

Abstract
In lead discovery, libraries of 106 molecules are screened for biological activity. Given the over 1060 drug-like molecules thought possible, such screens might never succeed. That they do, even occasionally, implies a biased selection of library molecules. Here a method is developed to quantify the bias in screening libraries towards biogenic molecules. With this approach, we consider what is missing from screening libraries and how they can be optimized.
High-throughput screening (HTS) is the dominant method of lead discovery in pharmaceutical research and chemical biology. A plurality of the new chemical entities in clinical trials may have their origins in this technique, as do at least two drug.1 Whereas these screens have been productive against traditional drug targets, such as GPCRs, ligand-gated ion channels, and kinases, screening libraries of synthetic molecules has been problematic for others, such as antimicrobial targets and those identified from genomic studies. The reasons for these successes and failures have been widely debated.2-5 From a theoretical perspective, however, one might wonder not that screens of 106 molecules sometimes fail, but rather that they ever succeed.
Chemical space, i.e. all possible molecules, is estimated to be greater than 1060 molecules with 30 or fewer heavy atoms;6 10μg of each would exceed the mass of the observable universe. This figure will diminish if criteria for synthetic accessibility and drug-likeness are taken into account and increase steeply if up to 35 heavy atoms, about 500 Daltons, are allowed. Positing even a modest specificity of proteins for their ligand, the odds of a hit in a random selection of 106 molecules from this space seems negligible.
HTS nevertheless does return active molecules for many targets; how does it overcome the odds stacked against it? One might hazard two hypotheses. First, molecules that are formally chemically different can be degenerate to a target, and many derivatives of a chemotype may have little effect on affinity. This behavior, and the polypharmacology of small molecules,7-9 undoubtedly contributes to screening hit rates. Such chemical degeneracy seems unlikely, however, to overcome the long odds against screening. A second explanation is that screening libraries are far from random selections, but rather are biased toward molecules likely to be recognized by biological targets. This second hypothesis seems more plausible, as many accessible molecules are likely to resemble or derive from metabolites and natural products. Some of these will have been synthesized to resemble such biogenic molecules, while others will have used biogenic molecules as a starting material. The role of bias in screening has been mooted before,10-13 and indeed methods to measure metabolite- or natural product-likeness have been reported, permitting the design of these features into screening libraries.14,15 How such bias might be quantified relative to what one would expect for an unbiased collection, and thus its extent and impact on screening and discovery, has remained unexplored.
Quantifying library bias requires three sets of molecules: one that represents all of chemical space, one that represents molecules that proteins are intrinsically likely to recognize—defining the optimal bias, and one that represents screening libraries. The set representing chemical space previously seemed inaccessible. Recently, however, Fink and Reymond have calculated all of the synthetically accessible molecules with 11 or fewer non-hydrogen (heavy) atoms composed of first row elements (C, N, O, and F); there are over 26 million of these, not allowing for stereochemistry.16 Whereas these molecules are small compared to most biologically interesting compounds, this Generated DataBase (GDB) is comprehensive, giving us the full unbiased set within its boundary criteria. For the molecules that proteins are likely to bind—defining the bias—several sets are possible, such as those molecules that have become drugs. Indeed, several investigators have adopted this approach when asking “what is drug-likeness and how can libraries be biased towards it?”17,18 Here, however, we ask why one should expect to find any hits from screening, and so need a reference set that captures protein recognition in general. For this purpose drugs are imperfect, reflecting many other criteria, like bioavailability, and are backward-looking, capturing information only on a small number of targets. We therefore chose metabolites and natural products from the KEGG (2 018 molecules) and the Dictionary of Natural Products (141 985 molecules) databases, respectively. These molecules are recognized by at least one protein in the biosphere, often many, and are out-group molecules, uninfluenced by human invention. For the set of molecules representing screening libraries we use those molecules that are commercially available, reasoning that most HTS libraries, even in the pharmaceutical industry, are largely composed of molecules that have been purchased from commercial vendors, or closely resemble them (for the MLSMR, the US national screening collection, almost all of the ∼300 000 molecules are commercially sourced). To compare the commercially available molecules to those of the GDB, we restrict the former by the same criteria: only purchasable molecules with 11 or fewer heavy atoms composed of first row elements are considered. There are 25 810 such molecules in the ZINC database of commercially available molecules (http://zinc.docking.org); we refer to these as the purchasable-GDB (Fig. 1).
Figure 1.

Overlap between commercially available molecules and the GDB gives the purchasable GDB.
As we will show, when metabolites are compared to both the purchasable-GDB and the full GDB, the purchasable subset is almost 1000-fold more similar to metabolites than the overall GDB, our proxy to full chemical space. The same bias is observed when the two sets are compared to natural products. The bias grows dramatically with molecular size, suggesting that this bias will be greater still among larger “lead-like” or “drug-like” molecules in screening. This is consistent with the idea that these libraries are massively and productively biased toward biogenic molecules. We leverage this observation to ask what scaffolds occur among biogenic molecules but are absent from those commercially available. Almost 1300 ring-scaffolds are found among natural products that are missing from commercial libraries—these scaffolds provide criteria that could be used to further increase the bias in screening libraries toward those molecules that proteins have evolved to recognize.
RESULTS
We first compared the 26 million GDB and the 25 810 purchasable-GDB molecules to the metabolites and the natural products. With the widely used ECFP_4 topological fingerprints19 we calculated the similarity between each biogenic molecule and each GDB and purchasable-GDB molecule. We plotted the percentage of GDB and purchasable-GDB molecules that had at least one metabolite or, separately, one natural product within a certain similarity, expressed as Tanimoto score (where a similarity score of 1 indicates identity between the pair of molecules and a similarity score of 0.2 indicates a similarity so low as to be essentially meaningless). As expected, almost all GDB and purchasable-GDB molecules had a similarity coefficient of 0.2 or greater to at least one metabolite or natural product (Fig. 2a,b, and Supplementary Figs. 1a,b, and 2a,b online). After this plateau, the percentages decreased rapidly as the similarity threshold became more stringent. Critically, it diminished orders of magnitude faster for the GDB than it did for the purchasable subset of GDB. Even by a similarity coefficient of 0.32 there were tenfold more molecules from the purchasable-GDB, expressed as a percentage, than GDB molecules that resembled at least one metabolite. By a similarity coefficient of 0.53 this ratio was 100-fold and by the time full identity was reached, considering only exact biogenic molecules in the purchasable subset of the GDB, 0.83% (215 compounds) and 10.5% (2703 compounds) of the 25 810 purchasable GDB molecules were identical to metabolites and natural products, respectively—an enrichment of 922-fold and 618-fold compared to the full GDB. Since there are 1000-fold more GDB than purchasable-GDB molecules this ratio implies that most of the natural products or metabolites represented in the 26 million compounds of the GDB are captured by the 25 810 molecule subset that has actually been synthesized and may be purchased. This bias was unaffected by the size of the molecules used in the reference databases, and was the same whether we used the full set of metabolites and natural products or only those compliant with the GDB rules (Supplementary. Fig 3 online). A bias towards metabolites and natural products of similar magnitude was also found among commercially-available compound sets specifically designed for screening (Fig. 2c,d), while for the US National MLSMR library the molecules subject to the GDB restrictions (MLSMR-GDB) were even more biased toward metabolites and natural products (Fig. 2c,d). In all libraries this bias increases exponentially with molecular size (Fig 3a,b>) and so we expect that most screening collections, made up of larger molecules than considered here, will be even orders of magnitude more biased towards biogenic molecules.
Figure 2. Compounds in screening libraries are biased toward biogenic molecules.

Percentage of the GDB and purchasable-GDB databases as a function of the Tanimoto similarity to their nearest neighbor in [a] the KEGG and [b] the Dictionary of Natural Compound databases. Percentage of the GDB, the purchasable-GDB, Asinex (360 042 compounds − 815 GDB compliant compounds), Chembridge (473 745 compounds − 389 GDB compliant compounds); IBS (424 806 compounds − 884 GDB compliant compounds), Life Chemicals (285 581 compounds − 172 GDB compliant compounds), Otava (121 657 compounds − 287 GDB compliant compounds) databases as a function of the Tanimoto similarity to their nearest neighbor to the [c] KEGG and the [d] Dictionary of Natural Products databases.
Figure 3.

Ratio of the percentage of compounds in the purchasable-GDB and GDB databases that had a similarity ≥ 0.75 to their nearest neighbor in [a] the KEGG and [b] the Dictionary of Natural Products databases versus the number of heavy atoms up to which the database compound (in purchasable-GDB and GDB) are considered.
We investigated if there were particular chemical features that were most responsible for this bias. Whereas we do not pretend to undertake a comprehensive analysis of particular chemotypes here, a preliminary study may interest some investigators. We identified 871 bits from unfolded fingerprints that occurred in more than 1% of the metabolites or the natural products (there were 174 823 different bits among these molecules before folding). 28 of those bits (functional groups) also occurred in more than 0.5% of the purchasable-GDB database and in less than 0.05% of the GDB database, representing at least a ten-fold bias of the purchasable-GDB relative to the entire GDB. The substructures corresponding to these bits illustrate some of the fragments contributing to the observed bias (Supplementary Table 1 online). For instance, meta-substituted aryl alcohols, such as 3-methylphenol, occur among 1.3% of metabolites, 2.2% of natural products and 0.6% of the purchasable-GDB, but only in 0.006% of the entire GDB. As an aside, notwithstanding the much greater similarity of the purchasable-GDB to metabolites and natural products than the overall GDB, some functional groups were actually under-represented among the purchasable-GDB. Thus, 38 bits occurred in more than 0.5% of the GDB and more than 1% of metabolites or natural products but in less than 0.2% of the purchasable-GDB (Supplementary Table 2 online). For instance, 1,2-trisubstituted epoxides occur in 2.6% of natural products and 1.7% of the GDB but in only 0.01% of the purchasable-GDB. Were such substructures included in future commercial compounds they would increase the already substantial bias still further.
If it is true that bias towards biogenic molecules contributes to the success of screening, it seems interesting to ask whether this bias might be increased, productively, by adding scaffolds present among biogenic molecules that are currently unexplored in our libraries. We again turned to the metabolites, natural products, and commercially available molecules, this time considering each without limit to molecular size or composition. To find scaffolds present among the two biogenic sets but absent from the commercially available molecules, we represented the molecules by their core rings (Supplementary Fig. 4 online).20 Each core ring scaffold among the biogenic molecules was matched to its counterpart among commercially available molecules, when available. There were 173, 15 637 and 29 496 unique rings among the 2 018 metabolites, 141 985 natural products and 9 131 254 commercially available molecules, respectively. 34 (20%) of the ring scaffolds among the metabolites and 12 977 (83%) of the ring scaffolds among the natural products were unrepresented among the commercial molecules. Even if one restricts this set to molecular weight ≤350 Da and two or fewer stereocenters, there remain 1 891 ring scaffolds represented among natural products that have no counterpart among commercially available molecules, and by extension screening libraries.
DISCUSSION
Returning to our motivating question, a major reason why the screening of synthetic compounds ever finds interesting hits is that our libraries are biased towards the sort of molecules that proteins have evolved to recognize. Thus, there are almost as many metabolites and natural products among the 25 810 purchasable GDB molecules as there are among the 26 million GDB molecules overall. This bias increases rapidly as molecules grow in size (Fig. 3), and the bias among larger “lead-like” and “drug-like” molecules is expected to be many orders of magnitude more still than that measured for the very small molecules explored here, where full enumeration16 allowed us to compare to a complete chemical space.
From this observation two opposite inferences might be drawn. Since our libraries are already biased, then perhaps we should look for new screening molecules that are dissimilar to metabolites and natural products. Whereas this will certainly explore new chemotypes and ensure novel scaffolds, we do not draw this conclusion. Chemical space is so large that, unless proteins are highly promiscuous, the likelihood of finding anything biologically interesting is remote. Instead, we suggest that screening libraries may be improved by increasing the bias toward biogenic molecules further still, by adding to libraries molecules resembling biogenic scaffolds that are now absent from them. After all, the bias in our current libraries is largely unintentional, the product of what organic chemists have synthesized since the birth of the field with urea in 1828 (though see refs 21-24). This leaves room for intentional optimization. Indeed, 83% of the core ring-scaffolds present among natural products are simply absent among commercially available molecules, and by extension screening libraries.
It is interesting to compare these missing scaffolds with those from earlier studies that sought rings most common among drug-like molecules.20,25-27 An example are the six rings highlighted as characteristic of drug-like molecules by Ertl and colleagues (Table 1). Comparing these to those scaffolds found among natural products but unavailable commercially reveals molecules that are so similar to the drug-like rings that their absence from screening libraries is startling (a few examples are given in Table 2). Earlier studies have suggested that scaffolds characteristic of drug-like molecules be sought when purchasing new molecules for screening; here we suggest that molecules containing scaffolds present in natural products but absent from commercial collections are places to begin expanding the biogenic chemistry available for screening. Biasing future screening libraries to fill these systematic absences in our current collections will help address the new genomic targets with which we are increasingly confronted, and against which screening has had such mixed success.
Table 1.
Core ring structures common among drugs and related molecules.25
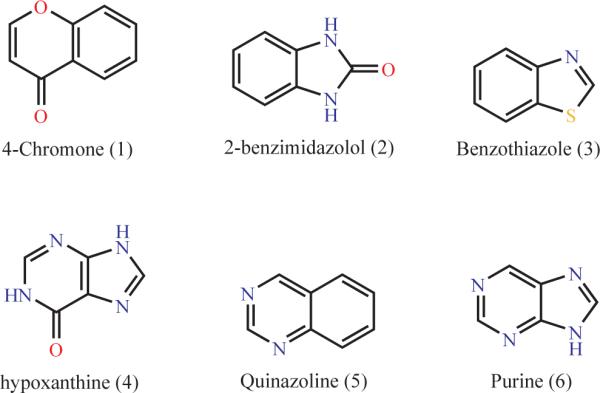
|
Table 2.
Characteristic scaffolds present among natural products but missing from commercially available molecules. The core ring scaffold is shown in green for the natural products.

|
METHODS
Chemical Space
We used the Generated DataBase (GDB) of Reymond et al. as a proxy for chemical space.16,28 The GDB was obtained by exhaustively enumerating all the possible topologies for molecules composed of only first row elements (C, N, O, and F), taking into account the stability, synthetic accessibility, and drug likeness of the resulting molecules. GDB contained 26 429 328 unique compounds with no consideration of stereochemistry.
Biogenic Space
We used two databases to approximate the space of molecules that occur in natural organisms: the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and the Dictionary of Natural Products (DNP). The KEGG (Ligand) database contained 11 434 unique compounds,29 but many of these were xenobiotics such as hexachlorohexane. A xenobiotic-free subset of the KEGG database was generated by only considering primary metabolic pathways (Supplementary Table 3). This subset contained 2 018 unique compounds and was used as the reference to metabolic space. The CRC Dictionary of Natural Products30 (DNP version 16.2) contained 141 985 unique structures and was used as the source of natural products.
Screening library space
ZINC31 contains 9 131 254 unique, commercially available compounds after adding the latest vendor catalogs and discarding some of the physical property filters normally used for docking purposes, such as molecular weight. ZINC was used as an approximation to a general-purpose screening library. To compare commercial compounds to the GDB, we filtered ZINC for molecules that conformed to the same rules as those used to generate the GDB. ZINC contained 25 810 compounds with 11 or less C, N, O, or F atoms, the “purchasable-GDB.”
The National Institute of Health (NIH) Molecular Libraries Small Molecule Repository (MLSMR) contained 298 794 unique compounds (downloaded from http://pubchem.ncbi.nlm.nih.gov on 2008/06/18). The MLSMR contained 866 compounds with 11 or less C, N, O, and F heavy atoms, i.e. subject to the same restrictions as GDB. This subset was referred to as MLSMR-GDB.
Comparison of the chemical and screening space to the biogenic spaces
Each molecule of the GDB and purchasable-GDB databases was, in turn, compared to each molecule in the KEGG and DNP databases. Compounds were represented by their Scitegic Extended Connectivity FingerPrints32 (ECFP_4) which encodes the presence or absence of topological fragments (with no stereochemistry consideration) in the form of an extended connectivity string centered on a specific atom and calculated using a modification of the Morgan Algorithm.33 The initial code assigned to each molecule's atom is based on the number of connections, the element type, and the mass. This code is hashed to produce the next order code, which is mapped into an address space of size 232 and the process iterates twice to describe features up to four bonds in diameter.34 The resulting fingerprint was further folded into a bit-string of length 1 024 bits. The similarity between two molecules was measured by comparing their respective ECFP_4 bit-string with the Tanimoto coefficient. If a denotes the number of bits set to “on” for molecule A, b the number of bits set to “on” for molecule B, and c the number of bits set to “on” in both molecules, the Tanimoto similarity between these two molecules is:
Analysis of the bit frequencies
Each bit's exact substructure was exported as a SMARTS pattern (before folding) using the built-in function of the standard molecular fingerprint component of Scitegic Pipeline Pilot.32 There were cases where a single bit corresponded to more than one substructure; often these substructures were related. For the frequency calculations or even for illustration purposes (see Supplementary Tables 1 and 2 online), one of these substructure was chosen arbitrarily.
Generation of the core ring scaffolds
Several approaches to extract the scaffold of a molecule are available;20,26,35 here we use the approach of Bemis & Murcko.20 Each molecule in the KEGG, DNP, and ZINC databases was decomposed into its core ring scaffold using Pipeline Pilot.32 Core ring scaffolds consist of individual contiguous ring systems keeping atom types, bond orders, aromaticity information, and exocyclic double bonds but discarding stereochemistry and charges (see Supplementary. Fig. 4 online). A canonical SMILES string was generated for each resulting core ring structure. The presence (or absence) of a particular KEGG or DNP scaffold was evaluated by matching its SMILES string to those obtained from the ZINC database.
Supplementary Material

Acknowledgements
Supported by GM59957 (to B.K.S.). J.H. was supported by a Marie Curie fellowship from the 6th Framework Program of the European Commission, M.J.K. by a National Science Foundation graduate fellowship and C.L. by a fellowship from the Max Kade Foundation.
Footnotes
Author Contributions: The project was conceived of by J.H. and B.K.S. J.H. undertook most of the calculations, with molecular proof-checking by J.J.I. and C.L. and algorithmic assistance from M.J.K. J.H. and B.K.S. wrote the manuscript, which was read and commented on by the other authors.
REFERENCES
- 1.Wilhelm S, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 2.Spencer RW. High-throughput screening of historic collections: observations on file size, biological targets, and file diversity. Biotechnol. Bioeng. 1998;61:61–67. [PubMed] [Google Scholar]
- 3.Fox S, Farr-Jones S, Sopchak L, Boggs A, Comley J. High-Throughput Screening: Searching for Higher Productivity. J. Biomol. Screen. 2004;9:354–358. doi: 10.1177/1087057104265290. [DOI] [PubMed] [Google Scholar]
- 4.Macarron R. Critical review of the role of HTS in drug discovery. Drug Discov. Today. 2006;11:277–279. doi: 10.1016/j.drudis.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Pereira DA, Williams JA. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007;152:53–61. doi: 10.1038/sj.bjp.0707373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohacek R, McMartin C, Guida W. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996;16:3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Roth B, Sheffler D, Kroeze W. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discovery. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 8.Paolini G, Shapland R, van Hoorn W, Mason J, Hopkins A. Global mapping of pharmacological space. Nat. Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 9.Yildirim M, Goh K-I, Cusick M, Barabasi A-L, Vidal M. Drug–target network. Nat. Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 10.Martin YC. Diverse viewpoints on computational aspects of molecular diversity. J. Comb. Chem. 2001;3:231–250. doi: 10.1021/cc000073e. [DOI] [PubMed] [Google Scholar]
- 11.Breinbauer R, Vetter IR, Waldmann H. From protein domains to drug candidates-natural products as guiding principles in the design and synthesis of compound libraries. Angew. Chem., Int. Ed. 2002;41:2879–2890. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 12.Koehn F, Carter G. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discovery. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 13.Arve L, Voigt T, Waldmann H. Charting Biological and Chemical Space: PSSC and SCONP as Guiding Principles for the Development of Compound Collections Based on Natural Product Scaffolds. QSAR Comb. Sci. 2006;25:449–456. [Google Scholar]
- 14.Ertl P, Roggo S, Schuffenhauer A. Natural Product-likeness Score and Its Application for Prioritization of Compound Libraries. J. Chem. Inf. Model. 2007;48:68–74. doi: 10.1021/ci700286x. [DOI] [PubMed] [Google Scholar]
- 15.Gupta Sunil, Aires De S, Joao Comparing the chemical spaces of metabolites and available chemicals: models of metabolite-likeness. Mol. Diversity. 2007;11:23–36. doi: 10.1007/s11030-006-9054-0. [DOI] [PubMed] [Google Scholar]
- 16.Fink T, Reymond JL. Virtual Exploration of the Chemical Universe up to 11 Atoms of C, N, O, F: Assembly of 26.4 Million Structures (110.9 Million Stereoisomers) and Analysis for New Ring Systems, Stereochemistry, Physicochemical Properties, Compound Classes, and Drug Discovery. J. Chem. Inf. Model. 2007;47:342–353. doi: 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- 17.Sadowski J, Kubinyi H. A scoring scheme for discriminating between drugs and nondrugs. J. Med. Chem. 1998;41:3325–3329. doi: 10.1021/jm9706776. [DOI] [PubMed] [Google Scholar]
- 18.Good AC, Hermsmeier MA. Measuring CAMD technique performance. 2. How “druglike” are drugs? Implications of Random test set selection exemplified using druglikeness classification models. J. Chem. Inf. Model. 2007;47:110–114. doi: 10.1021/ci6003493. [DOI] [PubMed] [Google Scholar]
- 19.Glen RC, et al. Circular fingerprints: flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs. 2006;9:199–204. [PubMed] [Google Scholar]
- 20.Bemis GW, Murcko MA. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber S. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 22.Haggarty S, Clemons P, Wong J, Schreiber S. Mapping Chemical Space Using Molecular Descriptors and Chemical Genetics: Deacetylase Inhibitors. Comb. Chem. High Throughput Screen. 2004:669–676. doi: 10.2174/1386207043328319. [DOI] [PubMed] [Google Scholar]
- 23.Shang S, Tan DS. Advancing chemistry and biology through diversity-oriented synthesis of natural product-like libraries. Curr. Opin. Chem. Biol. 2005;9:248–258. doi: 10.1016/j.cbpa.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Gregori-Puigjané E, Mestres J. Coverage and bias in chemical library design. Curr. Opin. Chem. Biol. 2008;12:359–365. doi: 10.1016/j.cbpa.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Ertl P, Jelfs S, Mühlbacher J, Schuffenhauer A, Selzer P. Quest for the rings. In silico exploration of ring universe to identify novel bioactive heteroaromatic scaffolds. J. Med. Chem. 2006;49:4568–4573. doi: 10.1021/jm060217p. [DOI] [PubMed] [Google Scholar]
- 26.Wester MJ, et al. Scaffold topologies. 2. Analysis of chemical databases. J. Chem. Inf. Model. 2008;48:1311–1324. doi: 10.1021/ci700342h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wetzel, et al. Cheminformatic Analysis of Natural Products and their Chemical Space. CHIMIA International Journal for Chemistry. 2007;61:355–360. [Google Scholar]
- 28.Fink T, Bruggesser H, Reymond J-L. Virtual Exploration of the Small-Molecule Chemical Universe below 160 Daltons. Angew. Chem., Int. Ed. 2005;44:1504–1508. doi: 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- 29.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buckingham J. Dictionary of Natural Products. Chapman & Hall/CRC; 2008. [Google Scholar]
- 31.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scitegic Pipeline Pilot is available from Accelrys Software, Inc. at http://accelrys.com.
- 33.Morgan HL. Generation of a unique description for chemical structures-A technique developed at Chemical Abstract Service. J. Chem. Doc. 1965;5:107–113. [Google Scholar]
- 34.Hert J, et al. Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org. Biomol. Chem. 2004;2:3256–3266. doi: 10.1039/B409865J. [DOI] [PubMed] [Google Scholar]
- 35.Koch M, et al. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proc. Natl. Acad. Sci. U. S. A. 2005;102:17272–17277. doi: 10.1073/pnas.0503647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.