

Abstract
MAX dimerization protein 1 (MAD1) is a transcription suppressor that antagonizes MYC-mediated transcription activation, and the inhibition mechanism occurs mainly through the competition of target genes' promoter MYC binding sites by MAD1. The promoter binding proteins switch between MYC and MAD1 affects cell proliferation and differentiation. However, little is known about MAD1's regulation process in cancer cells. Here, we present evidence that AKT inhibits MAD1-mediated transcription repression by physical interaction with and phosphorylation of MAD1. Phosphorylation reduces the binding affinity between MAD1 and its target genes' promoter and thereby abolishes its transcription suppression function. Mutation of the phosphorylation site from serine to alanine rescues the DNA-binding ability in the presence of activated AKT. In addition, AKT inhibits MAD1 mediated target genes (hTERT and ODC) transcription repression and promotes cell cycle and cell growth. However, mutated S145A MAD1 abrogates the inhibition by AKT. Thus, our results suggest that phosphorylation of MAD1 by AKT inhibits MAD1-mediated transcription suppression and subsequently activates the transcription of MAD1 target genes.
Keywords: AKT, MAD1, hTERT
Introduction
The MYC/MAX/MAD family is a group of basichelix-loop helix-leucine-zipper (bHLHZip) transcription factors that play important roles in cell physiology, including proliferation, transformation, differentiation, and apoptosis. Among these factors, MYC is the most well known factor because of its frequent dysregulation in many types of cancer [1]. MYC is a transcription activator that forms a heterodimer complex with MAX and interacts with the consensus 5′-CACGTG-3′ DNA sequence, referred to as E-box [2]. Subsequently, the transcription activation domain (TAD) in MYC recruits transcription activators such as transformation/transcription domain-associated protein (TRRAP) and creb-binding protein (CBP) to promote expression of its target genes [3].
While MYC stimulates target genes expression, MAD1 serves an opposite role by repressing their transcription. MAD1 has the same common bHLHZip motif as MYC. However, MAD1 possesses a Sin3-interacting domain (SID) in the N terminal, thereby recruiting the mSin3-HDAC transcription repressor complex [4,5]. Similar to MYC, formation of the heterodimer complex with MAX is required for MAD1 to interact with the E-box region on the promoter. Thus, the occupation of a target gene's promoter by MYC or MAD1 determines the positive or negative expression, respectively [6].
The expression levels of MYC and MAD1 are considered opposite in normal cells. A general observation is that a high expression level of MYC is found in proliferating cells, whereas the MAD1 expression level is elevated in differentiated cells [7]. Thus, one aspect of antagonism between MAD1 and MYC is their distinct expression patterns. Interestingly, however, it has been reported that MAD1 is expressed in several different types of cancer, including leukemia, gastric cancer, and breast cancer [8-10]. Therefore, alternate types of regulation, such as post-translation modifications may occur in cancer cells to suppress MAD1 function.
The phosphoinositide 3-kinase (PI3K)-AKT signaling pathway plays an important role in preventing cells from undergoing apoptosis and contributes to the pathogenesis of malignancy, including proliferation [11,12]. Furthermore, AKT is frequently dysregulated in cancer cells and activates several MYC and MAD1 target genes such as human telomerase reverse transcriptase (hTERT) and ornithine decarboxylase (ODC) [13-15]. The activations of these target genes occur mainly through transcriptional activation, but the regulation mechanisms remain obscure. It has been reported that MAD1 is phosphorylated at serine residue 145 (Ser145) by RSK and S6K. The phosphorylation inhibits MAD1 function by promoting MAD1 degradation [16]. In this study, we confirmed that the phosphorylation of Ser145 inhibits MAD1 function. However, in addition to RSK and S6K, we demonstrated that MAD1-mediated transcriptional repression is also suppressed by AKT and that this suppression is correlated with MAD1 DNA-binding affinity. We found that AKT can physically associate with MAD1 and phosphorylates a consensus serine residue (Ser145). Phosphorylated MAD1 reduces its binding affinity to the promoter DNA E-box site. Consequently, the E-box on the promoter is dominantly occupied by MYC while MAD1 is phosphorylated. Thus, our study shows that AKT plays an important role in regulating MAD1 target genes by inhibiting MAD1 inhibition.
Materials and Methods
Cell Culture and Transfection Conditions
All cell lines were provided by ATCC and maintained in Dulbecco's modified Eagle's medium/F-12 with 10% fetal calf serum and 100 μg/ml penicillin and streptomycin. For transient transfection, cells were transfected with DNA using either SN liposome [17] or lipofectamine plus reagent. Briefly, cells were grown on Petri dishes and incubated with a plasmid-liposome complex in serum-free medium for 4h, after which the medium was replaced by complete medium and the cells incubated at 37°C for 24–48h.
Plasmids, Antibodies, and Chemicals
DNA plasmids encoding CA-AKT and DN-AKT were described previously [18]. The full-length MAD1 cDNA was then subcloned into the pCMV5-FL, pGEX-6P-1, and pEGFP-C2 vectors. Mutations of Ser-145 to Ala in MAD1 and GST-MAD1 were generated using a Quickchage site-directed mutagenesis kit (Strategene) according to the manufacturer's instructions and mutations were confirmed by DNA sequencing. We used antibodies to Flag (Sigma, F3165), HA (Roche, 11666606001), MAD1 (Santa Cruz Biotechnology, C-19), MAX (Santa Cruz Biotechnology, C-17), c-MYC (Cell Signaling Technology, 9402), pS6K1(T389) (Cell Signaling Technology, 9205), pAKT(S473) (Cell Signaling Technology, 9271), AKT (Cell Signaling Technology, 9272, 2966), Phospho-(Ser/Thr)AKT Substrate antibody (Cell Signaling Technology, 9611), ERK1/2 (Upstate Biotechnology, 06-182), pERK1/2(T202/Y204) (Cell Signaling Technology, 4377), and ACTIN (Sigma, A2066). Antibodies to the Ser145 phosphorylation sites of MAD1 were generated in collaboration with the Center for Molecular Medicine, China Medical University Hospital. Synthetic phosphorylated peptides representing portions of MAD1 around serine 145 was used as an antigen for producing antibodies. Rapamycin, IGF, and cycloheximide were purchased from Sigma. PD98059 was purchased from Cell Signaling.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP analysis was performed as previously described [19]. Cells were lysed with sodium dodecyl sulfate(SDS) lysis buffer (1% SDS, 10mM EDTA, and 50mM Tris, pH 8.1) and then sonicated and subjected to immunoprecipitation with indicated antibodies. The resulting samples were purified by phenol/chloroform extraction followed by ethanol precipitation. Polymerase chain reactions for ChIP were performed and analyzed on ethidium bromide-stained agarose gels.
In Vitro Pull-down Assay
Recombinant AKT proteins (Upstate Biotechnology, 14-276) were incubated with in vitro transcription and translation lysates of Flag-tagged MAD1, which was produced using a TNT-coupled reticulocyte lysate system (Promega), in binding buffer (25 mM Tris-HCl, pH 7.5, 125 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 μg of leupeptin/ml, 1 μg of aprotinin/ml, and 1 μg of pepstatin/ml) for 1 h at 4°C. Anti-AKT antibody was then added and incubation was continued at 4°C overnight. The resulting immunocomplexes were precipitated with protein A-Sepharose beads (Roche) at 4°C for 4 h and washed extensively with Tris-buffered saline. The bound proteins were eluted with sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer and then analyzed.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting were performed as previously described [20].
DNA Affinity Protein-binding Assay (DAPA)
The biotin-labeled double-stranded oligonucleotides were synthesized by the company we obtained them from (Sigma); Biotin-5′-CCGCGCTTCCCACGTGGCGGAGGGAC-3′ and 5′ GTCCCTCCGCCAC GTGGGAAGCGCGG-3′; according to the hTERT promoter MAD1 binding sequence. The binding assay was performed by mixing 400 μg of nuclear proteins, 2 μg biotin-labeled DNA oligonucleotides, and 20 μl streptavidin-agarose beads (4%) with 70% slurry. The mixture was incubated at room temperature for 1 h with shaking. The beads were pelleted and washed with cold phosphate buffered saline PBS three times. The binding proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by Western blot analysis, and probed with specific antibodies.
Identification of Phosphorylation Sites by Mass Spectrometry
After protein gel electrophoresis, bands corresponding with MAD1 phosphorylated by AKT in vitro were identified, excised from the gels, and subjected to tryptic digestion. Subsequently, samples were isolated by immobilized metal affinity chromatography, the enriched phosphopeptides were analyzed by micro-liquid chromatography/tandem mass spectrometry. The raw data were searched against the National Center for Biotechnology Information (NCBI) protein sequence database using the Mascot search engine.
In Vitro Phosphorylation
In vitro phosphorylation was assessed as described previously [20]. The AKT kinase (Upstate Biotechnology, 14-276) was incubated at 30°C for 30 min in 20 μl of kinase reaction mixture containing 10 μM [γ-32P[ATP and 1 μg of bacterial expressed GST-MAD1 as AKT substrate. The reaction was stopped by the addition of an equal volume of protein sample buffer. Proteins were separated by electrophoresis on 10% SDS polyacrylamide gels, and phosphorylated GST-fusion proteins were visualized by autoradiography.
Quantitative RT-PCR
MCF7 cells were transfected with GFP-MAD1 and sorted for GFP positive cells. Total RNA was extracted from GFP-positive cells by TRIzol (Invitrogen, Carlsbad, CA) and was quantified by SmartSpec Plus Spectrophotometer (BIORAD, Hercules, CA). The reverse transcription reaction was performed using the Superscript III first-strand synthesis system (Invitrogen) according to the manufacturer's instructions. Quantitative RT-PCR analyses of human hTERT and ODC were performed using designed primers (5′-AACCTTCCTCAGCTATGCCC, 5′-GTTTGCGACGCATGTTCCTC for hTERT and 5′-TCGATGAAGGTTTTACTGCCAAG, 5′-AGAGCTTTTAACCACCTCAGATG for ODC) and iQ SYBRTM Green Supermix (BIORAD, Hercules, CA). Assays were performed using the ABI StepOne Plus Real-Time PCR system (Applied Biosystems, Foster City, CA). GAPDH was used for normalization.
Genome Structure Analyses
For SNP-based copy number analysis, we obtained Affymetrix 10K SNP microarray genotype data for 104 breast cancers clinical samples and cell lines from the Cancer Genome Project of the Wellcome Trust Sanger Institute. The SNP data of the MAD1 loci were analyzed and visualized using Cluster and TreeView software (Eisen, MB), and the results were presented in heat maps. For array comparative genomic hybridization (aCGH) analysis, we used SIGMA software to examine the genome gain and loss of the MAD1 loci.
Cell Cycle, Cell Proliferation, Reporter, and Immunofluorescence Assays
For cell cycle analysis, MCF7 cells were transfected with GFP-MAD1 with or without AKT for 30 h. Cells were harvested and stained with propidium iodide, and subjected to FACS analysis. For cell proliferation assay, Hela cells were transfected with wild-type or S145A mutant MAD1 with or without constitutive active AKT. The cell proliferation rate was measure by MTT assay. Luc assays were performed using Dual-Luciferase Assay (Promega). For immunofluorescence, MCF7 was co-transfected with Flag-MAD1 plus the indicated AKT constructs. After fixation, the cells were stained with anti-Flag and anti-HA antibodies, and the cellular localizations of MAD1 and AKT were determined using a fluorescent microscope.
Results
MAD1 Expresses in Cancers in the Presence of MYC
MYC is deregulated in many types of cancer, and deregulation is also responsible for tumorigenesis [3,21,22]. However, clinical pathology studies have shown that MAD1 is expressed in various types of cancers [9,10]. To confirm the expression level of MAD1 in cancers and its correlation with MYC, we first searched for the MAD1 mRNA level and its correlation with MYC in 295 cases of breast carcinoma from the Oncomine database (http://www.oncomine.org). Positive expression of MAD1 was found in several cases but with a low correlation between MYC and MAD1 (R < 0.1) (Supplementary Figure 1A). Protein levels of MAD1 and MYC were also analyzed in several breast cancer cell lines, and low correlation between MAD1 and MYC was found (Supplementary Figure 1B). In addition, when the copy numbers of MAD1 loci were evaluated by SNP arrays in 104 breast cancer samples from the Cancer Genome Project database at Wellcome Trust Sanger Institute, no obvious genomics loss was found (Supplementary Figure 2). These results suggest that alternative regulation in suppressing MAD1 function in cancers.
AKT Suppresses MAD1 Transcription Repression Function
Several reports have indicated that AKT could regulate MYC and MAD1 target genes at the transcriptional level [13-15,23]; however, the mechanism for regulation is obscure. Because AKT is frequently dysregulated in cancers [24], we speculated that AKT activates MAD1 target genes by suppressing MAD1 function. To investigate this hypothesis, we transiently co-transfected pGL3-CM2 (4 × E-box luciferase reporter plasmid) and MAD1, along with dominant-negative or constitutively active AKT, into the cancer cell line MDA-MB-435 [25] and a rat fibroblast cell line Rat1. After 48 h of transfection, cells were lysed for the reporter assay. As shown in Figure 1A, dominant-negative AKT leads to a two-fold improvement in MAD1-mediated transcription repression, but the constitutively active AKT relieved such repression in the MDA-MB-435 cell line. We also observed similar effects in the Rat-1 cell line (Figure 1B), suggesting that MAD1 function is inhibited by AKT. Although there has been debated regarding whether MDA-MD-435 is a breast cancer or melanoma cell line [26], one report indicated that MDA-MD-435 indeed originates from a breast cancer cell line that expresses melanocyte proteins [25].
Figure 1.
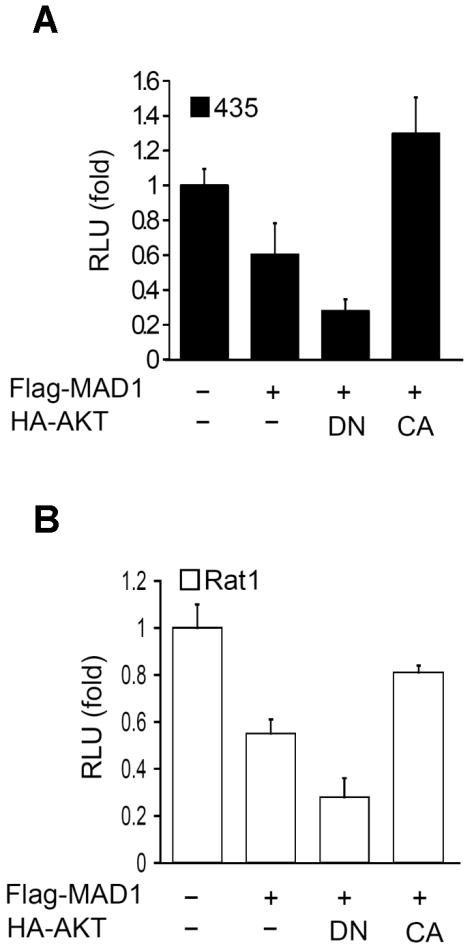
AKT suppresses MAD1 transcription repression function. (A) The dominant-negative AKT leads to a twofold increase in MAD1 repression, whereas constitutively active AKT relieves MAD1-mediated repression. Lysates of MDA-MB-435 cancer cells co-transfected with MAD1 and AKT were subjected to Ebox luciferase reporter assays. The relative activity was normalized by Renilla luciferase. (B) Similar effects were also observed in Rat1 cells subjected to the same experimental conditions. CA, constitutively active; DN, dominant negative.
MAD1 Is Physically Associated with AKT
The observation of AKT-mediated MAD1 inhibition raised a question; what is the mechanism of this negative regulation? Because AKT is a serine/-threonine kinase, we hypothesized that MAD1 is a substrate of AKT and that the phosphorylation of MAD1 suppresses its function. To address this hypothesis, the physical interaction between MAD1 and AKT was investigated. Co-immunoprecipitation assays showed that AKT physically interacted with MAD1 when both were overexpressed in the 293 cell line (Figure 2A) and this interaction was also observed endogenously in the MCF7 cell line (Figure 2B). The in-vitro pull-down assay further suggested that AKT and MAD1 have a direct interaction (Figure 2C). In addition, MAD1 showed an obvious interaction with constitutively active AKT but less with dominant-negative AKT (Figure 2D), suggesting that activation of AKT is important for the MAD1/AKT association.
Figure 2.
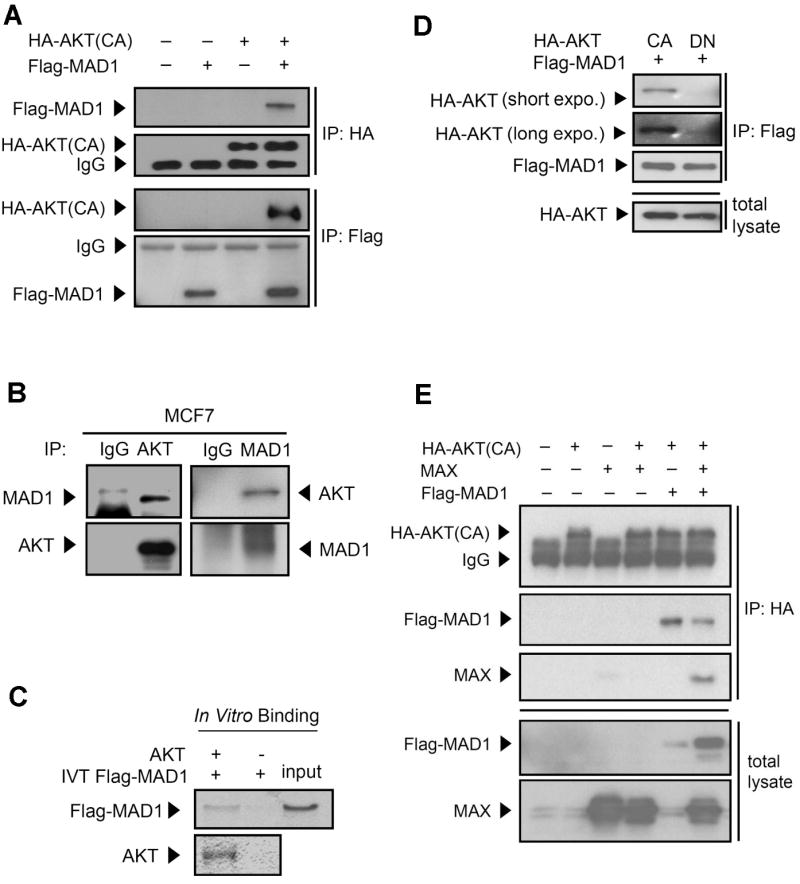
MAD1 physically interacts with AKT in vitro and in vivo. (A) Exogenous MAD1 interacted with AKT. Lysates of 293 cells co-transfected with HA-AKT and Flag-MAD1 were subjected to immunoprecipitation and immunoblotting with either anti-HA or anti-Flag antibodies. (B) Endogenous MAD1 interacted with AKT. Lysates of MCF7 breast cancer cells were analyzed by reciprocal co-immunoprecipitation and immunoblotting with anti-MAD1 and anti-AKT antibodies. (C) MAD1 directly interacted with AKT. In vitro transcribed Flag-MAD1 proteins, which were produced using a TNT-coupled reticulocyte lysate system, were incubated with AKT protein and then pulled down by antibody to AKT. IVT, in vitro transcription and translation. (D) Kinase activity is required for AKT and MAD1 interaction. CA and DN AKT were co-transfected with Flag-MAD1 in 293 cells. Lysates were immunoprecipitated by anti-Flag antibody and immunoblotting with anti-HA and anti-Flag antibodies. expo. is the abbreviation of “exposure time” (E) AKT physically interacted indirectly with MAX in the presence of MAD1. Lysates of 293 cells transfected with HA-AKT, Flag-MAD, and/or MAX were subjected to immunoprecipitation with the anti-HA antibody and immunoblotting with anti-MAX, anti-Flag, and anti-HA antibodies.
Since MAD1 forms a heterodimer complex with MAX to interact with the target genes' promoter and suppresses target gene transcription, the physical interaction between AKT and the MAD1/MAX complex was examined. Co-transfection of only MAX and AKT showed no obvious physical interaction (Figure 2E, lane 4). However, MAX showed a physical association with AKT in the presence of MAD1 (Figure 2E, lane 6). Thus, MAX indirectly associates with AKT, i.e., AKT interacts with the MAD1/MAX complex through MAD1.
AKT Phosphorylates MAD1 Predominately at Ser145
Given the physical interaction between AKT and MAD1, we examined whether MAD1 is a physical substrate of AKT-mediated phosphorylation. Co-transfection of MAD1 and AKT showed that AKT caused the accumulation of a slow-migrating form of MAD1, and this migrating form was entirely eliminated by treatment with calf intestinal alkaline phosphatase (CIAP)(Figure 3A). This mobility shift was caused by AKT-mediated post-translation modification on MAD1, which could be phosphorylation. Amino acid sequences revealed that MAD1 has a putative AKT phosphorylation site at Ser145 (Figure 3B). In addition, mass spectrometric analysis of GST-MAD1 showed that the Ser145 was phosphorylated by AKT in vitro (Figure 3C). To further confirm that MAD1 is phosphorylated at Ser145, the in vitro kinase assay demonstrated that MAD1 was phosphorylated by purified AKT kinase, and the mutation of Ser145 residue to Ala (S145A) abolished the phosphorylation (Figure 3D). These results suggest that AKT directly phosphorylates MAD1 at Ser145.
Figure 3.
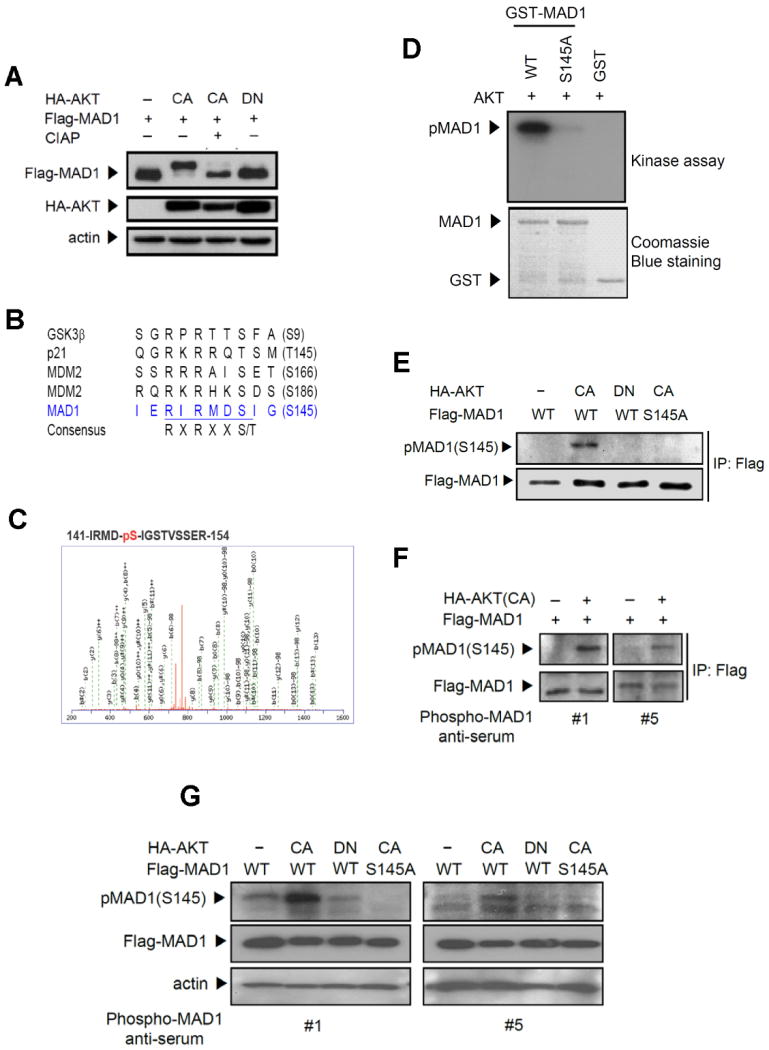
AKT phosphorylates MAD1 both in vitro and in vivo. (A) AKT induced a mobility shift of MAD1, and calf intestine alkaline phosphatase (CIAP) reversed it. Lysates from 293 cells transfected with HA-AKT and Flag-MAD1 were treated with CIAP and subjected to immunoblotting. (B) MAD1 has a putative AKT phosphorylation site at Ser145 (RIRMDS145). R, arginine; S, serine; T, threonine; X, any amino acid. (C) Mass spectrometric analysis of AKT-mediated MAD1 phosphorylation site on GST-MAD1 at Ser145. (D) MAD1 phosphorylation site Ser145 was identified by in vitro kinase assays in which recombinant AKT was incubated with GST-MAD1 and mutant GST-MAD1 (S145A) fusion proteins. (E) AKT-mediated phosphorylation of MAD1 was detected by phospho-(Ser/Thr) AKT substrate antibody and showed in vivo phosphorylation in the presence of AKT. Lysates from 293 cells transfected with the indicated AKT and MAD1 were subjected to immunoblotting. (F) Characterization of antibodies to pMAD1 (S145) by immunoprecipitation. Lysates from 293 cells transfected with MAD1 and AKT were immunoprecipitated by anti-Flag antibody and immunoblotting was performed using phospho-MAD1 anti-serum #1 and #5. (G) MAD1 Ser145 in vivo phosphorylation was detected by direct immunoblotting. Lysates were prepared as in (E).
To assess whether this phosphorylation also occurs in vivo, phospho-AKT substrate antibody was used to detect the AKT-mediated phosphorylation. Wild-type MAD1 was phosphorylated in the presence of constitutively active AKT but not in the presence of the dominant-negative AKT (Figure 3E). In addition, phosphorylation of MAD1 Ser145 was abrogated when it mutated to Ala. To further confirm MAD1 Ser145 phosphorylation in vivo, mouse polyclonal antibodies were raised to specifically detect MAD1 Ser145 phosphorylation. Two of the five anti-sera detected phosphorylation of MAD1 in the presence of constitutively active AKT (Figure 3F). Similar results were also shown in direct immunoblotting, but with S145A abrogating the AKT-mediated phosphorylation (Figure 3G). A recent report indicated that MAD1 Ser145 is also phosphorylated by RSK and S6K [16]. We therefore tested AKT-mediated MAD1 phosphorylation in the presence of inhibitors that inhibit either RSK or S6K activity. The inhibition of S6K activity partially decreased MAD1 S145 phosphorylation, which confirmed that S6K can also phosphorylate MAD1 S145. However, combination inhibition of MAPK-ERK pathway downstream kinase RSK and S6K did not further suppress S145 phosphorylation levels compared with inhibition of S6K alone (Supplementary Figure 3). This result suggested that activation of AKT can phosphorylate MAD1 S145, while other MAD1 kinases are inhibited. Taken together, MAD1 is phosphorylated by AKT both in vitro and in vivo.
Ser145 Phosphorylation Suppresses the Interaction Between MAD1 and Its Target Promoter
Protein phosphorylation was reported to inhibit protein function largely through translocation, degradation, and functional disabling [12,20,27,28]. To determine the possible mechanism of AKT-mediated MAD1 inhibition, MAD1's location, stability, and function were investigated when activated AKT appeared. Neither constitutively active AKT nor dominant-negative AKT affected MAD1's nuclear localization (Supplementary Figure 4), indicating that AKT-mediated phosphorylation of MAD1 does not alter its localization. To investigate whether AKT causes MAD1 degradation, MAD1 protein half-life was measured in the presence or absence of AKT. The half-life of MAD1 was around 10 to 20 minutes, and constitutively active AKT did not significantly affect the MAD1 half-life (Supplementary Figure 5A). Further half-life assays were performed to compare the MAD1 S145A mutant with wild-type MAD1 and also found no significant difference (Supplementary Figure 5B). In addition, endogenous MAD1 was detected in some AKT highly activated cancer cell lines such as MDA-MB-436 and MDA-MB-453 (Supplementary Figure 1B). Thus, degradation might not be the major mechanism of AKT-mediated MAD1 inhibition. Since MAD1 has to form a heterodimer complex with MAX to bind to the target promoter site, the dimerization was investigated upon MAD1 phosphorylation. Constitutively active AKT could be present or absent, and the interaction between MAX and MAD1 remained comparable (Supplementary Figure 6). Therefore, phosphorylation of MAD1 does not affect its dimerization with MAX.
Since MAD1 is a DNA-binding transcription factor, the DNA-binding ability of phosphorylated MAD1 was investigated. Interestingly, the in vitro DNA-binding assay demonstrated that constitutively active AKT reduced the interaction between MAD1 and the target gene promoter (MAD1 target gene hTERT Ebox element, Figure 4A), but this reduction of interaction was abolished in the set of MAD1 S145A mutants. To confirm this result, chromatin immunoprecipitation (ChIP) assays were performed. The results showed that constitutively active AKT suppressed the interaction between MAD1 and the target genes promoter (hTERT and ODC). However, the MAD1 S145A mutant abrogated this inhibition (Figure 4B and 4C). Similar effects also were demonstrated in endogenous ChIP that endogenous MAD1 reduced the interaction to both hTERT and ODC promoters upon the IGF-1 stimulation, while the stimulation of IGF-1 increased the interaction between MYC and target genes promoters (Figure 4D and 4E). Consequently, we found that AKT-mediated phosphorylation of MAD1 causes the promoter to switch from the transcription repression MAD1/MAX complex to the transcription activation MYC/MAX complex and activates the target genes' expression.
Figure 4.
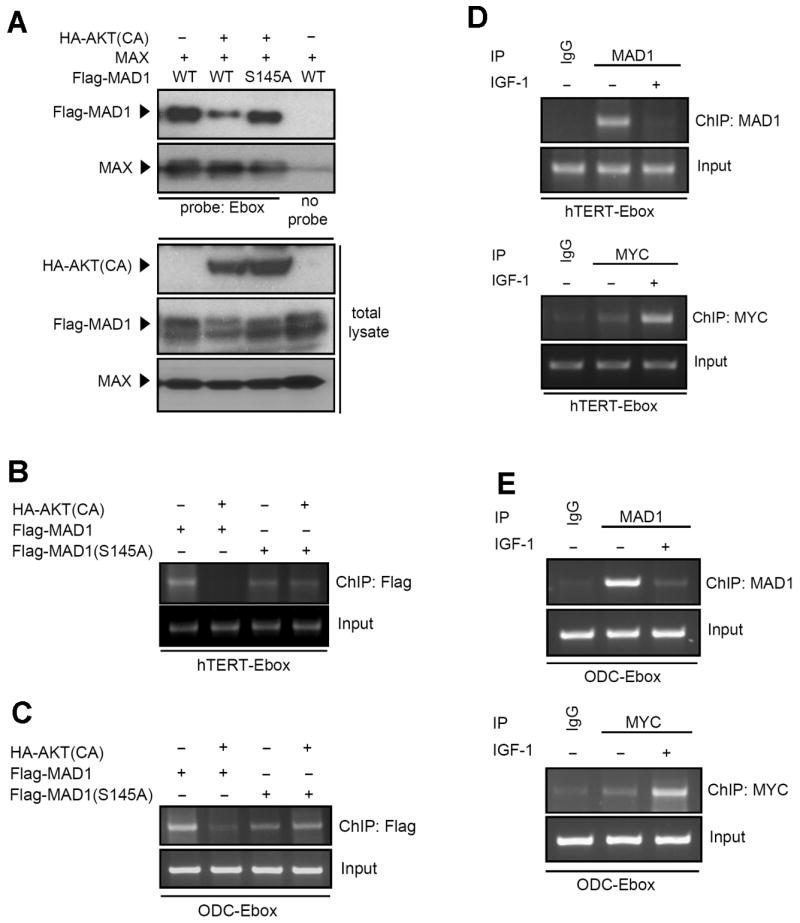
Phosphorylation of MAD1 affects its target DNA binding. (A) DNA binding affinity assay showed that MAD1 phosphorylation affects its target DNA binding. Lysates of 293 cells transfected with designed plasmids were incubated with biotin-conjugated hTERT-Ebox DNA probe and pulled down by streptavidin beads. Probes pulled down proteins were subjected to immunoblotting by anti-Flag, anti-HA, and MAX antibodies. (B) AKT-mediated phosphorylation affects hTERT promoter binding to the MAD1 target gene but not to the MAD1 S145A mutant. Lysates of HeLa cells transfected with the indicated plasmids were subjected to ChIP analysis with anti-Flag antibody. (C) Phosphorylation mediated MAD1 promoter binding alternation was also shown in ChIP assay with anti-Flag antibody and ODC promoter. The experimental setup was the same as for (B). (D) IGF-1 stimulation suppressed MAD1 and induced MYC binding on the hTERT promoter. Lysates of MCF7 cells, treated as indicated, were subjected to the ChIP assay with anti-MAD1 and anti-MYC antibodies. (E) Transcription factors that switched from MAD1 to MYC upon IGF-1 stimulation were also shown in the ODC promoter ChIP assay with anti-MAD1 and anti-MYC antibodies.
AKT-Mediated Phosphorylation of MAD1 Increases MAD1 Target Gene Expression and Promotes the Cell Cycle
Based on the results of the reduced interaction between phosphorylated-MAD1 and the target genes' promoter, we speculated that the expression level of MAD1 target genes can be elevated by AKT. To test this theory, we compared the MAD1 S145A mutant with the wild-type form in several functional assays. In the reporter assay, AKT abolished the wild-type MAD1 inhibition effect but not in the MAD1 S145A mutant (Figure 5A). We further analyzed the MAD1 target genes' expression by real-time polymerase chain reaction (PCR) and found that the MAD1 S145A mutant transcription suppression function was not affected by active AKT, which is similar to the result of reporter assay (Figure 5B).
Figure 5.
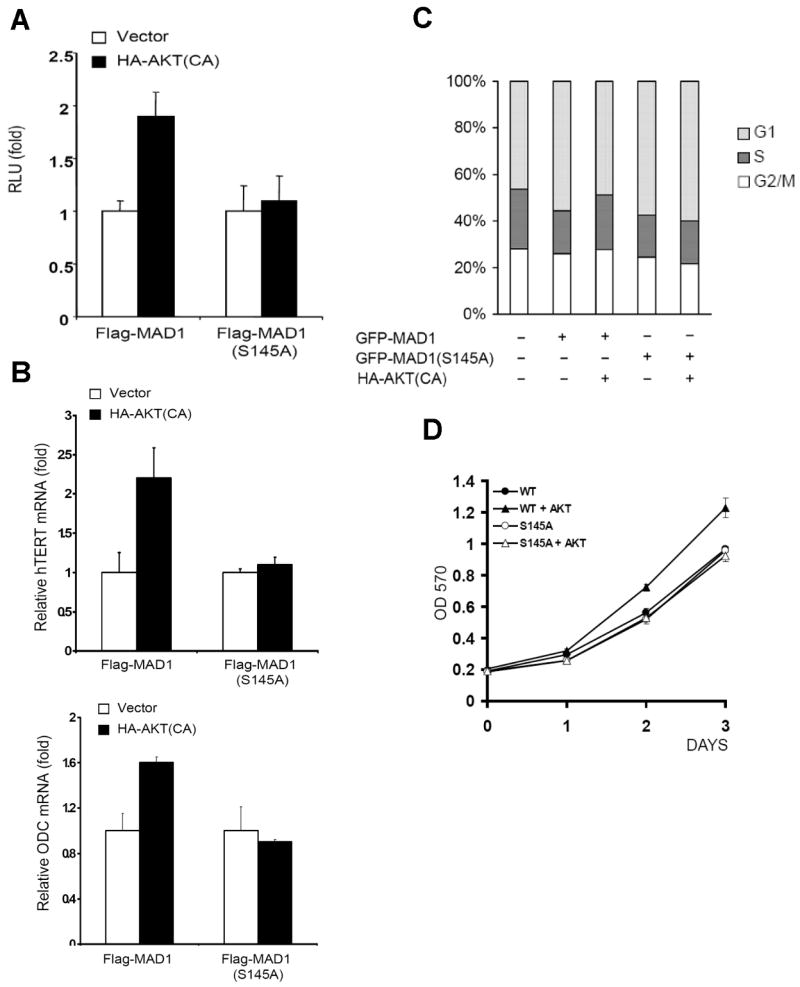
Ser145 Phosphorylation of MAD1 inhibits its transcription repression to target genes. (A) MAD1(S145A) mutant transcription repression was not affected by AKT. Lysates of MDA-MB-435 cells transfected with Flag-MAD1 or Flag-MAD1(S145A) mutant with or without AKT were subjected to reporter assay. (B) mRNA levels of MAD1 target genes hTERT and ODC were affected by the presence of AKT in MAD1 but not in MAD1(S145A) mutant. RNA of MCF7 cells transfected with the indicated plasmids were subjected to quantitative PCR. (C) MAD1(S145A) mutant did not reduce its suppression of the cell cycle in the presence of AKT. MCF7 cells were transfected with the indicated plasmids, and GFP-positive cells were gated for cell cycle assay by flow cytometry. (D) The repression of cell growth by MAD1(S145A) mutant was not affected by AKT. Hela cells were transfected with wild-type MAD1 (WT) or mutant MAD1 (S145A) in the present or absence of AKT. The growth rate was measure by MTT assay.
Since MAD1 and MYC are highly related to cell cycle, we performed cell cycle assays to address the alteration caused by MAD1 phosphorylation. Fewer cells in S phase in the presence of MAD1 S145A compared with wild-type MAD1 cells in the presence of activated AKT (Figure 5C), suggesting that Ser145 phosphorylation inhibits MAD1-mediated cell cycle suppression. In addition, in vitro cell proliferation assay also showed higher growth rate in presence of both wild-type MAD1 and constitutive active AKT in compare to wild-type alone. However, this effect was abrogated when wild-type MAD1 was replaced by MAD1 S145A mutant (Figure 5D). Taken together, our findings indicate that AKT activates MAD1 target genes through phosphorylation of MAD1 and inhibits its transcription repression function, subsequently promoting cell cycle.
Discussion
MAD1 plays an important role in antagonizing MYC-mediated transcription activation by competing with the same target genes' promoter and recruiting transcription repressors. Differential expression has been well-documented using different cells and tissues [6,21]. During cell differentiation, MAD1 RNA expression is elevated, while MYC RNA decreases [22]. This unique expression pattern is the cause of the switch between MYC and MAD1 in the normal cell condition. However, the expression of MAD1 was found in various cancers in which cells are continuously proliferating [10]. Current concepts of cancer stem cells also suggest that cancer cells may also still undergo a differentiation process, although the proliferation status is preserved due to the dysregulation of oncogenes [23]. This might provide an explanation as to why MAD1 can be detected in cancer cells.
Since MAD1 is a tumor suppressor gene, what mechanism could cancer cells use to suppress MAD1 function? We hypothesized that post-translation modification is responsible for the functional alteration and identified that AKT-mediated MAD1 phosphorylation caused the abrogation of target promoter binding ability. AKT is frequently activated in cancer cells and inhibits various tumor suppressors such as Forkhead, p21, p27, p53, and GSK3β through post-translation phosphorylation modification [12,18,24,25]. Upon stimulation by growth factors, AKT detaches from the inner surface of the plasma membrane and re-localizes to the nucleus, suggesting that nuclear proteins may also be targets of AKT [25]. In this study, we identified that the function of the nuclear protein MAD1 is suppressed by AKT by physical interaction and phosphorylation. In vitro and in vivo assays identified Ser145 as the phosphorylation site on MAD1, and this Ser is also located in the AKT consensus phosphorylation motif (RXRXXS-145). Mutation of MAD1 Ser145 to Ala abolished AKT-mediated phosphorylation. Although the phosphorylation site is located at the C-terminal of MAD1 and is close to the leucine-zipper domain, the phosphorylation does not significantly affect the interaction with MAX. However, the phosphorylation somehow affects the interaction between MAD1 and its target genes' promoter. Consequently, AKT causes the molecules to switch from a MAD/MAX complex to a MYC/MAX complex to activate the expression of target genes.
It has been reported that MAD1 C-terminal Ser182 and Ser184 are phosphorylated by Casein Kinase II (CKII), and that phosphorylation also inhibits MAD1 DNA binding [26]. Additionally, a recent study reported that Ser145 of MAD1 is phosphorylated by S6K and RSK and that this phosphorylation mediates protein degradation [16]. Although in such study mentioned that MAD1 Ser145 phosphorylation is blocked by rapamycin and PD98059 under serum stimulation condition, which implies that AKT doesn't involved directly in the phosphorylation of MAD1 Ser145. However, we demonstrated that AKT physically interacts with MAD1 and phosphorylates MAD1 both in vitro and in vivo. In addition, in the presence of constitutive active AKT, rapamycin and PD98059 can only partially reduce MAD1 Ser145 phosphorylation. The possibilities for the discrepancy results could be due to the different cell lines and experimental conditions used. For instance, the cells were serum starved and followed by serum stimulation in their study. However, serum stimulation is able to trigger multiple pathways which might caused more complicated results; thus, in this study, we tried to identify the role of AKT in mediating MAD1 phosphorylation directly by presence of AKT under normal serum containing culture condition. Taken together, our results indicated that AKT is also directly involved in MAD1 Ser145 phosphorylation under our experimental conditions. Moreover, in addition to MAD1 degradation, AKT-mediated MAD1 phosphorylation and inhibition of function seem to occur mainly through the suppression of DNA binding. It is not clear why the same Ser145, which can be phosphorylated by different enzymes, results in a different outcome. One possibility is that other unknown molecules are involving the complex, which may affect the recruitment of different enzymes and lead to different outcomes. In this regards, it is worthwhile to mention that p53 has similar effects in which same serine or threonine sites were phosphorylated by different enzymes and followed by different consequences [27]. Therefore, the detailed mechanisms of phosphorylated MAD1 in reducing interaction with promoter will need further investigation.
MYC/MAX and MAD1/MAX complexes are well regulated by the status of proliferation and differentiation under normal development. However, in cancer cells, dysregulation of oncogenic pathways may alter that status by inhibiting differentiation-related proteins such as MAD1. Thus, based on our findings, we propose a model of AKT for regulating MAD1 function. AKT-mediated phosphorylation on MAD1 affects its DNA-binding property and subsequently induces MAD1 target gene expression. Taken together, our results provide a model of AKT-mediated MAD1/MAX/MYC network regulation.
Supplementary Material
Supplementary Figure 1. Comparison of MAD1 and MYC expression levels in breast cancer samples. (A) mRNA levels of MAD1 and MYC were plotted according to their expression level and less correlation was identified (R < 0.1). Data are from the public database Oncomine (www.oncomine.org). (B) MAD1 was detected in the presence of MYC in several breast cancer cell lines. Lysates of breast cancer cell lines were subjected to immunoblotting in detecting MAD1, MYC, and phosphor-AKT protein levels by using specific antibodies.
Supplementary Figure 2. SNP array assay showed that MAD1 genes did not lose or gain copies of the gene.
Supplementary Figure 3. Blockage of S6K activation partially suppresses AKT-mediated MAD1 phosphorylation. Lysates of HeLa cells transfected with Flag-MAD1 and HA-AKT were treated with rapamycin and PD98059 to block S6K and ERK activities, repectively. Samples were subjected to immunoblotting with the indicated antibodies.* indicated Phospho-MAD1 anti-serum #1; ** indicated phospho-(Ser/Thr) AKT substrate antibody.
Supplementary Figure 4. Phosphorylation of MAD1 does not affect its localization. MAD1 was transfected with constitutively active AKT or dominant-negative AKT to MCF7 cells. Cells were fixed with 3.7% formaldehyde and stained with the indicated antibodies.
Supplementary Figure 5. AKT does not significantly affect the MAD1 half-life. (A) No obvious difference in the MAD1 half-life was seen in the presence of AKT. 293 cells co-transfected with the indicated plasmids were harvested after treatment with cycloheximide (CHX) at the indicated time points. (B) The MAD1(S145A) mutant showed a similar half-life compared with wild-type MAD1. Treatment of samples was the same as in (A).
Supplementary Figure 6. AKT-mediated phosphorylation of MAD1 does not affect its binding with MAX. Lysates of HeLa cells transfected with the indicated plasmids were immunoprecipitated using anti-Flag antibody and subsequently subjected to immunoblotting.
Acknowledgments
We thank Z.B. Han, X.Y. Cheng, H.-H. Lee, and J.-F. Lee for their technical support. The manuscript was supported by the following grants: NIH CA099031 and Cancer Center Support Grant CA16672 to M.-C.H.; a pre-doctoral fellowship from the US Army Breast Cancer Research Program, grant W81XWH-05-1-0252, and the Presidents' Research Scholarship, T.C. Hsu Endowed Memorial Scholarship, Andrew Sowell-Wade Huggins Scholarship, and Alfred Knudson Jr. Outstanding Dissertation Award from The University of Texas Graduate School of Biomedical Sciences at Houston to D.-F.L.; a pre-doctoral fellowship from the US Army Breast Cancer Research Program, grant W81XWH-08-1-0397, and Andrew Sowell-Wade Huggins Scholarship from The University of Texas Graduate School of Biomedical Sciences at Houston to H.-P.K.; a pre-doctoral fellowship from the US Army Breast Cancer Research Program and grant W81XWH-06-1-0709 to C.-K.C. We would also like to thank National Cheng-Kung University Proteomics Research Core Laboratory for the assistance in mass spectrometry analysis.
Abbreviations
- MAD1
MAX Dimerization Protein 1
- MAX
MYC-Associated Factor X
- hTERT
Human Telomerase Reverse Transcriptase
References
- 1.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6(8):635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 2.Nair SK, Burley SK. Structural aspects of interactions within the Myc/Max/Mad network. Curr Top Microbiol Immunol. 2006;302:123–143. doi: 10.1007/3-540-32952-8_5. [DOI] [PubMed] [Google Scholar]
- 3.Cole MD, Nikiforov MA. Transcriptional activation by the Myc oncoprotein. Curr Top Microbiol Immunol. 2006;302:33–50. doi: 10.1007/3-540-32952-8_2. [DOI] [PubMed] [Google Scholar]
- 4.Ayer DE, Lawrence QA, Eisenman RN. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell. 1995;80(5):767–776. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- 5.Ayer DE, Laherty CD, Lawrence QA, Armstrong AP, Eisenman RN. Mad proteins contain a dominant transcription repression domain. Mol Cell Biol. 1996;16(10):5772–5781. doi: 10.1128/mcb.16.10.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rottmann S, Luscher B. The Mad side of the Max network: antagonizing the function of Myc and more. Curr Top Microbiol Immunol. 2006;302:63–122. doi: 10.1007/3-540-32952-8_4. [DOI] [PubMed] [Google Scholar]
- 7.Cultraro CM, Bino T, Segal S. Regulated expression and function of the c-Myc antagonist, Mad1, during a molecular switch from proliferation to differentiation. Curr Top Microbiol Immunol. 1997;224:149–158. doi: 10.1007/978-3-642-60801-8_15. [DOI] [PubMed] [Google Scholar]
- 8.Han S, Park K, Kim HY, Lee MS, Kim HJ, Kim YD. Expression of Mad1 protein inhibits proliferation of cancer cells and inversely correlated with Myc protein expression in primary gastric cancer. Oncol Rep. 1999;6(3):569–574. [PubMed] [Google Scholar]
- 9.Guo XL, Pan L, Zhang XJ, et al. Expression and mutation analysis of genes that encode the Myc antagonists Mad1, Mxi1 and Rox in acute leukaemia. Leuk Lymphoma. 2007;48(6):1200–1207. doi: 10.1080/10428190701342018. [DOI] [PubMed] [Google Scholar]
- 10.Han S, Park K, Kim HY, et al. Clinical implication of altered expression of Mad1 protein in human breast carcinoma. Cancer. 2000;88(7):1623–1632. doi: 10.1002/(sici)1097-0142(20000401)88:7<1623::aid-cncr17>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 11.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10(2):262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 12.Zhou BP, Hung MC. Novel targets of Akt, p21(Cipl/WAF1), and MDM2. Semin Oncol. 2002;29(3 Suppl 11):62–70. doi: 10.1053/sonc.2002.34057. [DOI] [PubMed] [Google Scholar]
- 13.Zhou C, Bae-Jump VL, Whang YE, Gehrig PA, Boggess JF. The PTEN tumor suppressor inhibits telomerase activity in endometrial cancer cells by decreasing hTERT mRNA levels. Gynecol Oncol. 2006;101(2):305–310. doi: 10.1016/j.ygyno.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 14.Wetterau LA, Francis MJ, Ma L, Cohen P. Insulin-like growth factor I stimulates telomerase activity in prostate cancer cells. J Clin Endocrinol Metab. 2003;88(7):3354–3359. doi: 10.1210/jc.2002-021326. [DOI] [PubMed] [Google Scholar]
- 15.Flamigni F, Marmiroli S, Capanni C, Stefanelli C, Guarnieri C, Caldarera CM. Phosphatidylinositol 3-kinase is required for the induction of ornithine decarboxylase in leukemia cells stimulated to growth. Biochem Biophys Res Commun. 1997;239(3):729–733. doi: 10.1006/bbrc.1997.7543. [DOI] [PubMed] [Google Scholar]
- 16.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105(18):6584–6589. doi: 10.1073/pnas.0802785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou Y, Peng H, Zhou B, et al. Systemic tumor suppression by the proapoptotic gene bik. Cancer Res. 2002;62(1):8–12. [PubMed] [Google Scholar]
- 18.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3(11):973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 19.Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26(1):75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130(3):440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 21.Larsson LG, Pettersson M, Oberg F, Nilsson K, Luscher B. Expression of mad, mxi1, max and c-myc during induced differentiation of hematopoietic cells: opposite regulation of mad and c-myc. Oncogene. 1994;9(4):1247–1252. [PubMed] [Google Scholar]
- 22.Xu D, Popov N, Hou M, et al. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci U S A. 2001;98(7):3826–3831. doi: 10.1073/pnas.071043198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 24.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 25.Kumar R, Hung MC. Signaling intricacies take center stage in cancer cells. Cancer Res. 2005;65(7):2511–2515. doi: 10.1158/0008-5472.CAN-05-0189. [DOI] [PubMed] [Google Scholar]
- 26.Barrera-Hernandez G, Cultraro CM, Pianetti S, Segal S. Mad1 function is regulated through elements within the carboxy terminus. Mol Cell Biol. 2000;20(12):4253–4264. doi: 10.1128/mcb.20.12.4253-4264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Comparison of MAD1 and MYC expression levels in breast cancer samples. (A) mRNA levels of MAD1 and MYC were plotted according to their expression level and less correlation was identified (R < 0.1). Data are from the public database Oncomine (www.oncomine.org). (B) MAD1 was detected in the presence of MYC in several breast cancer cell lines. Lysates of breast cancer cell lines were subjected to immunoblotting in detecting MAD1, MYC, and phosphor-AKT protein levels by using specific antibodies.
Supplementary Figure 2. SNP array assay showed that MAD1 genes did not lose or gain copies of the gene.
Supplementary Figure 3. Blockage of S6K activation partially suppresses AKT-mediated MAD1 phosphorylation. Lysates of HeLa cells transfected with Flag-MAD1 and HA-AKT were treated with rapamycin and PD98059 to block S6K and ERK activities, repectively. Samples were subjected to immunoblotting with the indicated antibodies.* indicated Phospho-MAD1 anti-serum #1; ** indicated phospho-(Ser/Thr) AKT substrate antibody.
Supplementary Figure 4. Phosphorylation of MAD1 does not affect its localization. MAD1 was transfected with constitutively active AKT or dominant-negative AKT to MCF7 cells. Cells were fixed with 3.7% formaldehyde and stained with the indicated antibodies.
Supplementary Figure 5. AKT does not significantly affect the MAD1 half-life. (A) No obvious difference in the MAD1 half-life was seen in the presence of AKT. 293 cells co-transfected with the indicated plasmids were harvested after treatment with cycloheximide (CHX) at the indicated time points. (B) The MAD1(S145A) mutant showed a similar half-life compared with wild-type MAD1. Treatment of samples was the same as in (A).
Supplementary Figure 6. AKT-mediated phosphorylation of MAD1 does not affect its binding with MAX. Lysates of HeLa cells transfected with the indicated plasmids were immunoprecipitated using anti-Flag antibody and subsequently subjected to immunoblotting.