | Exon | Predicted effect on protein | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JBS | 1 | JBS-006 | 4 | Algeria | no | - | - | OMA, myopia | AT, HT, CVA, MR | - | - | c.2161C>T hmz | 18 | p.Pro721Ser |
| 2 | JBS-008 | 19 | France | no | - | - | OMA, AEM, ERG & optic disc | AT, HT, MR MTS |
- | - | c.3341C>T htz c.4667A>T htz |
27 37 |
p.Thr1114Met p.Asp1556Val |
|
| MKS | 1 | MKS-84A | 33w | Algeria | yes | MKS | - | « Dandy-Walker » | - | - | No DNA | |||
| MKS-84B | 21w | MKS | - | Coloboma | Occipital defect | - | No DNA | |||||||
| MKS-84C | 19w | MKS | - | Retinal cyst | OM, Arh | U L | Epididymal cysts | No DNA | ||||||
| MKS-84D | 21w | MKS | HF, BDP | OM « Dandy-Walker » |
- | Left heart hypoplasia femoral bowing | c.3399_3975del hmz | 28–31 | p.Ala1134AsnfsX9 | |||||
| 2 | MKS-54 | 17,5w | Mauritania | yes | MKS | BDP | Optic nerve dysplasia | OE | 4 L | IUGR Cleft palate |
c.517C>T hmz | 8 | p.Arg173X | |
| 3 | MKS-160 | 23w | France | yes | MKS | ? - | OE | U L | c.3399-3C>A hmz | IVS27 | potential missplicing | |||
| 4 | MKS-410 | 23w | Turkey | yes | MKS | BDP | Normal | OE, microcephaly | 4 L | Cleft palate, bifide tongue, epididymal & pancreatic cysts | ||||
| MKS-413 | 11,5w | MKS | BDP | ND | OE, microcephaly | 4L | ND | |||||||
| MKS-414 | 12,5w | MKS | ? | Coloboma | OE, microcephaly | 4L | Cleft palate | c.3584delT, hmz | 29 | p.Phe1195SerfsX11 | ||||
| 5 | MKS-977 | 34w | Turkey | yes | CK | ND | No autopsy | OE | - | ND | c.3084delG hmz | 25 | p.Lys1029ArgfsX3 | |
| 6 | MKS-010 | 19w | Guadeloup | no | MKS | BDP | OM, CVA | U L | Bell chest | |||||
| MKS-011 | 14w | MKS | BDP | OM | U L | c.3145C>G, hmz | 25 | p.Arg1049X | ||||||
| 7 | MKS-142 | 25w | France | no | MKS | ? | OM, arh, CCA, CVA | 4 L | Bicornuate Uterus | |||||
| MKS-143 | 25w | MKS | ? | OM, Arh, CCA | 4 L | Cleft palate, micropenis, femoral bowing | c.3522_3523insTG htz c.4496+2 T>A htz |
29 IVS36 |
p.His1175CysfsX13 potential missplicing |
|||||
| 8 | MKS-982 | 13w | France | no | MKS | BDP | Vertex M anencephaly |
4 L | Cleft palate | c.1538T>A htz c.4179+1delG htz |
15 IVS33 |
p.Trp513X, pat potential missplicing, mat |
||
| 9 | MKS-362 | 20w | France | no | MKS | BPD | OM | U L | - | No DNA | ||||
| MKS-363 | 13w | ? | ? | OM | ? | No DNA | ||||||||
| MKS-364 | 13W | ? | ? | ? | U L | No DNA | ||||||||
| MKS-365 | 16w | MKS | BDP | OM | yes | c.2673C>T htz c.2486+1G>C htz |
22 IVS20 |
p.Arg925X potential missplicing |
||||||
| 10 | MKS-987 | 15w | no | MKS | BDP, HF | - | OE, CCA, Arh, CVA, HH | 4 L | Epididymal cysts | c.1339delG htz | 13 | p.Ala447ArgfsX11 | ||
| 11 | MKS-692 | 20w | USA | no | MKS | BDP | « Dandy-Walker » | 4 L | Hypospadias, ulnar bowing, accessory spleen, gonadal dysgenesis | c.834delG htz | 10 | p.Leu279CysfsX40 | ||
Fetal cases : age is given in gestational weeks.
C: consanguinity. AEM: abnormal eye movements, Arh: arhinencephaly, AT : ataxia, BDP : bile duct proliferation of liver, CCA: corpus callosum agenesis. CVA : cerebellar vemis agenesis, ERG: electroretinogramm, HH: hypothalamic hamrtoma, HF : hepatic fibrosis, HT : hypotonia, IUGR: intrauterine growth retardation, MKS: cystic kidneys characteristic of Meckel syndrome, MR : mental retardation, MTS : molar tooth sign, ND: non determined, OE: occipital encephalocele, OM: occipital meningocele, OMA : oculomotor apraxia, PD: polydactily, UL : upper limbs. Mutation numbering is based on cDNA reference sequences (EU450799)
In MKS-84D case (family 1), we failed to amplify exons 28 to 31 of the CC2D2A gene. This suggests that the fetus was carrying a homozygous deletion of 4 exons. In order to confirm the deletion, three multiplex PCRs were then performed including exon 13 (as an internal control) and each of the exons 28 to 30 (Figure 2). While the normal control showed 2 bands, only exon 13 was amplified in fetus MKS-084D. A simplex PCR is shown for exon 31 (no amplification, while exons 27 (not shown) and 32 amplified correctly. This confirms that this fetus carries a homozygous exons 28 to 31 intragenic deletion.
Figure 2. PCR analysis of fetus MKS-084D.

Multiplex PCR were performed with primers that amplify exon 13 (expected size : 647pb) and exons 28 (484pb), 29 (384pb), 30 (482pb) respectively. Both exons are amplificated with a control DNA (C+), but only exon 13 for the MKS-084D DNA confirming the deletion of exons 28 to 30. For exon 31 (687bp) and 32 (355bp) a simplex PCR is shown. MKS84D fails to amplify exon 31 but correctly amplifies exon 32. C− is a negative control (no DNA in reaction).
In families 2–5, originating respectively from Mauritania (family 2), France (family 3), and Turkey (families 4 and 5), parents were related and fetuses were homozygous at the locus. Consistent with the linkage data, we identified a homozygous mutation in each of these families: c.517C>T/p.Arg173X; c.3399-3C>A; c.3584delT/p.Phe1195SerfsX11 and c.3084delG/p.Lys1029ArgfsX3. All mutations predicted a truncated protein, and segregated with the disease in multiplex families. In MKS-160, with the homozygous c.3399-3C>A mutation in intron 27, no cDNA was available from the fetus to confirm the effect on splicing of this mutation. In silico analysis of the splice site sequences by HSF showed a reduction in efficacy of the acceptor splice site (89% to 80%), while the mutation completely suppressed the same splice site using the NNSPLICE algorithm.
Interestingly, in family 6 Guadeloupian parents were unaware to be related. Linkage analysis showed homozygosity for the intragenic polymorphic markers in the 2 fetuses, and indeed a c.3145C>G, p.Arg1049X homozygous mutation was identified in this family, suggesting a founder effect.
Three French cases were compound heterozygous for CC2D2A mutations (families 7–9). In family 7 with two affected fetus (twins), both had characteristic Meckel syndrome. In this family, 2 mutations predicting truncated proteins were identified: a 2 bases insertion in exon 29 leading to a frameshift c.3522_3523insTG/p.His1175CysfsX13 and a splice mutation at the donor splice site of intron 36: c.4496+2T>A. In family 8, MKS-982 was diagnosed at the first 12w ultrasound, and the brain was severely affected as a vertex defect resulted in an anencephaly phenotype. The fetus carried compound heterozygous mutations with a nonsens paternally inherited mutation c.1538T>A/p.Trp513X in exon 15, and a maternal donor splice site mutation c.4179+1delG in intron 33. In family 9, 4 fetuses were affected with MKS but DNA was available for only one. In this case also, 2 mutations: a nonsens c.2673C>T/p.Arg925X and a donor splice site c.2486+1G>C were found.
In two sporadic cases, only one CC2D2A mutation could be identified (families 10–11). Altogether, the mutational screening in our MKS cohort identified 11 families carrying CC2D2A mutations. All predict null alleles. These data confirm that the CC2D2A gene is a MKS-causing gene, and shows a major contribution to the disorder of at least 10%. In addition, it suggests that truncating alleles lead to this severe MKS phenotype. Clinical findings of MKS cases are summarized in Table 1. Interestingly, polydactyly is common in CC2D2A-mutated fetuses (16/21), and femoral bowing is noted in at least 2 cases (Figure 1-l). Cerebral malformations belong to the spectrum of neural tube defects (encephalocele and meningocele) and “Dandy Walker” malformations. Anencephalic phenotype secondary to an upper encephalocele (vertex) was found in one case. Eyes were involved in 3 examined cases and showed optic nerve coloboma (Figure 1-k). Interestingly, both twin fetuses fetuses of family 7 had arhinencephaly and corpus callosum agenesis in addition to the occipital meningocele.
Then, we analysed CC2D2A in a series of 10 JBS patients excluding other known JBS loci either by the analysis of polymorphic markers in familial or consanguineous cases (8) and/or by direct sequencing of the known genes in 2 sporadic cases. The CC2D2A intragenic polymorphic markers were analysed in the 6 consanguineous cases (one was homozygous), and in 2 families where unaffected sibs were available for linkage analysis (the 2 did not exclude linkage). These 3 families and 2 sporadic cases were then sequenced, and CC2D2A mutations were identified in 2 families.
In family 1, the patient was homozygous at the CC2D2A locus while his healthy brothers were heterozygous. JBS-006 is an 8 year-old male born to parents originating from the same village in Algeria and unaware of being related. He presented mental retardation, hypotonia with ataxia, an oculomotor apraxia, vermian agenesis giving rise to a radiological molar tooth sign (Figure 3). Two homozygous changes were identified: one in exon 18, p.Pro721Ser and one in exon 20, p.Lys800Glu. The amino acid Lys800 is not conserved and the variation is predicted as benign according to Polyphen software, while the amino acid Pro721 is evolutionary conserved; Polyphen predicts that the Pro721Ser change is probably damaging (PSIC score > 2). This might be due to the fact that this conserved amino acid is close to the C2 domain of the CC2D2A protein and this change might interfere with the currently little-known function of CC2D2A or protein-protein interactions.
Figure 3. Brain MRI of a normal control (C), 6 month-old affected boy (JBS-006) and 13 year-old affected boy (JBS-008) carrying CC2D2A mutations.
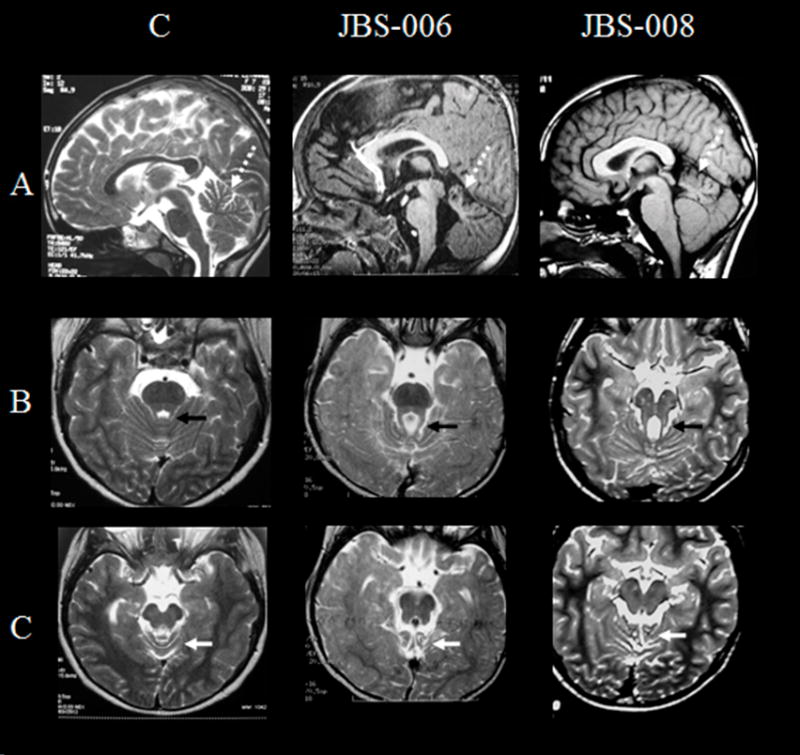
A: Sagittal images show in both affected patients a superior vermian dysgenesis. The middle and inferior segments of the vermis are hypoplastic or absent.
B: Axial T2-weighted FSE images demonstrate the abnormally thickened and elongated superior cerebellar peduncles and the molar tooth sign in all affected patients (black arrow)
C: Axial T2-weighted FSE images show superior vermian dysgenesis (white arrow) at the level of the cerebral peduncles in all affected patients.
The second patient (case JBS-008) is a sporadic case who presented mild mental retardation, hypotonia, ataxia, oculomotor apraxia, abnormal eye movements and a “molar tooth” sign (Figure 3). We identified two heterozygous missense mutations: one in exon 27, p.Thr1114Met inherited from his mother, and another in exon 37, p.Asp1556Val inherited from his father. These two changes are not known to be single nucleotide polymorphisms (SNPs), and they were not observed 250 control chromosomes. In silico analysis of the two amino acid substitutions on the protein structure and function using the Polyphen software predicts that these changes are deleterious, with a high PSIC score equal to 1.9 for the former and greater than 2 for the latter (Ramensky et al., 2002). These data further demonstrate that mutations of the CC2D2A gene are causing JBS, defined as JBTS9.
In order to assess the role of CC2D2A in these disorders, we investigated the pattern of gene expression at 4 different stages of early human development. At CS13 (Figure 4A, A′) and CS14 (not shown) CC2D2A is ubiquitously expressed, with a distinct signal in the spinal cord and limb buds. At CS17 (d, Figure 4B–D′), CC2D2A transcripts continue to be widely expressed in particular throughout the central nervous system, lung and digestive tract epithelia (Figure 4D,D′). At C22 (day, Figure 4E–G′), expression continues to be intense within the CNS, where strong and specific expression is observed in the eye and in external granular layer of cerebellum (Fig 4F,F′). CC2D2A expression is also observed in the costal perichondrum (Fig 4G,G′).
Figure 4. In situ hybridization studies.

In situ hybridization of the CC2D2A gene during human embryonic development. Adjacent sections were treated with antisense (A–G) or with control (sense, A′–G′) riboprobes. Hybridization with sense probes did not give any signal. CC2D2A expression is found in multiple organs during development including brain, upper limbs and kidneys. A-A′). A-A′: at Carnegie stage (CS)13, after 4 weeks of development, CC2D2A is ubiquitously expressed within embryonic tissues in transverse section, including spinal cord (sc), limb bud (lb) and mesonephros (Mn). By CS17 (41d, B–D′), the staining was slightly more intense within the entire central nervous system (prosencephalon (pr), mesencephalon (mes), rhombencephalon (rh), spinal cord) and the peripheral dorsal root ganglia (drg). At CS22 (54d, E–G′), CC2D2A is still ubiquitously expressed with a stronger signal in the cerebellar primordium (cb) and costal perichondrum (co). ey: eye; H: heart; Li: liver; Md: mandible, Rp: Rathke poutch; T: trachea; To: tongue, v: vertebrae.
DISCUSSION
The identification of CC2D2A mutations in 11 MKS fetuses confirms that CC2D2A as a gene for MKS and adds a major contribution of this gene to the disease reaching up to 10% of cases. In addition, novel mutations in two JBS cases are reported, confirming CC2D2A as the 9th JBS locus (JBTS9). Moreover, a genotype-phenotype correlation was observed (Supp. Figure S1). All mutations found in the MKS in this study (14) and the previously reported Finnish mutation (Tallila et al., 2008) predicted null alleles (15/15 mutations). By contrast, all but two patients with JBS and CC2D2A mutations reported here and in previous studies (Gorden et al., 2008; Noor et al., 2008) have at least one missense mutation (10/12). Indeed only the homozygous stop mutation described by Gorden et al. and the homozygous splice site mutation reported by Noor et al. in JBS patients do not fit with this correlation. The former one might be explained by the exon skipping caused by a nonsens mutation. If this is the case, exon 23 in wich the nonsense mutation is located is in frame (93bp), and the exon skipping would give rise to a protein lacking 31 aminoacids outside the functional C2 and coiled coil domains. The effect of the mutation on cDNA should be tested on the cDNA of the patient. However, this hypothesis can not be applied to the homozygous splice site mutation reported by Noor as it results in exon19 skipping that is not inframe and disturb the reading frame predicting a premature stop codon. One can hypothesis that while this mutation retains RNA splicing and some residual CC2D2A function, MKS mutations could lead to nonsens mediated decay or other post transcriptional effects. Another explanation would be other factors, such as extragenic modifiers modulating the phenotype as discussed below.
Phenotypic overlap between JBS and MKS has been reported for TMEM67, CEP290 and RPGRIP1L genes and is also illustrated by the considerable phenotypic variability observed in JBTS2-linked patients with multsystemic involvement, including occipital encephalocele, polydactyly, microphtalmia, and kidney disease (Keeler et al., 2003; Valente et al., 2003). Including CC2D2A, four genes have been shown to be involved in JBS as well as MKS. It is likely that the allelic nature of these conditions will extend to the MKS2 locus, which has been mapped to chromosome 11q13 (Roume et al., 1998) and may therefore be allelic to JBTS2, which has been mapped to 11p12-11q13.3. With respect to RPGRIP1L, mutations identified in MKS fetuses are truncating mutations on both alleles, while most JBS patients harbour at least one missense mutation. “Hypomorphic mutations leading to a viable phenotype were also observed for the MKS1 gene in Bardet Biedl syndrome”. While all reported mutations in MKS1 are truncating/splice mutations in MKS fetuses, two missense mutations were reported in a patient diagnosed with Bardet-Biedl syndrome (Leitch et al., 2008), suggesting that only hypomorphic mutations in the MKS1 gene may lead to another viable, postnatal ciliopathy.
In a recent study, we analysed MKS1 and MKS3 (TMEM67) genes in 54 MKS fetuses and we were able to establish phenotype/genotype correlations according to the mutated gene: whilst the occipital encephalocele is constant in MKS1-mutated fetuses, vermian hypoplasia without neural tube defects is observed in some MKS3 cases whereas polydactyly is significantly less frequent in MKS3-mutated cases (Khaddour et al., 2007). Taking into account affected siblings, clinical data of 21 fetuses with CC2D2A mutations are summarized in Table 1. All had characteristic kidney histological features of MKS. Intrahepatic biliary duct proliferation was absent in only fetuses from family 1. Skeletal anomalies including polydactyly and bone dysplasia were frequent: 16/21 fetuses presented polydactyly, 2 fetuses femoral bowing (Figure 1-l) and one had a bell-shaped chest (Figure 1-m). Interestingly, in one case, NTD affects the calvarium leading to a rostrally located encephalocele, resulting in anencephalic phenotype. This rare feature was also observed in all RPGRIP1L mutated fetuses reported thus far (Delous et al., 2007).
In 2 MKS families (10 and 11), we found only one truncating mutation. One possibility is that we failed to find the second molecular CC2D2A event because it is located in the regulatory regions of CC2D2A, or in the intron. A heterozygous intragenic deletion would also be missed by PCR-sequencing and quantitative PCR or RNA analysis could give the clue. It is also possible that two pathogenic mutations will be found in another gene (not yet identified), and that the CC2D2A mutation is acting as a modifier in an oligogenic context. By contrast in one JBS case (JBS-006), two homozygous missense mutations were identified and no one is a known SNP: p.Lys800Glu and p.Pro721Ser. In silico analysis predict that the p.Pro721Ser is the pathogenic mutation because it concerns a conserved aminoacid, located in addition close to the C2 domain of the protein. The possibility that the disease is caused by the combined effect of the 2 identified variants is also a possibility.
As far as phenotype/genotype correlation is concerned, JBS shows highly variable expressivity and genotype-phenotype correlations allowed prioritizing disease genes to be tested in each patient (Valente et al., 2008). For exemple, AHI1 mutations usually lead to JBS with retinal involvement and sometimes polymicrogyria (Dixon-Salazar et al., 2004). Only few JBS patients have kidney involvement (Utsch et al., 2006). Also, NPHP1 is responsible for isolated NPHP in the vast majority of cases, and only few JBS patients with a mild form of brain malformation were found to carry the NPHP1 deletion (Caridi et al., 2006; Parisi et al., 2004). One can hypothesize that oligogenic or epistatic inheritance is responsible for the additional signs found in these rare patients. Such evidence of oligogenic inheritance has been given in nephronophtisis (Tory et al., 2007; Hoefele et al., 2007). Clinical variability reflected by modifier genes, was also recently demonstrated for RPGRIP1L and the retinal phenotypes (Khanna et al., 2009). In the present study, both CC2D2A-mutated patients had oculomotor apraxia (Cogan syndrome), but neither retinal nor kidney involvement respectively at the age of 8 and 19 years. In a previous report, ten JBS patients with MTS and CC2D2A mutations were reported (Gorden et al., 2008). Most had also abnormal eye movement (7/10 cases), but only two of them showed retinal dystrophy, renal disease was also infrequent (3/10).
Genes involved in NPHP/JBS and MKS encode proteins involved in ciliary function. These syndromes and similar conditions such as Bardet-Biedl syndrome are collectively known as ciliopathies (Badano et al., 2006). Little is known about the function of CC2D2A. We show that CC2D2A is ubiquitously expressed during early human development. These results are consistent with data in adult tissues of Noor et al. who showed using RT-PCR that the CC2D2A mRNA is present in many tissues (Noor et al., 2008). Recently, using a yeast two-hybrid assay it was shown that CC2D2A interacts with CEP290/NPHP6 (Gorden et al., 2008). Among several domains tested for affinity with other ciliopathy proteins, the N-term domain (amino acids 1-998) of CC2D2A is responsible for the CEP290/NPHP6 interaction. Interestingly, the homozygous mutation found in patient JBS-06 affects Pro721, located near the C2 domain of the CC2D2A protein and within this CEP290-interacting domain.
In conclusion, the present study confirms the involvement of CC2D2A in MKS and shows that CC2D2A is mutated in patients throughout the world.
Our data also clearly indicates a correlation between the phenotype and the genotype, as null alleles lead to all MKS cases, while a missense/hypomorphic mutation in at least one allele is found in all but two JBS cases reported to date.
Supplementary Material
Acknowledgments
We thank the patients and their families for participation. We thank Pr. Valerie Cormier-Daire for helpful discussion, Dr Daniela Buzas for referring cases, Dr. Heather Etchevers for editing and the SOFFOET for clinical data and material support. This work was supported by grants from ANR (ANR-06-MRAR-034-02), NIH “Hereditary Basis of Neural Tube Defects” N° NS039818-07 to Marcy Speer, and Telethon Italy (GGP08145) to EMV. SZ is supported by INSERM-DGRSRT (CS/RN/2008 n°56 and CS/RN/2008 n°87).
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge MC, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrere AM, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler MC, Genin E, Johnson CA, Vekemans M, Encha-Razavi F, Attie-Bitach T. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007a;81(1):170–9. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007b;80(1):186–94. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attie-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM, Woods CG, Gleeson JG International Joubert Syndrome Related Disorders Study Group. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83(2):170–9. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caridi G, Dagnino M, Rossi A, Valente EM, Bertini E, Fazzi E, Emma F, Murer L, Verrina E, Ghiggeri GM. Nephronophthisis type 1 deletion syndrome with neurological symptoms: prevalence and significance of the association. Kidney Int. 2006;70(7):1342–7. doi: 10.1038/sj.ki.5001768. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16(2):173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U, Schneider-Maunoury S, Attie-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39(7):875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75(6):979–87. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36(9):1008–13. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topçu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D. CC2D2A Is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83(5):559–71. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, Attanasio M, Utsch B, Antignac C, Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18(10):2789–95. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology. 1968;18(3):302–3. [PubMed] [Google Scholar]
- Keeler LC, Marsh SE, Leeflang EP, Woods CG, Sztriha L, Al-Gazali L, Gururaj A, Gleeson JG. Linkage analysis in families with Joubert syndrome plus oculo-renal involvement identifies the CORS2 locus on chromosome 11p12-q13.3. Am J Hum Genet. 2003;73(3):656–62. doi: 10.1086/378206. Epub 2003 Aug 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaddour R, Smith U, Baala L, Martinovic J, Clavering D, Shaffiq R, Ozilou C, Cullinane A, Kyttälä M, Shalev S, Audollent S, d’Humières C, Kadhom N, Esculpavit C, Viot G, Boone C, Oien C, Encha-Razavi F, Batman PA, Bennett CP, Woods CG, Roume J, Lyonnet S, Génin E, Le Merrer M, Munnich A, Gubler MC, Cox P, Macdonald F, Vekemans M, Johnson CA, Attié-Bitach T SOFFOET (Société Française de Foetopathologie) Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: a genotype-phenotype correlation. Mutation in brief #960. Online. Hum Mutat. 2007;28(5):523–4. doi: 10.1002/humu.9489. [DOI] [PubMed] [Google Scholar]
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, Macdonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attie-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, Gibbs RA, Koenekoop RK, Swaroop A, Katsanis N. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41(6):739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38(2):155–7. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40(4):443–8. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- Noor A, Windpassinger C, Patel M, Stachowiak B, Mikhailov A, Azam M, Irfan M, Siddiqui ZK, Naeem F, Paterson AD, Lutfullah M, Vincent JB, Ayub M. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am J Hum Genet. 2008;82(4):1011–8. doi: 10.1016/j.ajhg.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75(1):82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Barkovich AJ. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002;23(7):1074–87. [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roume J, Genin E, Cormier-Daire V, Ma HW, Mehaye B, Attie T, Razavi-Encha F, Fallet-Bianco C, Buenerd A, Clerget-Darpoux F, Munnich A, Le Merrer M. A gene for Meckel syndrome maps to chromosome 11q13. Am J Hum Genet. 1998;63(4):1095–101. doi: 10.1086/302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar K, Al-Gazali L, Sztriha L, Rueschendorf F, Nur EKM, Reis A, Bayoumi R. Homozygosity mapping in families with Joubert syndrome identifies a locus on chromosome 9q34.3 and evidence for genetic heterogeneity. Am J Hum Genet. 1999;65(6):1666–71. doi: 10.1086/302655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH, Harris PC, Johnson CA. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38(2):191–6. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. 2008;82(6):1361–7. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, Antignac C, Salomon R, Saunier S. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18(5):1566–75. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, Hennies HC, Otto EA, Hildebrandt F. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21(1):32–5. doi: 10.1007/s00467-005-2054-y. [DOI] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008;51(1):1–23. doi: 10.1016/j.ejmg.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Valente EM, Salpietro DC, Brancati F, Bertini E, Galluccio T, Tortorella G, Briuglia S, Dallapiccola B. Description, nomenclature, and mapping of a novel cerebello-renal syndrome with the molar tooth malformation. Am J Hum Genet. 2003;73(3):663–70. doi: 10.1086/378241. Epub 2003 Aug 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PEM. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature Checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.