

Abstract
A general method has been developed for in situ trapping of arylmetal intermediates by halogen, sulfur, ketone, and aldehyde electrophiles affording the functionalization of the most acidic position in arene. Pentafluorobenzene, benzothiazole, and benzoxazole can be functionalized by using K3PO4 base. For less acidic arenes, tBuOLi base is required. Arenes with DMSO pKa’s of 35 or less are reactive.
1. Introduction
Arene deprotonation followed by quench with electrophiles is a powerful method for introducing functionality in organic molecules (Scheme 1A). Typically, such reactions require cryogenic conditions and limited functional group tolerance is observed due to the possibility of aryl(alkyl)lithium reaction with electrophilic groups in arene molecule. Alternatively, in situ trapping techniques allow simultaneous presence of base and electrophile in the reaction mixture (Scheme 1B).1 This concept was developed by Martin for arene deprotonation/silylation sequences.1a,b LiTMP/TMSCl mixtures were employed for functionalization of aromatic nitriles, esters, and pyridines. Low concentrations of intermediate aryllithium species result in increased functional group tolerance. Other examples of in situ trapping sequences include trifluorobenzene metalation by tBuLi in the presence of TMSCl that has been described by Gilman and reinvestigated by Schlosser1c,d as well as nitrofluorobenzene, N-Boc-indole, and substituted bromobenzene lithiation/silylation.1e,f,g Exhaustive bromination of cyclopentadiene by a mixture of KOH/KOBr was reported in 1930.1h Bunnett has shown that tribromobenzene can be brominated by treatment with a mixture of tBuOK and tBuOBr.1i Several groups have demonstrated that in situ metalation/boronation sequences are successful for protected indoles, neopentyl benzoates, benzonitriles, halobenzenes, and pyridylamides.1f,2 Metalation of pentafluorobenzene by KOH/liquid NH3 in the presence of methyl iodide results in the formation of pentafluorotoluene.3 Organic bases have also been used for in situ generation/trapping of aryl anions. 2-Acylimidazoles have been prepared by the reaction of imidazoles and benzimidazoles with acid chlorides in the presence of triethylamine base.4 Presumably, N-acylation is followed by deprotonation of the acidic C-2 proton and subsequent acyl migration affords the final product. Silylations under similar conditions have also been successful.5 Finally, Kondo has shown that electron-rich and electron-deficient arenes can be functionalized by employing tBu-P4 base/non-enolizable ketone and aldehyde mixtures.6
Scheme 1.

Arene Deprotonation/Electrophilic Quench
We have recently developed a method for copper-catalyzed arylation of sp2 C-H bonds possessing DMSO pKa’s below 35.7a The reaction was shown to proceed via an aryllithium or arylpotassium intermediate formed in situ by the reaction of the arene with either K3PO4 or tBuOLi metalating agent (Scheme 2A). It occurred to us that electrophilic trapping of arylmetals formed under the copper-catalyzed arylation conditions may result in a convenient method for functionalization of acidic arenes (Scheme 2B). Preliminary results describing arene in situ halogenation were recently disclosed.7b We report here a method for arylmetal in situ trapping with halogen, sulfur, and carbonyl electrophiles.
Scheme 2.

Deprotonative Functionalization of Arene Carbon-Hydrogen Bonds
2. Results and Discussion
2.1 Halogenation
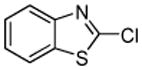
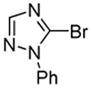
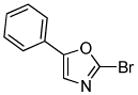
Minor modifications of the copper-catalyzed arylation conditions7b allowed the development of an efficient process for in situ sp2 C-H bond halogenation. The reactions are carried out in DMF or mixed DMF/xylenes solvent at 50–130 °C (Table 1). Halogen electrophile needs to be optimized for each reaction. Carbon tetrachloride and tetrabromide (entries 1, 2, 6, 7, 9, 10) as well as dibromotetrafluoroethane (entries 3–5), iodine (entries 11–12, 15), and ICl (entries 8, 13, 14) can be employed. Electron rich heterocycles such as benzothiazole (entries 1, 2), N-phenyltriazole (entry 3), caffeine (entry 4), and 5-phenyloxazole (entry 5) are halogenated in the most acidic positions.8 Electron-poor pyridine oxides are also halogenated under the optimized conditions (entries 6 and 7). For pyridine oxide reaction with carbon tetrabromide, dibromination-deoxygenation product is obtained in 30% yield.
Table 1.
In Situ Halogenation
 | ||||
|---|---|---|---|---|
| reagent | ||||
| entry | arene | base | product | yield |
| 1 | 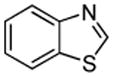 |
CCl4 tBuOLi |
 |
80% |
| 2 |  |
CBr4 K3Po4 |
 |
82% |
| 3 |  |
(BrCF2)2 tBuOLi |
 |
77% |
| 4 | 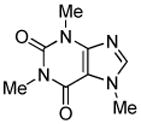 |
(BrCF2)2 tBuOLi |
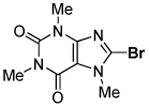 |
65% |
| 5 | 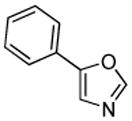 |
(BrCF2)2 tBuOLi |
 |
80% |
| 6 | 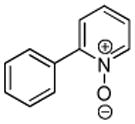 |
CBr4 tBuOLi |
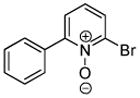 |
56% |
| 7b | 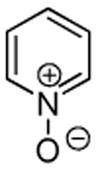 |
CBr4 tBuOLi |
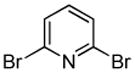 |
30% |
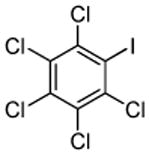
| 8 | C6Cl5H | ICl tBuOLi |
 |
90% |
| 9 |  |
CBr4 tBuOLi |
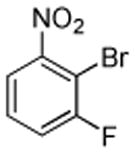 |
55% |
| 10 | 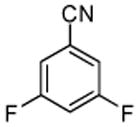 |
CBr4 tBuOLi |
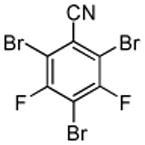 |
40% |
| 11 | C6F5H | I2 K3Po4 |
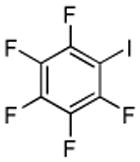 |
85% |
| 12 | 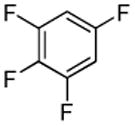 |
I2 tBuOLi |
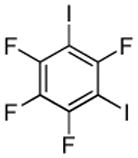 |
95% |
| 13c | 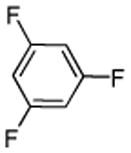 |
ICl tBuOLi |
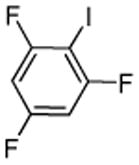 |
58% |
| 14d | 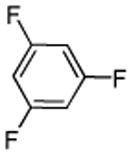 |
ICl tBuOLi |
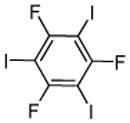 |
32% |
| 15 |  |
I2 tBuOLi |
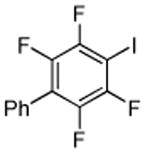 |
97% |
Substrate (1 equiv), base (2.0–4.0 equiv), halogenating reagent (1.0–3.0 equiv). Yields are isolated yields. See the Supporting Information for details.
m-Xylene solvent.
1,3,5-Trifluorobenzene (3 equiv), ICl (1 equiv).
1,3,5-Trifluorobenzene (1 equiv), ICl (3 equiv).
Halogenation of electron-poor arenes is also successful. Typically, such reactions require either harsh electrophilic conditions or use of strong alkylithium bases and cryogenic conditions.9 By employing in situ trapping conditions, either tBuOLi or weak K3PO4 bases can be used under relatively mild temperatures (50–130 °C). Polychloro- and polyfluoroarenes can be halogenated in moderate to high yields (entries 8, 11–15). Cyano- (entry 10) and nitro (entry 9) groups are tolerated. Selective mono (entry 13) or polyhalogenation (entries 10, 12, 14) is possible by changing the reaction stoichiometry. The following substrates were not efficiently halogenated: 1,3-dinitrobenzene, 1,3,5-trinitrobenzene, 1,3-difluorobenzene, pyrimidine, and pyridazine. Either low conversions (<10%) or substrate decomposition was observed.
2.2 Reactions with Sulfur Electrophiles
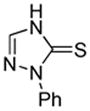
We also show that sulfur electrophiles can be employed for in situ trapping of arylmetals (Table 2). One can employ either elemental sulfur or diphenyldisulfide as the trapping agent. The reaction scope is somewhat limited compared with the halogenation reaction. Electron-rich heterocycles such as benzoxazole and N-phenyl-1,2,4-triazole react with sulfur in the presence of tBuOLi base to afford the respective thiones (entries 1, 2). Phenylsulfenylation can be effected by the reaction with diphenyl disulfide (entries 3–7). Benzothiazole, N-phenylbenzimidazole, N-methyl-1,2,4-triazole, and N-phenylpyrazole can be converted to the corresponding S-phenyl derivatives. The reaction of 2-chlorothiophene with PhSSPh afforded 2,5-bis(phenylthio)thiophene. Less acidic pyrazole derivative requires the use of a stronger tBuOK base for high conversion to the product. The limitations of the reaction are as follows. Dibutyl disulfide reacts with benzothiazole to form the benzothiazolethione by butylsulfenylation/dealkylation. Alkyl aryl ether containing substrates such as tetrafluoroanisole are dealkylated by RS− that is formed in the reaction mixture. Polyfluoroarenes such as 1,3,5-trifluorobenzene undergo nucleophilic substitution of fluorides in addition to the desired reaction.
Table 2.
Reactions with Sulfur Electrophilesa
 | ||||
|---|---|---|---|---|
| reagent | ||||
| entry | arene | base | product | yield |
| 1 | 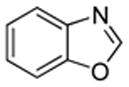 |
S tBuOLi |
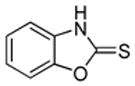 |
90% |
| 2 | 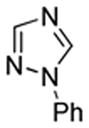 |
S tBuOLi |
 |
74% |
| 3 | 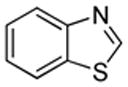 |
PhSSPh K3PO4 |
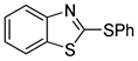 |
85% |
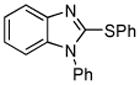
| 4 | 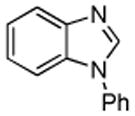 |
PhSSPh tBuOLi |
 |
84% |
| 5 | 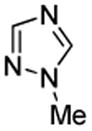 |
PhSSPh tBuOLi |
 |
55% |
| 6 | 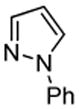 |
PhSSPh tBuOK |
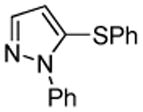 |
81% |
| 7 | 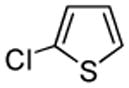 |
PhSSPh tBuOLi |
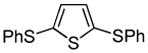 |
82% |
Substrate (1 equiv), base (1.5–3.0 equiv), sulfur (8 equiv) or PhSSPh (1.5–2.0 equiv). Yields are isolated yields. See the Supporting Information for details.
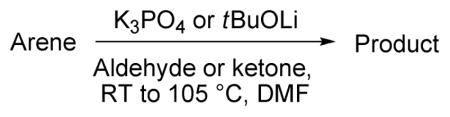
2.3. Reactions with Carbonyl Electrophiles
An impressive advance in aryl anion in situ trapping by nonenolizable ketones and aldehydes has been demonstrated previously by Kondo.6 However, expensive phosphazene bases were employed and only benzophenone and pivalaldehyde electrophiles were employed. A thermal reaction of N-alkylimidazoles with aldehydes affording addition products at the 2-position has been reported by Roe.10 A similar iridium-catalyzed reaction between imidazoles, aldehydes, and dimethylethylsilane producing 2-substituted imidazoles was published by Murai.11 Hlasta has shown that imidazoles, thiazoles, and triazoles can react with aldehydes in the presence of acid chlorides and triethylamine to give 2-substituted azoles.12 A more general method that would allow functionalization of a variety of substrates by employing cheaper reagents would be useful.
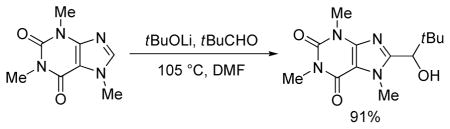
The results of in situ trapping with aldehydes and ketones are presented in Table 3. Electron-rich heterocycles such as 2-chlorothiophene, benzothiazole, N-phenyltriazole, caffeine, and benzoxazole are reactive (entries 1–4, 8). In situ trapping can be extended to electron-deficient arenes such as substituted tetrafluorobenzenes (entries 5–7). Both ketones and aldehydes can be employed as the carbonyl component. For the most acidic substrates such as benzoxazole, the reaction with enolizable cyclohexanecarbaldehyde is successful producing the addition product in acceptable yield (entry 8). Reactions fail with primary enolizable aldehydes due to aldol condensation. It is known that base-catalyzed self-condensation of secondary aldehydes is reversible, in contrast to acetaldehyde self-aldol that has been described as “sufficiently irreversible” reaction.13
Table 3.
In Situ Trapping with Aldehydes and Ketonesa
 | ||||
|---|---|---|---|---|
| reagent | ||||
| entry | arene | base | product | yield |
| 1 | 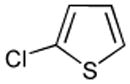 |
Ph2CO tBuOLi |
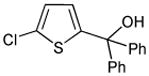 |
41% |
| 2 | 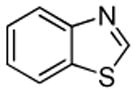 |
Ph2CO tBuOLi |
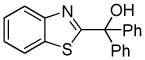 |
77% |
| 3 | 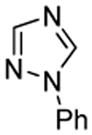 |
tBuCHO tBuOLi |
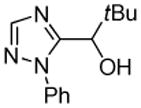 |
66% |
| 4 | 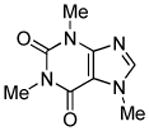 |
tBuCHO tBuOLi |
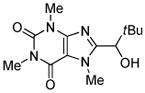 |
91% |
| 5 | 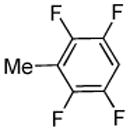 |
tBuCHO tBuOLi |
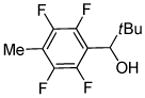 |
68% |
| 6 | 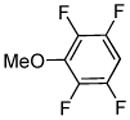 |
p-Chloro-benzaldehyde tBuOLi |
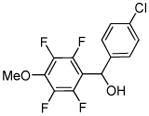 |
93% |
| 7 | 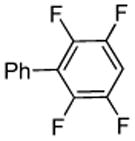 |
p-Chloro-benzaldehyde tBuOLi |
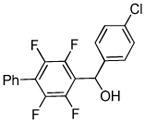 |
85% |
| 8 | 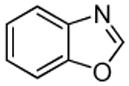 |
C6H11CHO K3PO4 |
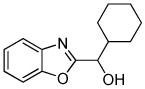 |
50% |
Substrate (1 equiv), base (1.5–3.0 equiv), ketone or aldehyde (3.0 equiv). Yields are isolated yields. See the Supporting Information for details.
3. Conclusions
We have developed a method for in situ trapping of aryllithium and potassium species with halogen, sulfur, ketone, and aldehyde electrophiles. For halogen and carbonyl electrophiles, both five-membered ring heterocycles and electron-deficient arenes are reactive. Heterocycle anion trapping is successful also with sulfur electrophiles. In the case of most acidic arenes such as pentafluorobenzene, benzothiazole, and benzoxazole, K3PO4 base can be employed instead of the currently used alkyllithiums for metalation/functionalization sequences.
Experimental Section
General procedure for halogenation
Outside the glovebox a 1-dram or 2-dram vial equipped with a magnetic stir bar was charged with substrate (1.0 or 2.0 mmol) and halogenating reagent (1.0–3.0 equiv). The vial was flushed with argon, capped and placed inside a glovebox. To this mixture anhydrous DMF or a mixture (1/1) of DMF and xylenes (1.0 mL) was added, followed by base (K3PO4 or tBuOLi, 2.0–4.0 equiv). The sealed vial was taken out of the glovebox and placed in a preheated oil bath (50–130 °C) for the indicated time. The reaction mixture was allowed to cool to room temperature and subjected to flash chromatography on silica gel. After concentrating the fractions containing the product, the residue was dried under reduced pressure to yield pure halogenation product.
8-Bromo-3,7-dihydro-1,3,7-trimethyl-1H-purine-2,6-dione (Entry 4, Table 1)
Caffeine (194 mg, 1.0 mmol), dibromotetrafluoroethane (780 mg, 3.0 mmol), tBuOLi (320 mg, 4.0 mmol), and DMF (2.0 mL), 100 °C, 13 hours. After column chromatography (gradient 10%–15% AcOEt in CH2Cl2) 176 mg (65%) of a white solid was obtained. Rf=0.25 (SiO2, hexanes/AcOEt 1/1). This compound is known.14 1H NMR (300 MHz, CDCl3) δ 3.40 (s, 3H), 3.56 (s, 3H), 3.97 (s, 3H).
General procedure for reaction with sulfur electrophiles
Outside the glovebox a 1-dram or 2-dram vial equipped with a magnetic stir bar was charged with substrate (1.0–2.0 mmol) and sulfur electrofile (1.5–8.0 equiv). The vial was flushed with argon, capped and placed inside a glovebox. To this mixture anhydrous DMF or DMPU (1.0–2.0 mL) was added, followed by base (K3PO4, tBuOK or tBuOLi, 1.5–3.0 equiv). The sealed vial was taken out of the glovebox and placed in a preheated oil bath (80–130 °C) for the indicated time. The reaction mixture was allowed to cool to room temperature, quenched with 15% aqueous citric acid, extracted with AcOEt, washed with brine, dried over anhydrous MgSO4, filtered and subjected to flash chromatography on silica gel or preparative TLC. After concentrating the fractions containing the product, the residue was dried under reduced pressure to yield pure product.
1-Phenyl-2-(phenylthio)-1H-benzimidazole (Entry 4, Table 2)
1-Phenylbenzimidazole (388 mg, 2.0 mmol), diphenyl disulfide (873 mg, 4.0 mmol), tBuOLi (320 mg, 4.0 mmol), and DMF (2.0 mL), 130 °C, 12 hours. After column chromatography (30% CH2Cl2 in hexanes, CH2Cl2, then 30% AcOEt in CH2Cl2) 512 mg (84%) of a light tan solid was obtained. Rf=0.52 (SiO2, hexane/AcOEt 3/1). Analytical sample was recrystallized from hexanes/AcOEt, mp 92–94 °C. 1H NMR (300 MHz, CDCl3) δ 7.09–7.15 (m, 1H), 7.17–7.54 (m, 12H), 7.56–7.80 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 110.2, 119.7, 122.9, 123.5, 127.6, 128.4, 129.1, 129.4, 129.7, 131.1, 132.4, 135.7, 137.5, 143.4, 149.5. FT-IR (neat, cm−1) υ 1497, 1424, 1343, 1260, 1220. Anal calcd for C19H14N2S (302.39 g/mol): C, 75.47; H, 4.67; N, 9.26; Found. C, 75.41; H, 4.60; N, 9.21.
General procedure for reaction with carbon electrophiles
Outside the glovebox a 1-dram or 2-dram vial equipped with a magnetic stir bar was charged with substrate (1.0–2.0 mmol) and aldehyde or ketone (3.0 equiv). The vial was flushed with argon, capped and placed inside a glovebox. To this mixture anhydrous DMF (1.0–2.0 mL) was added, followed by base (K3PO4 or tBuOLi, 1.5–3.0 equiv). The sealed vial was taken out of the glovebox and was placed in a preheated oil bath (60–105 °C) or stirred at RT for the indicated time. The reaction mixture was allowed to cool to room temperature, quenched with 15% aqueous citric acid, extracted with AcOEt, washed with brine, dried over anhydrous MgSO4, filtered and subjected to flash chromatography on silica gel or preparative TLC. After concentrating the fractions containing the product, the residue was dried under reduced pressure to yield pure product.
(4-Chlorophenyl)(2,3,5,6-tetrafluoro-4-methoxyphenyl)methanol (Entry 6, Table 3)
2,3,5,6-Tetrafluoroanisole (180 mg, 1.0 mmol), 4-chlorobenzaldehyde (422 mg, 3.0 mmol), t-BuOLi (120 mg, 1.5 mmol), and DMF (1.0 mL), RT, 2 hours. After column chromatography (gradient 30%–60% CH2Cl2 in hexanes) 300 mg (93%) of a colorless oil was obtained. Rf=0.45 (SiO2, hexanes/CH2Cl2 1/4). 1H NMR (300 MHz, CDCl3) δ 2.64 (dt, J=7.5 Hz, 1.1 Hz, 1H), 4.08 (t, J=1.4 Hz, 3H), 6.18 (d, J=7.5 Hz, 1H), 7.33 (s, 4H). 19F NMR (282 MHz, CDCl3) δ −160.6– −160.4 (m, 2F), −148.2 – −148.0 (m, 2F). 13C NMR (75 MHz, CDCl3) δ 62.6 (t, JC-F=3.6 Hz), 67.4 (quintet, JC-F=2.4 Hz), 114.5–115.4 (m), 127.4, 129.3, 134.4, 138.3–140.0 (m), 140.1, 142.8–143.9 (m), 146.6–147.3 (m). FT-IR (neat, cm−1) υ 3387, 1651, 1491, 1438, 1416, 1196, 1132, 1092. Anal calcd for C14H9ClF4O2 (320.67 g/mol): C, 52.44; H, 2.83; Found. C, 52.40; H, 2.88. At elevated temperatures, base-catalyzed formation of p-chlorobenzyl p-chlorobenzoate is observed.
Supplementary Material
Acknowledgments
We thank the Welch Foundation (Grant E-1571), NIGMS (Grant R01GM077635), A. P. Sloan Foundation, and Camille and Henry Dreyfus Foundation for supporting this research.
Footnotes
Supporting Information Available: Detailed experimental procedures and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Krizan TD, Martin JC. J Am Chem Soc. 1983;105:6155. [Google Scholar]; (b) Taylor SL, Lee DY, Martin JC. J Org Chem. 1983;48:4156. [Google Scholar]; (c) Dua SS, Gilman H. J Organomet Chem. 1974;64:C1–C2. [Google Scholar]; (d) Schlosser M, Guio L, Leroux F. J Am Chem Soc. 2001;123:3822. doi: 10.1021/ja0032733. [DOI] [PubMed] [Google Scholar]; (e) Black WC, Guay B, Scheuermeyer F. J Org Chem. 1997;62:758. doi: 10.1021/jo961839t. [DOI] [PubMed] [Google Scholar]; (f) Vazquez E, Davies IW, Payack JF. J Org Chem. 2002;67:7551. doi: 10.1021/jo026087j. [DOI] [PubMed] [Google Scholar]; (g) Lulińsky S, Serwatowski J. J Org Chem. 2003;68:9384. doi: 10.1021/jo034790h. [DOI] [PubMed] [Google Scholar]; (h) Straus F, Kollex L, Heyn W, Kuhnel R. Chem Ber. 1930;63B:1868. [Google Scholar]; (i) Mach MH, Bunnett JF. J Am Chem Soc. 1974;96:936. [Google Scholar]
- 2.(a) Kristensen J, Lysén M, Vedsø P, Begtrup M. Org Lett. 2001;3:1435. doi: 10.1021/ol015598+. [DOI] [PubMed] [Google Scholar]; (b) Caron S, Hawkins JM. J Org Chem. 1998;63:2054. [Google Scholar]; (c) Alessi M, Larkin AL, Ogilvie KA, Green LA, Lai S, Lopez S, Snieckus V. J Org Chem. 2007;72:1588. doi: 10.1021/jo0620359. [DOI] [PubMed] [Google Scholar]
- 3.Shtark AA, Chuikova TV, Shteingarts VD. Russ J Org Chem. 1984;20:960. (English) [Google Scholar]
- 4.Regel E, Büchel KH. Liebigs Ann Chem. 1977;1:145. [Google Scholar]
- 5.Zarudnitskii EV, Pervak II, Merkulov AS, Yurchenko AA, Tolmachev AA, Pinchuk AM. Synthesis. 2006:1279. [Google Scholar]
- 6.Imahori T, Kondo Y. J Am Chem Soc. 2003;125:8082. doi: 10.1021/ja0342300. [DOI] [PubMed] [Google Scholar]
- 7.(a) Do H-Q, Khan RMK, Daugulis O. J Am Chem Soc. 2008;130:15185. doi: 10.1021/ja805688p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Do HQ, Daugulis O. Org Lett. 2009;11:421. doi: 10.1021/ol802411f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Shen K, Fu Y, Li J-N, Liu L, Guo Q-X. Tetrahedron. 2007;63:1568. [Google Scholar]; (b) Bordwell FG. Acc Chem Res. 1988;21:456. [Google Scholar]
- 9.Review: Slocum DW, Shelton P, Moran KM. Synthesis. 2005:3477.Prakash GKS, Mathew T, Hoole D, Esteves PM, Wang Q, Rasul G, Olah GA. J Am Chem Soc. 2004;126:15770. doi: 10.1021/ja0465247.Deacon GB, Smith RNM. Aust J Chem. 1982;35:1587.Hellmann M, Bilbo AJ, Pummer WJ. J Am Chem Soc. 1955;77:3650.Mongin F, Marzi E, Schlosser M. Eur J Org Chem. 2001:2771.Burukin AS, Vasil’ev AA, Struchkova MI, Kachala VV, Zlotin SG. Russ Chem Bull. 2005;54:964.Boga C, Del Vecchio E, Forlani L, Todesco PE. J Organomet Chem. 2000;601:233.
- 10.Roe AM. J Chem Soc. 1963:2195. [Google Scholar]
- 11.Fukumoto Y, Sawada K, Hagihara M, Chatani N, Murai S. Angew Chem, Int Ed Engl. 2002;41:2779. doi: 10.1002/1521-3773(20020802)41:15<2779::AID-ANIE2779>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 12.Deng Y, Hlasta DJ. Tetrahedron Lett. 2002;43:189. [Google Scholar]
- 13.Hine J, Houston JG, Jensen JH. J Org Chem. 1965;30:1184. [Google Scholar]
- 14.Vollmann K, Muller CE. Heterocycles. 2002;57:871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.