

Abstract
Coregulators (coactivators and corepressors) occupy the driving seat for actions of all nuclear receptors, and consequently, selective receptor modulator drugs. The potency and selectivity for subreactions of transcription reside in the coactivators, and thus, they are critically important for tissue-selective gene function. Each tissue has a ‘quantitative finger print’ of coactivators based on its relative inherited concentrations of these molecules. When the cellular concentration of a coactivator is altered, genetic dysfunction usually leads to a pathologic outcome. For example, many cancers overexpress ‘growth coactivators’. In this way, the cancer cell can hijack these coactivator molecules to drive proliferation and metastasis. The present review contains summaries of selective coactivators and corepressors that have been demonstrated to play important roles in the malignant process and emphasizes their importance for future therapeutic interventions.
It has been only 12 years since the first nuclear receptor coactivator, SRC-1, was cloned and discovered to be a new form of transcription factor that, rather than binding to DNA, binds to nuclear receptors (NRs) and mediates the transcriptional potency of NRs (1, 2). The NR coactivators belong to a class of molecules generally termed ‘coregulators’ (2). We define coactivators as molecules which are recruited by DNA-binding transcription factors (TFs) to enhance transcription, and corepressors as molecules which are recruited by TFs to repress transcription. Current evidence indicates that this operational definition can be modified by gene and cell and signaling context for any one coregulator. Our current understanding is that NRs search out the target genes to be regulated by binding to specific DNA sequences (or other TFs at such sequences) termed NR-response elements; the second job of NRs is to recruit the coregulators that perform all of the subsequent biochemical reactions required for induction or repression of gene transcriptions. In fact, most coregulators are either enzymes needed for gene expression, or they regulate the enzymatic activities of other co-coregulators that are present in the high molecular weight transcriptional complexes at the gene. Over the past decade, the mechanistic importance of coregulators has expanded logarithmically, and we now realize that they perform virtually ‘all’ of the reactions needed for control of TF-dependent gene expression (3, 4).
Coactivator Mechanisms
In the steady-state cell, coregulators exist and function in large multiprotein complexes (3). For example, coactivator complexes are recruited by NRs to target genes in an ordered sequence to provide the many enzyme capacities required for transcription (5). Subreactions of transcription mediated by coactivator complexes include chromatin modification and remodeling, initiation of transcription, elongation of RNA chains, mRNA splicing, and even, proteolyic termination of the transcriptional response. Surprisingly, recent reports show that coactivators can influence cellular reactions outside the nucleus such as mRNA translation, mitochondrial function, and motility (6). The expanding importance of coactivators (and corepressors) to mammalian cancer merits reflection on selected historical advances that have promoted the development of this field. In the following mini-review, there is insufficient space to discuss all of the numerous excellent studies that substantiate the importance of coregulators to cancer, so only a limited number of selected cases will be employed to illustrate their relevance to cancer biology.
When considering the plethora of functions that NRs play in tissues such as breast, ovary, prostate, intestine, pancreas and lungs, and the interest in the cancers of these organs, we anticipate a relatively large number of coregulators to be involved in cancer. However, this spectrum of tissues indicates that the oncogenic or tumor suppressive activities of coregulators play key roles in diverse tissues, well beyond the classic endocrine targets of NRs.
The concentrations of coactivators and corepressors in cells are critical to their functional potential. Indeed, the cellular levels of coactivators are frequently regulated by altering their posttranslational degradation rates (24). A high cellular concentration of a coactivator will lead to an amplified signal within a downstream transcription factor action pathway, as well as a more rapid response to environmental signals. However, available data from our labs and a variety of other sources suggest that the key to understanding the true diversity of coregulator functions lies in first understanding the surprisingly extensive ‘posttranslational coding’ that has been discovered to exist in the coregulator proteins (7). In the case of a coregulator, it has been observed that a combination of such posttranslational modifications (PTMS) can lead to functionally distinct activities for the same primary sequence protein. The ‘differentially coded’ coactivator is now predisposed to form different multimeric coactivator complexes, and thereby, to interact with distinct genes and to perform different compartmental functions in a cell. For example, over 40 separate modifications of the SRC-3 coactivator have been determined to date (8). These include phosphorylations, methylations, acetylations, ubiquitinations and SUMOylations. This plethora of 40 PTMs creates an enormous combinatorial ‘potential’of (2)40 (8). The vast majority of this PTM ‘potential complexity’ likely is never actually used in a given cell. Nevertheless, since every single PTM can result in a protein with ‘some’ altered function, it is an inescapable conclusion that multiple combinations of PTMs can bestow a huge array of distinct functions to any one coregulator protein.
SRC-3 Coactivator is an Oncogene and a Target for Epigenetic Signals
SRC-3/AIB1/ACTR/pCIP/RAC3/TRAM1/NCOA3 is the third member of the SRC/p160 coactivator family, and since it is the most studied of NR coactivators, it serves as a prototype example for defining the range of mechanisms and functions of such molecules. This gene was characterized as AIB1 (Amplified in breast cancer-1) and originally reported to be amplified in approximately 10% and overexpressed in 64% of primary breast tumor cells (9). It was subsequently found to be amplified and overexpressed in numerous other human cancers such as bladder, nasopharyngeal, endometrial, prostate etc. Clinical studies in breast cancer patients show that overexpression of SRC-3 correlates with tamoxifen resistance, p53 and HER2 expression status, and leads to markedly negative clinical outcomes (10–14).
The oncogenic potential for SRC-3 is substantiated by studies in transgenic mice that overexpress SRC-3 and develop spontaneous malignant mammary tumors (15). In contrast, SRC-3−/− mice are resistant to chemical carcinogen-induced and viral-induced mammary tumorigenesis (16, 17). SRC-3−/− mice also show resistance to induced prostate cancer progression (18). These results are consistent with the idea that SRC-3 is a potentially powerful oncogene. In the lymphatic system, however, SRC-3 can act paradoxically as a tumor suppressor because B-cell lymphomas develop in gene-deleted mice (19). These two faces of SRC-3 highlight the fact that SRC-3 is a versatile protein, allowing the cell to decide between proliferation or growth suppression in a cell and signal context-dependent manner.
Although the majority of studies have been directed toward SRC-3, SRC-1 and SRC-2 are not without relevance to cancer. SRC-1 has been shown to be necessary for tumor metastasis in mice (30) and has been suggested as a reliable marker for disease reoccurrence in human breast cancer (31). SRC-2 also has been implicated in leukemic translocations (32).
Together with SRC-1 and SRC-2 (GRIP1/TIF2), SRC-3 completes the structurally related p160 family of coactivator proteins, whose C-terminal domains mediate interactions with histone acetyltransferases and the coregulators CBP and p300, while the A-terminal basic helix-loop-helix/PAS-containing domains interact with additional co-coregulators such as CoCoA (coiled-coil coactivator), GAC63 (Grip1-associated coactivator 63) and CARM1 (Coactivator-associated arginine methyltransferase 1) (20–22). These interactions and SRC-3 activation are modulated by posttranslational phosphorylation, whereby different combinations of site-specific phosphorylations provide interaction specificity for different coregulators and transcription factors (6). Phosphorylation has been directly linked to the oncogenic-proliferative potential of SRC-3 (23).
Ubiquitinylation is another posttranslational modification that is essential for the regulation of SRC-3 by cellular signaling (24). It was recently demonstrated that transcriptional activation and turnover of SRC-3 are events controlled by phosphorylation-dependent ubiquitinylation. More specifically, GSK3 kinase-mediated phosphorylation of a specific peptide sequence in SRC-3 precedes non-proteolytic activation of SRC-3 through mono-ubiquitinylation; multiple mono-ubiquitinylation events promote SRC-3 transcription factor specificity and NR coactivator function. Ultimately, the transition from mono-ubiquitinylation to long-chain polyubiquitinylation leads to SRC-3 degradation. Because the course of polyubiquitinylation is processive during the transcriptional activation of transcription factors, this ‘phosphorylation-dependent ubiquitinylation’ functions as a ‘transcriptional time clock’ to first initate coactivator activation, and then ultimately, to limit the lifetime of the PTM-activated coactivator (25).
Posttranslational modifications of SRC-3 influence its structural association dynamics with other co-coactivators. CARM1 methylates SRC-3 and dissociates it from its active coactivator complex (6). Peptidyl-prolyl isomerase 1 (Pin1) catalyzes the cis/trans isomerization of proline residues adjacent to phosphorylated serine/threonine-P bonds to induce conformational changes in the SRC-3 protein and enhance the interactions between SRC-3 and other coactivators such as CBP/p300 (26,27). Experimentally induced down-regulation of Pin1 in breast cancer cells was shown to reduce ligand-dependent transcription by nuclear receptors and Pin1 overexpression has been implicated in oncogenesis (33).
Metastatic tumor antigen 1 (MTA1) – a coregulator with dual function
MTA1 was initially identified as an upregulated gene through differential screening of a cDNA library from rat metastatic breast tumors (34). Subsequent studies found a widespread upregulation of MTA1 in multiple human tumors, including breast, prostate, colorectal, gastric, esophageal, ovarian, pancreatic, non–small cell lung, pancreatic, lymphoma, thymoma, and liver (35, 36). Forced overexpression of MMTV-MTA1 in the mouse mammary gland epithelium leads to ductal hyperbranching and proliferation in virgin glands and mammary gland adeno-carcinomas in older animals (37). In spite of a strong correlation between MTA1 upregulation and human cancer, the molecular functions of the MTA family remained a mystery until the proteomic analysis of the NuRD complex identified MTA1 as integral to the complex (38), and the first direct target of MTA proteins estrogen receptor-alpha (ER) was discovered (39). MTA1 directly interacts with ERα and HDACs, represses estrogen receptor element (ERE)-transactivation activity in a HDAC-sensitive manner, and promotes the development of hormone-independent growth of breast cancer cells. In addition to deacetylation of histones, MTA1-HDAC complexes interact and deacetylate non-histone proteins such as HIF1α, p53 and ERα, and affect the biology of these molecules (35).
In addition to a well-documented co-repressing role of MTA1 due to its interaction with HDACs and NuRD components, there are examples of a coactivating function of MTA1 on the breast cancer-amplified sequence 3 (BCAS3) (40) or Pax5 (41) owing to MTA1 interaction with RNA polymerase PolII and co-recruitment of MTA1-containing PolII complex to the target chromatin. In addition to BCAS3 and Pax5, MTA1 acts as a coactivator on genes important in transformation, epithelial-to-mesenchymal transition, and inflammation (R. Kumar, unpublished findings). Furthermore, the goal of gene stimulation also could be accomplished by a MTA family member by de-repression due to inhibition of a corepressor. For example, another MTA family member MTA3 stimulates the transcription of Wnt4 by repressing expression of Six3 which negatively controls Wnt4 expression (42).
These examples reinforce the notion that the functional MTA1-coactivator and corepressor complexes contain different core components. A larger issue relates to how exactly the same coregulator chooses to be part of complexes with opposing functions. In principle, this could result from one or more of the following possibilities: a) differential posttranslational modifications of MTA1 leading to distinct protein-protein interactions; b) relative abundance and/or stability of corepressor versus coactivator complexes; c) tempo-spatial influence of chromatin in the vicinity of a target gene; d) physical interactions with proteins that participate in conferring target promoter specificity; e) displacement or competition of activating multi-protein complexes on the target gene chromatin with HDACs complexes; and finally, f) signal-dependent alterations in the nature of coregulatory complexes.
While the scientific community waits for a precise experimental validation of these possibilities, recent data support the idea that the posttranslational acetylation of MTA1 on lysine 626 is essential for its coactivator function on BCAS3 transcription (40). The lysine 626, a site not conserved among other MTA family members, undergoes acetylation by histone acetyltransferase p300; this modification is required for MTA1 to interact with the RNA polymerase II (Pol II) complex and for its interaction with the BCAS3 promoter chromatin. Interestingly, BCAS3 itself is a bona-fide coactivator of ERE-driven transcription (43). Overexpression of BACS3 (44) as well as MTA1 (G. Landberg and R. Kumar, unpublished observations) in breast tumors has been observed to be associated with a reduced response of breast cancer patients to tamoxifen. These observations indicate that corepressor versus coactivator activity of MTA1 may be influenced by MTA1-binding partners in addition to PTMs. Indeed, in addition to BCAS3, MTA1 interacts with other coactivators such as MAT1 (a component of cyclin-dependent kinase-activating kinase (CAK), and MTA1-interacting coactivator 1 (MICoA), and NRIF3 (35). With the exception of MAT1, MTA1/HDAC complexes interfere with the recruitment of MICoA or NRIF3 coactivator complexes to the ERE-target gene chromatin. It is likely that the functionality of the ERα pathway is influenced by the balance between coactivator and corepressor complexes on ER target genes. Additional significance of the MTA1-K626 modification is revealed by the finding that elevated levels of MTA1 but not its lysine 626 acetylation-inactive mutant are transforming in nature.
A close analysis of MTA1 structure suggests that we are likely to discover additional posttranslational modifications on this protein. If this turns out to be the case, then there will be more layers of regulatory possibilities, not only dependent on individual site-specific modification(s) but also upon the resulting combinatorial PTMs. Each of a potential combination of PTMs could lead to altered, protein-protein interactions and eventually to a differential composition of MTA1-repressor and MTA1-coactivator complexes at a given target gene. In a physiologic-setting, these possibilities are driven by environmental signals which provide ultimate challenges to the cell to formulate and respond with appropriate molecular responses to achieve a desired biologic outcome.
PCBP1, an ultimate triple-threat coregulator
In addition to ‘selective coregulators’ that have dual function capacities, recent data point to the existence of molecules such as poly-C binding protein 1 (PCBP1) that possess multiple inherent functions in transcription, splicing, and translation (45). PCBP1 was originally discovered as a RNA-binding protein with a highly conserved triple repeat of the KH domain, and a protein that participates in mRNA processing at multiple steps (46). Interestingly, these PCBP1 functions are under the control of phosphorylations on Thr60 and Thr127 by the p21-activated kinase 1 (Pak1), a signaling nodular kinase stimulated by extracellular signals in cancer cells (47).
Under normal conditions, unphosphorylated PCBP1 binds to a subset of mRNA templates containing the DICE sequence and represses their translation (45). Once phosphorylated, PCBP1 dissociates from its target mRNAs, leading to the loss of translational repression followed by active translation of the mRNA targets. In parallel, phosphorylated PCBP1 translocates to the nucleus where it can participate in transcription and splicing events; the phospho-activated PCBP1 is recruited to oncogenic eIF4E gene chromatin, resulting in transcriptional stimulation. The coactivator function of PCBP1 is absolutely dependent on its phosphylation status as well as its nuclear localization. This event highlights a strategy wherein different pools of PCBP1 coregulator can act as a corepressor as well as a coactivator, and acquisition of the coactivator function is at the expense of simultaneous loss of corepressor activity. Accordingly, PCBP1 is capable of switching among its various functions depending upon its state of phosphorylation as well as its cellular localization, conferring a needed flexibility to PCBP1 to optimize its utility in eukaryotic gene expression in response to rapidly changing environmental signals.
An additional layer of coregulatory activity was uncovered by the finding that nuclear phosphorylated PCBP1 interacts with another coregulatory protein, called Caper-alpha (45); this interaction affects the recruitment of the spliceosome complex to the splice site to increase the production of properly spliced mature mRNAs. Although the resulting mRNAs are transported to the cytoplasm for translation by ribosomes, some of these mRNAs appear to remain under translational surveillance by PCBP1 until the cell is exposed to another round of mitogenic stimulation. PCBP1 represents an excellent example of a coregulator, which under appropriate signals, has the capacity to choose diverse functions – translation, transcription, or splicing. The ability to make such a choice is dependent on the signaling to one node (such as transcription or splicing) coupled with attenuation of another node (such as translation), to ensure a high biologic efficiency and economy. Available transcription profiling public data sets suggest that PCBP1 is upregulated in human advanced gastric cancer tissues (E-GEOD-2685), urinary superficial transitional cell carcinoma (E-GEOD-3167), malignant melanoma (E-GEOD-3189), and breast tumors (E-TABM-276).
Coregulators and Cancer Pathologies
Since the coregulators can be considered to be ‘master genes’ that have evolved to coordinate multiple diverse cellular reactions in order to implement major physiologic processes (4), they also contain the potential to efficiently promote cellular pathologies by coordinately misdirecting multiple independent functions. Evidence indicates that PTMs of coactivators can influence pathologic inflammation, aberrant adipogenesis, cell replication abnormalities and oncogenesis, etc. Impressively, of the ~300 coregulators identified by August 2007, >165 of them already had been associated with some disease state in humans4 (4, 11) (also see www.NURSA.org). Most of these associations have been published only in recent years, and we suspect it represents only the tip of the iceberg of what remains to be learned of these master genes in terms of human diseases.
NR coregulator misexpression represents the largest group of related pathologies in the list of pathologies and cancer is no exception. Comparing expression data reported in the literature with array data from Oncomine (http://www.oncomine.org) reveals that many instances of NR coregulator expression in human cancer tissues have been published. More NR coregulators are over- rather than under-expressed in cancers; in leukemia and lymphoma, the number of NR coregulators reported to be misexpressed are close to the total number of NR coregulator genes identified (11). Of course, over- or under-expression of NR coregulators may represent the consequence of the pathology rather than playing a causal role in the genesis of cancer. Still, misexpression of key regulatory factors that integrate functional signaling networks in a cancer cell provides the potential for amplification of temporal disease progression and also can counteract the biological activities of therapeutic drugs. Perturbations of the coregulator-transcription network due to misexpression restate the concept of coregulator competition as a regulatory strategy in the eukaryotic cell. For example, for β-catenin it was shown that elevated levels of homeodomain-containing proteins compete with TCF/LEF for nuclear β-catenin, thus switching transcriptional events to dictate cell-lineage determination (48).
Some NR coregulator target genes display a plethora of mutations that co-orchestrate human pathologies. The cyclin D1 gene, for example, was found to be translocated to immunoglobulin gene regions [t(11;14)(q13;q32)] in certain B-lymphocytic malignancies, particularly mantle-cell lymphoma (49); a common A/G polymorphism at nucleotide 870 (codon 242) alters mRNA splicing to produce two transcripts, with the consequence of increased susceptibility to colorectal cancer (50) and breast cancer (51). Amplification of the cyclin D1 gene was identified in oral squamous cell carcinoma cases (52), and the gene is generally overexpressed in many different tumors. Examples of other NR coregulators with gene amplifications in human diseases are BCAS2 and BCAS3, Caveolin 1, CoAA/CoAM, the cyclins A2, D1, D3, MLL2, PPMiD and SRC-3.
Both breast cancer amplified sequence 2 (BCAS2) and 3 (BCAS3) were identified as overexpressed and amplified genes in certain breast cancer cell lines and subsequently shown to be coactivators for ERα (53, 43). Despite the similar terminology, these genes do not share sequence similarity. The BCAS2 protein was found to be a component of the spliceosome and also to associate with other nuclear receptors in a ligand-independent fashion (53). The BCAS3 gene can transpose to another breast carcinoma amplified sequence (BCAS4) with unknown function (54). BCAS3 needs the NR coregulator PELP1/MNAR to function as an ERα coactivator. Interestingly, BCAS3 is also an ERα target gene in addition to PELP1. MTA acts as a transcriptional activator of BCAS3 expression, indicating a feed-forward mechanism whereby ER activation triggers a positive feedback loop through its ability to promote the expression of several of its own coregulators.
Caveolin-1 (CAV1), a principal component of plasma membrane vesicles (caveolae) and a platform protein for many signal transduction pathways, was shown to be a positive regulator of ERα signal transduction and a NR coactivator (55). CAV1 links integrin subunits to the tyrosine kinase FYN, which is an initiating step in coupling integrins to the Ras-ERK pathway and promoting cell cycle progression. Coactivator activator A (CoAA, also termed SYT-interacting protein, SIP; or RNA binding motif protein 14; RBM14) is an RRM-containing coactivator that promotes NR-mediated transcription through synergistic interactions with other coregulators such as thyroid hormone receptor-binding protein (TRBP) and CBP (39). An alternative splice form of CoAA, termed coactivator modulator CoAM, functions as a transcriptional repressor. Together, CoAA/CoAM regulates mRNA alternative splicing and affects transcription and splicing and cell growth in a promoter-preferential manner. With regard to cancer, the CoAA gene is overexpressed or amplified in certain non-small cell lung carcinomas, squamous cell skin carcinomas and lymphomas (57).
Protein phosphatase, PPM1D, was initially characterized as a p53-regulated phosphatase responsible for the inactivation of p38 MAPK and consequent inactivation of p53. It was shown to be a coregulator through its stimulation of the transcriptional activity of several NRs (58). Myeloid/lymphoid or mixed-lineage leukemia 2 (MLL2) is a component of a novel Set1-like complex in mammalian cells. The protein was shown to coactivate ERα via its two LXXLL motifs in breast cancer cells (59). MLL2 also has histone H3 Lys 4 methyltransferase activity that is dependent on menin, a protein mutated in multiple neoplasia type I (MEN1). MLL2 is amplified in some solid tumors (60) and was functionally linked to mixed-lineage leukemia (61). Finally, the protein phosphatase, PP1A, has been shown to modulate cancer cell growth by dephosphorylation of two sites in the N-terminal domain of SRC-3 that are required for its turnover and activity (28).
The numbers of NR coregulator genes disrupted because of chromosomal aberrations is large. For example, t (8;16) and inv (8) translocations of CBP and SRC2, are rearranged downstream of the monocytic leukemia zinc (MOZ) finger gene, resulting in a fusion protein consisting of the N terminus of MOZ fused to the C terminus of SRC2 (62). Chromosomal translocations in the genes BCL3 (B-cell lymphoma 3), CTIP-2 (COUP-TF-interacting protein 2), ELL (Eleven-nineteen lysine-rich leukemia), MN1 (Meningioma 1) and TRIP230 (Thyroid hormone receptor-interacting protein 230 kDa) are all associated with either lymphocytic (Bcl3) or acute myeloid leukemias. Translocation of the TIF-1α gene (Transcriptional intermediary factor 1α, or TRIM24 for tripartite motif-containing 24) with RET generates the TIF1/RET (PTC6) oncogene found in thyroid papillary carcinomas, while translocations involving either the Atro (Atrophin) or the PSF (Polypyrimidine tract-binding protein-associated splicing factor) genes contribute to neuroblastomas and papillary renal cell carcinoma, respectively. Translocation of acute myeloid leukemia 1 (AML1) gene to FOG-2 (Friend of GATA 2, officially termed ZFPM2 for Zinc finger protein, multitype 2) plays a central role in the pathogenesis of acute myeloid leukemia and myelodysplasia due to is interaction with COUP-TF NRs (63).
Finally, mutations in the two types of cancer genes discussed above – oncogenes and tumor suppressor genes – can occur in the germline as well, leading to hereditary predispositions to cancer. For example, individuals with germline mutations have a ‘head start’ in the cancer progression process by inherited possession of the first of two ‘hits’ to both alleles of a tumor suppressing gene. Inherited mutations in DNA repair genes (so called stability or ‘caretaker’ genes), rather than mutations in oncogenes or tumor suppressor genes, are often the major contributing factors for certain forms of breast and colon cancers (64). The BRCA genes are probably the most recognizable of these types of genes. BRCA1 and BRCA2 also are known to be coregulators, and it is thought that their coregulator role, at least in part, contributes to their tumor suppressor action. Some other well-known inherited cancer predisposition genes whose gene products also function as NR coregulators are p53, PTEN, TSC2, RB1, and SMAD4.
In summary, a greater understanding of these coregulator master genes should prove beneficial to the diagnosis and therapy of cancer. Assessments of coregulator levels and/or PTM status are already being utilized for diagnosis and prognosis of human cancers (29). In addition, coregulators could serve as potential new therapeutic targets for cancer. For example, inhibition of an oncogenic coactivator could simultaneously silence a cadre of downstream genes, which together, are responsible for the accelerated growth of the oncogenic cell (Fig. 1). In this manner, a small molecule drug designed against a powerful growth coactivator, such as SRC-3, could lead to simultaneous suppression of a host of downstream target genes (see Fig. 1 and legend). Also, since coregulator PTMs are a result of prior specific enzymatic reactions, the modifying enzymes are subject to inhibition/activation with drugs. Thus, it is only a matter of time before the fields of medicine and pharmacology realize the potential of this new information for therapeutic intervention and prognosis in cancers (12).
Figure 1. Targeting coactivators for therapy.
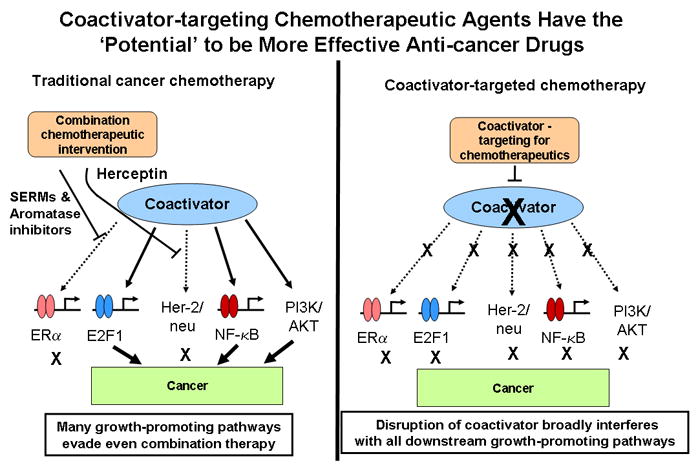
Left panel: Coactivator (e.g., SRC-3/AIB1) regulates the coordinate expression of multiple downstream genes (ER-responsive; E2F1-responsive; Her-2 responsive; NF-κB responsive; and protein kinase [P13K/AKT] responsive). These TF-responsive genes are all required for proliferation and metastasis of cancer cells. Targeting the individual pathways by using SERMS/aromatase inhibitors (for ER) or Herceptin (for Her-2/neu) only inhibits one or two pathways (for combinations). Cancer growth is then slowed, but the other pathways are upregulated to compensate, thereby producing eventual resistance to the therapy.
Right panel: If the coactivator is targeted and its function inhibited, simultaneous suppression of all pathways occurs. This scenario blocks alternate pathway compensatory upregulation, decreasing the onset of eventual drug resistance.
Acknowledgments
Research in authors’ laboratories is supported by NIH HD-08818, NIDDK DK059820, NIDDK DK062434-07/10, Welch Q-1521, and NCI P30CA125123 (BWO); and NCI CA98823 and CA90970, and CA80066 (RK).
Footnotes
Disclosure of Potential Conflicts of Interest. No potential conflicts of interests were disclosed.
References
- 1.Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–67. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 2.O’Malley BW. Coregulators: from whence came these “master genes”. Mol Endocrinol. 2007;21:1009–13. doi: 10.1210/me.2007-0012. [DOI] [PubMed] [Google Scholar]
- 3.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:466–74. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 4.O’Malley BW. Moloecular Biology - Little molecules with big goals. Science. 2006;313:1749–50. doi: 10.1126/science.1132509. [DOI] [PubMed] [Google Scholar]
- 5.Nagaich AK, Rayasam GV, Martinez ED, et al. Subnuclear trafficking and gene targeting by steroid receptors. Ann N Y Acad Sci. 2004;1024:213–20. doi: 10.1196/annals.1321.002. [DOI] [PubMed] [Google Scholar]
- 6.Lonard DM, O’Malley BW. Nuclear receptor coregulators: Judges, juries and executioners of cellular regulation. Molecular Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Lonard DM, O’Malley BW. SRC-3 Transcription-coupled activation, degradation and the ubiquitin clock: Is there enough coactivator to go around in cells? Science Signaling (STKE) 2008;1:pe16. doi: 10.1126/stke.113pe16. [DOI] [PubMed] [Google Scholar]
- 9.Anzick SL, Kononen J, Walker, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–68. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 10.Osborne CK, Bardou V, Hopp TA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 11.Lanz RB, Londard DM, O’Malley BW. Nuclear receptor coregulators in human diseases. In: Kumar R, O’Malley BW, editors. NR Coregulators and Human Diseases. World Scientific Publishing Co; 2008. pp. 1–133. [Google Scholar]
- 12.Hall JM, McDonnell DP. Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol Interv. 2005;5:343–57. doi: 10.1124/mi.5.6.7. [DOI] [PubMed] [Google Scholar]
- 13.Xie D, Sham JS, Zeng WF, et al. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol. 2005;36:777–83. doi: 10.1016/j.humpath.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Xu FP, Xie D, Wen JM, et al. SRC-3/AIB1 protein and gene amplification levels in human esophageal squamous cell carcinomas. Cancer Lett. 2007;245:69–74. doi: 10.1016/j.canlet.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Torres-Arzayus MI, Font de Mora J, Yuan J, et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;3:263–74. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 16.Kuang SQ, Liao L, Zhang H, Lee AV, O’ Malley BW, Xu J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875–85. doi: 10.1158/0008-5472.can-03-3745. [DOI] [PubMed] [Google Scholar]
- 17.Kuang SQ, Liao L, Wang S, Medina D, O’ Malley BW, Xu J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005;17:7993–8002. doi: 10.1158/0008-5472.CAN-05-1179. [DOI] [PubMed] [Google Scholar]
- 18.Tien JCY, Zhou S, Xu J. The role of SRC-1 in murine prostate carcinogenesis is nonessential due to a possible compensation of SRC-3/AIB1 overexpression. Int J Biol Sci. 2009;5:256–64. doi: 10.7150/ijbs.5.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coste A, Antal MC, Chan S, et al. Absence of the steroid receptor coactivator-3 induces B-cell lymphoma. EMBO J. 2006;25:2453–64. doi: 10.1038/sj.emboj.7601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen YH, Kim JH, Stallcup MR. GAG63, a GRIP1-dependent nuclear receptor coactivator. Mol Cell Biol. 2005;25:5965–72. doi: 10.1128/MCB.25.14.5965-5972.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JH, Li H, Stallcup MR. CoCoA, a nuclear receptor coactivator which acts through an N-terminal activation domain of p160 coactivators. Mol Cell. 2003;12:1537–49. doi: 10.1016/s1097-2765(03)00450-7. [DOI] [PubMed] [Google Scholar]
- 22.Lee YH, Campbell HD, Stallcup MR. Developmentally essential protein flightless I is a nuclear receptor coactivator with actin binding activity. Mol Cell Biol. 2004;24:2103–17. doi: 10.1128/MCB.24.5.2103-2117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu RC, Qin J, Yi P, et al. Selective phosphory-lations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–49. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Lonard DM, Nawaz Z, Smith CL, O’Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–48. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 25.Wu RC, Feng Q, Lonard DM, O’Malley BW. SRC-3 Coactivator Functional Lifetime Is Regulated by a Phospho-Dependent Ubiquitin Time Clock. Cell. 2007;129:1125–40. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–86. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 27.Yi P, Wu RC, Sandquist J, et al. Peptidyl-prolyl isomerase 1 (Pin1) serves as a coactivator of steroid receptor by regulating the activity of phosphorylated steroid receptor coactivator 3 (SRC-3/AIB1) Mol Cell Biol. 2005;21:9687–99. doi: 10.1128/MCB.25.21.9687-9699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Liang YY, Feng XH, Tsai SY, Tsai MJ, O’Malley BW. Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol Cell. 2008;31:835–49. doi: 10.1016/j.molcel.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonard DM, Lanz RB, O’Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28:575–87. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Yuan Y, Liao L, et al. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc Natl Acad Sci USA. 2009;106:151–6. doi: 10.1073/pnas.0808703105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redmond AM, Bane FT, Stafford AT, et al. Co-association of estrogen receptor and p160 proteins predicts resistance to endocrine treatment; SRC-1 is an independent predictor of breast cancer recurrence. Clin Cancer Res. 2009;15:2098–106. doi: 10.1158/1078-0432.CCR-08-1649. [DOI] [PubMed] [Google Scholar]
- 32.Sansone R, Sessarego M, Haupt R, Garré ML, Strigini P. Ins(6;1) in a patient with congenital leukemia. Cancer Genet Cytogenet. 1989;37:19–22. doi: 10.1016/0165-4608(89)90069-1. [DOI] [PubMed] [Google Scholar]
- 33.Wulf GM, Ryo A, Wulf GG, et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–72. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toh Y, Pencil SD, Nicolson GL. A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J Biol Chem. 1994;269:22958–963. [PubMed] [Google Scholar]
- 35.Manavathi B, Kumar R. Metastasis tumor antigens antigens, an emerging family of multifaceted master coregulators. J Biol Chem. 2007;282:1529–33. doi: 10.1074/jbc.R600029200. [DOI] [PubMed] [Google Scholar]
- 36.Manavathi B, Singh K, Kumar R. MTA1 family of coregulators in nuclear receptor signaling. Nuclear Receptor Signal. 2007:e010. doi: 10.1621/nrs.05010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagheri-Yarmand R, Talukder AH, Wang RA, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development. 2004;131:3469–79. doi: 10.1242/dev.01213. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–35. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mazumdar A, Wang RA, Mishra SK, et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3:30–37. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- 40.Gururaj AE, Singh RR, Rayala SK, et al. MTA1, a transcriptional activator of breast cancer amplified sequence 3. Proc Natl Acad Sci USA. 2006;103:6670–75. doi: 10.1073/pnas.0601989103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balasenthil S, Gururaj AE, Talukder AH, et al. Identification of Pax5 as a target of MTA1 in B-cell lymphomas. Cancer Res. 2007;67:7132–38. doi: 10.1158/0008-5472.CAN-07-0750. [DOI] [PubMed] [Google Scholar]
- 42.Zhang H, Singh RK, Talukder AH, Kumar R. Metastatic tumor antigen 3 is a direct corepressor of WNT4 pathway. Genes & Development. 2006;20:2943–48. doi: 10.1101/gad.1461706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gururaj AE, Peng S, Vadlamudi R, Kumar R. Estrogen induces expression of BCAS3, a novel estrogen receptor-alpha coactivator, through PELP1. Mol Endo. 2007;21:1847–60. doi: 10.1210/me.2006-0514. [DOI] [PubMed] [Google Scholar]
- 44.Gururaj A, Holmes C, Landberg G, Kumar R. Breast Cancer–Amplified Sequence 3, a target of MTA1, contributes to tamoxifen resistance in premenopausal patients with breast cancer. Cell Cycle. 2006;5:1407–10. doi: 10.4161/cc.5.13.2924. [DOI] [PubMed] [Google Scholar]
- 45.Meng Q, Rayala S, Gururaj AE, Talukder AH, O’Malley BW, Kumar R. Signaling-dependent and coordinated regulation of transcription, splicing, and translation resides in a single coregulator, PCBP1. Proc Nat Acad Sci USA. 2007;104:5866–71. doi: 10.1073/pnas.0701065104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi HS, Hwang CK, Song KY, Law PY, Wei LN, Loh HH. Poly(C)-binding proteins as transcriptional regulators of gene expression. Biochem Biophys Res Commun. 2009;380:431–36. doi: 10.1016/j.bbrc.2009.01.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar R, Gururaj A, Barnes CJ. p21-activating kinases in cancer. Nat Review Cancer. 2006;6:459–62. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 48.Olson LE, Tollkuhn J, Scafoglio C, et al. Homeodomain-mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell. 2006;125:593–605. doi: 10.1016/j.cell.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 49.Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science. 1984;224:1403–6. doi: 10.1126/science.6610211. [DOI] [PubMed] [Google Scholar]
- 50.Le Marchand L, Seifried A, Lum-Jones A, Donlon T, Wilkens LR. Association of the cyclin D1 A870G polymorphism with advanced colorectal cancer. J Am Med Associ. 2003;290:2843–48. doi: 10.1001/jama.290.21.2843. [DOI] [PubMed] [Google Scholar]
- 51.Shu XO, Moore DB, Cai Q, et al. Association of cyclin D1 genotype with breast cancer risk and survival. Cancer Epidemiol Biomarkers Prev. 2005;14:91–97. [PubMed] [Google Scholar]
- 52.Wong YK, Lin SC, Chang CS, et al. Cyclin D1 genotype in areca-associated oral squamous cell carcinoma. J Oral Pathol Med. 2003;32:265–70. doi: 10.1034/j.1600-0714.2003.00131.x. [DOI] [PubMed] [Google Scholar]
- 53.Qi C, Zhu YT, Chang J, Yeldandi AV, Rao MS, Zhu YJ. Potentiation of estrogen receptor transcriptional activity by breast cancer amplified sequence 2. Biochem Biophys Res Commun. 2005;328:393–98. doi: 10.1016/j.bbrc.2004.12.187. [DOI] [PubMed] [Google Scholar]
- 54.Bärlund M, Monni O, Weaver JD, et al. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer. 2002;35:311–17. doi: 10.1002/gcc.10121. [DOI] [PubMed] [Google Scholar]
- 55.Schlegel A, Wang C, Katzenellenbogen BS, Pestell RG, Lisanti MP. Caveolin-1 potentiates estrogen receptor alpha (ERalpha) signaling. caveolin-1 drives ligand-independent nuclear translocation and activation of ER-alpha. J Biol Chem. 1999;274:33551–56. doi: 10.1074/jbc.274.47.33551. [DOI] [PubMed] [Google Scholar]
- 56.Iwasaki T, Chin WW, Ko L. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM) J Biol Chem. 2001;276:33375–83. doi: 10.1074/jbc.M101517200. [DOI] [PubMed] [Google Scholar]
- 57.Sui Y, Yang Z, Xiong S, et al. Gene amplification and associated loss of 5′ regulatory sequences of CoAA in human cancers. Oncogene. 2007;26:822–835. doi: 10.1038/sj.onc.1209847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Proia DA, Nannenga BW, Donehower LA, Weigel NL. Dual roles for the phosphatase PPM1D in regulating progesterone receptor function. J Biol Chem. 2006;281:7089–101. doi: 10.1074/jbc.M511839200. [DOI] [PubMed] [Google Scholar]
- 59.Mo R, Rao SM, Zhu YJ. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem. 2006;281:15714–720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 60.Huntsman DG, Chin SF, Muleris M, et al. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene. 1999;18:7975–84. doi: 10.1038/sj.onc.1203291. [DOI] [PubMed] [Google Scholar]
- 61.Hess JL. Mechanisms of transformation by MLL. Crit Rev Eukaryot Gene Expr. 2004;14:235–54. doi: 10.1615/critreveukaryotgeneexpr.v14.i4.10. [DOI] [PubMed] [Google Scholar]
- 62.Troke PJ, Kindle KB, Collins HM, Heery DM. MOZ fusion proteins in acute myeloid leukaemia. Biochem Soc Symp. 2006;73:23–39. doi: 10.1042/bss0730023. [DOI] [PubMed] [Google Scholar]
- 63.Chan EM, Comer EM, Brown FC, et al. AML1-FOG2 fusion protein in myelodysplasia. Blood. 2005;105:4523–6. doi: 10.1182/blood-2004-07-2762. [DOI] [PubMed] [Google Scholar]
- 64.Friedberg EC. DNA damage and repair. Nature. 2003;421:436–40. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]