

Abstract
Results from clinical and imaging studies provide evidence for changes in schizophrenia with disease progression, however, the underlying molecular differences that may occur at different stages of illness have not been investigated. To test the hypothesis that the molecular basis for schizophrenia changes from early to chronic illness, we profiled genome-wide expression patterns in prefrontal cortex of schizophrenic subjects at different stages of illness, along with their age- and sex-matched controls. Results show that gene expression profiles change dramatically depending on the stage of illness, whereby the greatest number and magnitude of gene expression differences were detected in subjects with short-term illness (≤ 4 years from diagnosis). Comprehensive pathways analyses revealed that each defined stage of illness was associated with dysfunction in both distinct, as well as overlapping systems. Short-term illness was particularly associated with disruptions in gene transcription, metal ion binding, RNA processing and vesicle-mediated transport. In contrast, long-term illness was associated with inflammation, stimulus-response and immune functions. We validated expression differences of 12 transcripts associated with these various functions by real-time PCR analysis. While only four genes, SAMSN1, CDC42BPB, DSC2 and PTPRE, were consistently expressed across all groups, there was dysfunction in overlapping systems among all stages, including cellular signal transduction, lipid metabolism and protein localization. Our results demonstrate that the molecular basis for schizophrenia changes from early to chronic stages, providing evidence for a changing nature of schizophrenia with disease progression.
Keywords: Psychiatric disorder, schizophrenia, microarray, antipsychotic, chronic, transcription, inflammation
1. Introduction
Emil Kraepelin originally defined schizophrenia (“dementia praecox”) as a progressive brain disease (Kraepelin 1971 (original 1919)); however, the view of schizophrenia as a progressively deteriorating as opposed to a static, unchanging disorder remains controversial. Nonetheless, the results of major studies on the course of illness over 20-40 years of follow-up are consistent in reporting a chronic, generally persistent, course of illness for 50-70% of the patients who receive an initial diagnosis of schizophrenia (Huber et al., 1975; Steinmeyer et al., 1989; Fenton and McGlashan 1991; DeSisto et al., 1995; Stephens et al., 1997).
Much support for the argument that schizophrenia is a progressive disorder comes from magnetic resonance imaging and related studies. Such investigations have demonstrated progressive decreases in global gray matter volume in several brain regions as patients move from early to chronic stages of the disease (Mathalon et al., 2001; Wood et al., 2001; Sporn et al., 2003; van Haren et al., 2007). Significantly, it has been shown that excessive brain tissue loss occurs within the first 10 to 20 years of the illness (van Haren et al., 2008). In addition to changes in gray matter, progressive changes in white matter volume have been demonstrated (Ho et al., 2003; Whitford et al., 2007), as well as alterations in white matter integrity, as revealed by diffusion tensor imaging (Mori et al., 2007). Additionally, several studies have also demonstrated progressive ventricular enlargement in schizophrenic subjects with chronic illness (Saijo et al., 2001; DeLisi et al., 2004; DeLisi et al., 2006). While the causes of such structural changes are not known, it is possible that they could be triggered by changes in the expression of genes involved in critical cellular functions that maintain CNS structure; however, the extent to which prolonged antipsychotic drug exposure may contribute to such changes is not known.
Clinical studies have reported that the relative predominance of symptom presentation also changes with disease progression. Florid positive symptoms and deterioration of function, are most prevalent during the first few years of illness (Lieberman 1999; Lieberman et al., 2001). In contrast, the chronic form of schizophrenia is characterized by predominately negative symptoms, which appear to plateau and remain stable (McGlashan 1988; McGlashan 1998). There are conflicting reports on cognitive deficits, which are evident in patients with schizophrenia at the onset of illness. Longitudinal studies on chronic schizophrenic subjects show significant decline in cognitive deficits (Harvey et al., 1999; Friedman et al., 2001; Harvey et al., 2003), but one study has found no evidence for cognitive decline in at least a subgroup of subjects (Kurtz et al., 2005). It has been suggested that these variations may represent distinct pathological mechanisms of the disease during different stages of illness (Kurtz 2005).
Given the evidence for changing features of schizophrenia with disease illness, we hypothesize that the disease also changes on a molecular level at different stages of illness. Consistent with this idea, we have previously shown that increases in the expression of two genes, muscleblind protein 1 and protocadherin 17, in the prefrontal cortex are present in subjects with schizophrenia of short (< 7 years) but not long (> 22 years), duration of illness (DOI) (Dean et al., 2007). To test this hypothesis on a global scale, we sought to identify genome-wide expression changes in prefrontal cortex throughout the course of illness using carefully selected subject cohorts representing different durations of disease. While several previous studies have investigated global gene expression patterns in different CNS regions obtained post-mortem from subjects with schizophrenia (reviewed in (Katsel et al., 2005; Iwamoto and Kato 2006; Mirnics et al., 2006; Thomas 2006)), none focused on changes in gene expression from subjects at different durations of the illness. We find that indeed gene expression profiles change dramatically depending on the stage of illness, with the greatest number and magnitude of gene expression differences being detected in subjects with short-term illness (≤4 years from diagnosis). We further highlight similarities and differences in the underlying molecular patterns at different stages of schizophrenia.
2. Results
2.1 Gene expression profiles at different stages of illness
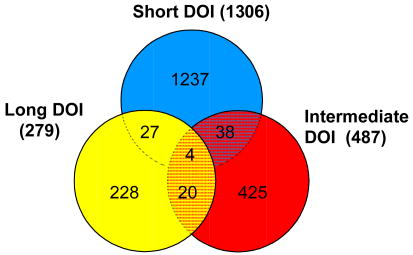
We analyzed genome-wide mRNA expression profiles from prefrontal cortical RNA samples isolated from schizophrenic subjects at three stages of illness, along with their age- and sex-matched controls (Table 1). We used both unpaired and pair-wise analyses to identify genes whose expression differed from matched controls at various durations of illness. The lists of differentially expressed genes from both analyses showed a high degree of overlap (83.4%, 65.6% and 82.5% for the short DOI cohort, intermediate DOI cohort and long DOI cohort, respectively). Gene lists generated at different levels of significance revealed that the absolute numbers of genes with altered expression greatly diminished with increased duration of the disease. At all significance levels, the greatest number of genes showing expression differences were detected in the subjects with short-term duration of illness (Table 2). The top 50 differentially expressed genes at each stage are shown in Table 3, with full lists provided in Supplementary Tables 1a-c. Notably, at the p<0.01 level, there was a ∼10-fold greater number of differentially expressed genes between schizophrenics and controls at short-term duration compared to long-term duration (Table 2). A Venn diagram of the gene lists from the unpaired analyses showed relatively low overlap in gene expression differences among the three cohorts: 42 gene expression differences were shared between subjects with short-term and intermediate-term DOI, 31 differences between subjects with short-term and long-term DOI subjects and 24 differences between subjects with intermediate and long-term DOI (Table 2). Only 4 genes demonstrated statistically significant changes in expression in all three groups: SAMSN1, CDC42BPB, DSC2 and PTPRE. There were also differences in the direction of the expression changes, with 67.8 and 65.5% of the genes showing decreased expression in subjects with short- and intermediate-term illness, respectively, but only 35% in chronic illness.
Table 1.
Demographic and array parameter data for all subjects.
| Short-DOI: | Short-controls: | Inter-DOI: | Inter-controls: | Long-DOI: | Long-controls: | |
|---|---|---|---|---|---|---|
| Age (yrs) | 26.1 ± 2.05 | 28.8 ± 2.55 | 41.9 ± 3.44 | 41.8 ± 2.47 | 65.3 ± 2.91 | 64.6 ± 3.26 |
| DOI (yrs) | 2.88 ± 0.29 | NA | 12.7 ± 0.97 | NA | 37.4 ± 2.56 | NA |
| Sex (ratio) | 6M/2F | 5M/2F | 8M/2F | 11M/3F | 7M/1F | 7M/1F |
| pH | 6.22 ± 0.04 | 6.26 ± 0.08 | 6.26 ± 0.05 | 6.33 ± 0.03 | 6.34 ± 0.06 | 6.32 ± 0.06 |
| PMI (hrs) | 46.1 ± 2.44 | 44.0 ± 4.0 | 39.3 ± 4.6 | 39.0 ± 3.85 | 32.9 ± 3.64 | 40.0 ± 5.62 |
| 3′/5′ ratio | 1.46 ± 0.17 | 1.19 ± 0.02 | 1.26 ± 0.08 | 1.18 ± 0.02 | 1.24 ± 0.02 | 1.30 ± 0.06 |
| %P | 46.6 ± 1.06 | 48.2 ± 0.82 | 44.9 ± 0.89 | 43.8 ± 0.81 | 49.2 ± 1.28 | 48.2 ± 1.34 |
| Drug dose* | 300.0 ± 92.6 | NA | 442.2 ± 73.2 | NA | 278.8 ± 83.2 | NA |
Chlorpromazine equivalents (mg); DOI, duration of illness; PMI, post-mortem interval; 3′/5′ ratio, microarray GAPDH 3′/5′ ratio; %P, microarray percent Present call.
Table 2.
Numbers of Differentially Expressed Genes in Subjects with Schizophrenia at Different Stages of Illness.
| Short DOI: | Inter DOI: | Long DOI: | ||||
|---|---|---|---|---|---|---|
| p-value: | U: | P: | U: | P: | U: | P: |
| 0.05 | 1306 | 1548 | 487 | 226 | 279 | 324 |
| 0.01 | 333 | 351 | 87 | 35 | 40 | 46 |
| 0.005 | 172 | 175 | 28 | 15 | 19 | 23 |
| 0.001 | 30 | 35 | 5 | 1 | 4 | 4 |
|
| ||||||
| ||||||
Numbers represent non-redundant, probeset IDs identified as differentially expressed in the subjects with the indicated duration of illness (DOI), using unpaired (U) and paired (P) analyses. Venn diagram shows overlap of differentially expressed genes from unpaired analysis at p<0.05.
Table 3.
Lists of the top 50 differentially expressed genes at each stage of illness.
| Short DOI | Intermediate DOI | Long DOI | ||||||
|---|---|---|---|---|---|---|---|---|
| UniGene Symbol: | FC | p-value: | UniGene Symbol: | FC | p-value: | UniGene Symbol: | FC | p-value: |
| SLC29A2 | 0.377 | 0.00003 | C17orf82 | 1.543 | 0.00041 | C14orf64 | 2.02 | 4.46E-05 |
| MBP | 0.341 | 0.00008 | AGBL1 | 1.663 | 0.00043 | IL1F5 | 2.23 | 0.000185 |
| TASP1 | 0.498 | 0.00033 | GPRIN2 | 0.583 | 0.00047 | FLJ13773 | 1.82 | 0.000623 |
| SCARB2 | 0.479 | 0.00035 | CD160 | 0.617 | 0.00065 | TAPBPL | 1.76 | 0.000668 |
| FLJ11155 | 0.408 | 0.00038 | RBPMS2 | 0.481 | 0.00079 | FLJ39743 | 0.59 | 0.001235 |
| TMEM64 | 0.505 | 0.0004 | SOST | 0.631 | 0.00143 | RIN3 | 0.54 | 0.001518 |
| NPY | 0.45 | 0.00062 | SLITRK2 | 1.551 | 0.00161 | PPIAL4 | 0.65 | 0.001769 |
| REST | 1.881 | 0.0008 | MESP1 | 0.492 | 0.00193 | DAND5 | 0.66 | 0.00203 |
| YTHDF3 | 0.492 | 0.00099 | NPIP | 0.616 | 0.00209 | VIL1 | 0.59 | 0.002341 |
| EGFR | 2.177 | 0.00138 | CLUL1 | 0.618 | 0.00241 | IQGAP3 | 0.61 | 0.003318 |
| MOG | 0.425 | 0.00171 | PDLIM5 | 1.703 | 0.00276 | FGF10 | 0.67 | 0.003706 |
| ELL2 | 0.474 | 0.00186 | GPR115 | 0.658 | 0.00411 | LTBR | 1.93 | 0.003873 |
| SORT1 | 0.499 | 0.00216 | LOC151658 | 0.661 | 0.00441 | SNTB2 | 0.67 | 0.004112 |
| UGT8 | 0.412 | 0.00249 | OXT | 0.705 | 0.00537 | FLJ14327 | 0.65 | 0.004442 |
| MAG | 0.489 | 0.00253 | ADAMTSL1 | 0.649 | 0.00546 | RAX | 0.63 | 0.004829 |
| BTLA | 1.83 | 0.00262 | ITGB8 | 0.657 | 0.00566 | GRIN2B | 0.62 | 0.005223 |
| ACP5 | 2.029 | 0.00292 | MR1 | 0.673 | 0.00597 | SLC18A3 | 1.62 | 0.006235 |
| C6orf52 | 1.904 | 0.00307 | SERPINB10 | 0.615 | 0.00608 | CA12 | 1.62 | 0.006752 |
| IL6ST | 0.487 | 0.00309 | RAC2 | 0.671 | 0.00669 | MTHFSD | 0.58 | 0.007097 |
| PPM1E | 0.509 | 0.00355 | CAPN3 | 1.491 | 0.00674 | HIST2H4A | 1.69 | 0.009234 |
| TRUB1 | 0.448 | 0.00413 | NR5A2 | 0.68 | 0.00737 | PLAC8 | 1.68 | 0.010338 |
| HRH3 | 1.848 | 0.00473 | IL17RE | 0.677 | 0.0092 | ALDOAP2 | 0.64 | 0.011149 |
| PI3 | 0.477 | 0.00484 | MGC12916 | 1.478 | 0.00932 | CREB3L4 | 1.67 | 0.011557 |
| GADD45G | 2.591 | 0.00498 | DHRS7C | 0.612 | 0.00949 | ITGB2 | 2.06 | 0.011657 |
| HHIP | 0.492 | 0.00527 | FGF5 | 0.465 | 0.01175 | TNFAIP8 | 0.68 | 0.01243 |
| LOC728411 | 2.266 | 0.0058 | ZFY | 0.671 | 0.012 | OR2W3 | 0.67 | 0.01281 |
| SNED1 | 1.895 | 0.00658 | PPP1R14A | 1.54 | 0.01259 | FKBP1B | 1.64 | 0.016035 |
| ZNF642 | 1.868 | 0.00697 | DDIT4 | 1.476 | 0.01275 | FLJ22655 | 0.65 | 0.016176 |
| H1FNT | 0.466 | 0.00849 | XYLT2 | 0.665 | 0.01416 | LOC149832 | 2.07 | 0.016799 |
| ZFP36 | 6.127 | 0.00902 | NEU3 | 0.666 | 0.01498 | SIX6 | 0.66 | 0.017077 |
| IL17B | 1.886 | 0.00928 | SLC25A42 | 0.643 | 0.01729 | TGFB1 | 1.69 | 0.017371 |
| KCTD14 | 1.871 | 0.0094 | DNAH17 | 1.523 | 0.01904 | C1QB | 2.31 | 0.017813 |
| TRIM59 | 0.421 | 0.00967 | SIRPD | 0.588 | 0.01937 | FPR1 | 2.14 | 0.019578 |
| LOC651600 | 2.002 | 0.01018 | LOC400713 | 1.52 | 0.01971 | HOXD8 | 0.65 | 0.019846 |
| CLCA4 | 0.487 | 0.01026 | RYR1 | 0.566 | 0.0198 | TLR10 | 1.76 | 0.020621 |
| CDKN1A | 2.326 | 0.01059 | TIGD3 | 0.666 | 0.02011 | RAC2 | 1.74 | 0.022982 |
| TCTE3 | 2.166 | 0.01089 | FAM112B | 0.647 | 0.02024 | MAGEA9 | 1.7 | 0.024677 |
| LOC731450 | 2.048 | 0.01167 | LOC144776 | 0.478 | 0.0209 | TLR2 | 1.76 | 0.027192 |
| COPA | 0.472 | 0.01258 | KL | 0.664 | 0.02165 | FKBP5 | 1.73 | 0.027792 |
| CYP2D6 | 1.877 | 0.0141 | TAGLN | 1.537 | 0.02444 | VSIG4 | 1.72 | 0.02959 |
| SYNJ1 | 0.469 | 0.01511 | NIBP | 0.576 | 0.02605 | C1QA | 2.02 | 0.030004 |
| ISL2 | 1.847 | 0.01554 | LOC129881 | 1.632 | 0.02904 | UST | 0.63 | 0.03252 |
| CTF1 | 1.916 | 0.01608 | ZNF174 | 0.647 | 0.0296 | C9orf71 | 1.7 | 0.03457 |
| NINJ2 | 0.49 | 0.01633 | TLR2 | 0.642 | 0.03195 | TLCD1 | 1.77 | 0.03753 |
| ZDHHC17 | 0.476 | 0.01773 | ENOSF1 | 1.491 | 0.03524 | F3 | 1.93 | 0.038307 |
| SCAMP1 | 0.494 | 0.02166 | PPP4R2 | 1.475 | 0.03764 | MGST1 | 1.63 | 0.03845 |
| PAPPA | 1.788 | 0.002445 | PGLYRP1 | 0.638 | 0.03836 | EBF | 0.53 | 0.039139 |
| SEMA3E | 0.458 | 0.04357 | RFPL3S | 1.596 | 0.03845 | TYROBP | 1.64 | 0.041762 |
| DKFZP434F0318 | 1.892 | 0.04465 | PEX6 | 1.524 | 0.04588 | SAMSN1 | 1.69 | 0.042864 |
| TGIF2 | 1.881 | 0.04711 | DTNBP1 | 0.644 | 0.04645 | SLC14A1 | 2.16 | 0.045121 |
Unigene symbols for each gene are shown, along with fold-change (FC) and p-value.
2.2. Gene coexpression patterns in subjects with short-term DOI
We next determined how the top differentially expressed genes from subjects early in illness changed with disease duration. From a one-way hierarchical clustering of the gene profiles, heat-maps depicting expression patterns of the top 337 differentially expressed mRNAs determined from the ANOVA (p<0.01) on the subjects with short-term DOI were generated. Heatmaps using these same genes were generated for subjects with intermediate and long duration illness (Figure 1). Distinct differences were observed in the expression of these genes in each cohort; this can be assessed visually from the heatmaps themselves or by the hierarchical clustering of samples shown beside the heatmaps in Figure 1. The subjects with short-term DOI show highly similar coexpression patterns for these genes, while the subjects with long-term DOI showed the opposite pattern, more resembling, in fact, the control subjects. This is consistent with the fact that ∼40% of the genes found to be differentially expressed in short-term DOI compared to those with long-term DOI were also identified when the short-term schizophrenic subjects were compared against their matched controls (data not shown). The greatest heterogeneity in the expression of this set of genes was evident in the subjects with intermediate-term DOI, with some subjects clustering together to resemble the expression profiles exhibited by the short-term subjects (Figure 1). This may reflect a transition from early stage to late stage schizophrenia. Heterogeneous coexpression patterns were also observed in subjects with intermediate- and long-term illness when hierarchal clustering was performed on gene lists generated from the ANOVAs in their respective cohorts (data not shown).
Figure 1.

Gene expression differences at multiple stages of illness in schizophrenia. Heatmaps allow visualization of the 337 most differentially expressed genes as revealed by ANOVA (p<0.01), conducted on subjects with short-term duration of illness (DOI), and how these changes in subjects with intermediate and long DOI. Each colored pixel represents an individual gene expression value from a single subject. Relative decreases in gene expression are indicated by yellow and increases in expression by blue. Log2-transformed expression values were subjected to two-dimensional hierarchical clustering in each cohort separately, as shown beside the heatmaps. Subjects shown are short-term DOI (Short-DOI, 1-8), and their controls (Short-C, 1-8); intermediate-term DOI, (Intermediate DOI, 1-10, and their controls (Intermediate-C, 1-14); and long-term DOI (Long DOI, 1-8) and their controls (Long-C, 1-8).
Also included in our lists of differentially expressed genes (Table 3; Supplementary Table 1) were many previously implicated in the pathology of schizophrenia based on post-mortem microarray studies. These include genes related to presynaptic machinery (Mirnics et al., 2000; Knable et al., 2001; Hemby et al., 2002; Vawter et al., 2002), ubiquitination (Vawter et al., 2001; Middleton et al., 2002; Vawter et al., 2002), myelination (Hakak et al., 2001; Tkachev et al., 2003; Sugai et al., 2004; Aston et al., 2005; Katsel et al., 2005; Haroutunian et al., 2007; Tkachev et al., 2007) and the GABAergic system (Lewis et al., 2005; Hashimoto et al., 2007), as well as other individual genes not associated with any particular system. The greatest significance for the expression differences of these genes between control and schizophrenic subjects was found in subjects with short-term illness.
2.3 Pathways dysfunction differs among stages of illness
To understand the potential biological relevance of the differentially expressed genes in each cohort, we used several functional annotation tools, including DAVID, which encompasses 40 annotation categories, and Ingenuity Pathway Analysis (IPA). One commonly used tool in DAVID describes gene products in terms of their associated biological processes, cellular components and molecular functions using Gene Ontology (GO) terms. We found that top significantly enriched GO terms were different in each stage of illness, although some overlap was observed between cohorts (Table 4). Using DAVID, we identified significantly enriched clusters of functional categories at each stage; these correlated well with the GO terms, with the addition of “metal-ion binding” representing the top functionally related category in subjects with short-term DOI (Supplementary Table 2). We next used the computational gene network prediction tool in the IPA database, which identifies known interactions between protein products of differentially expressed genes, referred to as “focus” genes. This analysis identified independent networks for each stage of illness demonstrating interactions of up to 32 coexpressed gene products within a given network. The top 4 networks in short-term illness, (from 16 networks identified linking together up to 23 coexpressed gene products), centered around gene expression, cellular signaling, development and carbohydrate metabolism (Table 5). The latter three categories were also found represented in subjects with intermediate-term DOI. Top networks in long-term DOI were centered around inflammation and injury abnormalities (Table 5). Despite the relatively low overlap of differentially expressed gene from each cohort, these comprehensive pathways analyses revealed that different stages of illness do have common systems dysfunction; all stages were associated with signal transduction mechanisms, lipid metabolism and protein transport/metabolism. These systems are summarized in Figure 2. Overall, short-term illness was particularly associated with disruptions in gene expression, metal ion binding, RNA processing and vesicle-mediated transport. Intermediate-term illness also shared gene expression deficits with early illness and, in addition, was significantly enriched for calcium signaling functions. Notably different from short-term illness, but sharing common features with intermediate stages, long-term illness was associated with inflammation, glycosylation, apoptosis and immune functions.
Table 4.
Gene Ontology (GO) terms associated with different stages of illness.
| Short-term DOI: | Inter-term DOI: | Long-term DOI: | ||||||
|---|---|---|---|---|---|---|---|---|
| GO Term: | # genes: | p-value: | GO Term: | # genes: | p-value: | GO Term: | # genes: | p-value: |
| vesicle-mediated transport | 48 | 1.65E-06 | biopolymer glycosylation | 9 | 0.0051 | immune response | 29 | 3.31E-04 |
| secretory pathway | 31 | 1.97E-06 | cell organization and biogenesis | 46 | 0.0115 | response to pathogen | 18 | 6.11E-04 |
| secretion | 34 | 1.29E-05 | endocytosis | 9 | 0.0129 | response to other organism | 18 | 0.0011 |
| transport | 219 | 1.41E-04 | calcium-mediated signaling | 4 | 0.0161 | inflammatory response | 10 | 0.0012 |
| localization | 235 | 1.93E-04 | detection of stimulus | 5 | 0.0302 | defense response | 29 | 0.0015 |
| intracellular signaling cascade | 100 | 2.08E-04 | receptor mediated endocytosis | 4 | 0.0353 | response to biotic stimulus | 29 | 0.0028 |
| protein transport | 57 | 2.93E-04 | regulation of apoptosis | 14 | 0.0364 | organismal process | 46 | 0.0034 |
| cell organization and biogenesis | 128 | 3.39E-04 | cell death | 20 | 0.0368 | response to wounding | 13 | 0.0037 |
| protein localization | 59 | 4.69E-04 | death | 20 | 0.0391 | signal transduction | 56 | 0.0043 |
| cellular localization | 60 | 6.56E-04 | detection of external stimulus | 4 | 0.0436 | response to external stimulus | 15 | 0.0048 |
| Golgi vesicle transport | 16 | 8.28E-04 | vesicle-mediated transport | 14 | 0.0436 | lymphocyte activation | 6 | 0.0055 |
| intracellular transport | 58 | 0.001 | intracellular signaling cascade | 33 | 0.0453 | cell communication | 59 | 0.0063 |
| exocytosis | 13 | 0.0012 | response to external stimulus | 18 | 0.0453 | response to stimulus | 47 | 0.0097 |
| protein kinase cascade | 32 | 0.0014 | protein amino acid glycosylation | 7 | 0.0457 | innate immune response | 5 | 0.0099 |
| RNA splicing | 21 | 0.0015 | apoptosis | 19 | 0.0472 | Ras protein signal transduction | 4 | 0.0101 |
| transcription | 48 | 0.0017 | cell adhesion | 22 | 0.0476 | immune cell activation | 6 | 0.0114 |
| mRNA metabolism | 27 | 0.0032 | programmed cell death | 19 | 0.0485 | cell activation | 6 | 0.0118 |
| membrane organization | 10 | 0.0046 | glycoprotein biosynthesis | 7 | 0.0535 | regulation of apoptosis | 11 | 0.0123 |
| phosphate metabolism | 78 | 0.0096 | positive regulation of apoptosis | 8 | 0.0562 | cell death | 15 | 0.0126 |
| primary metabolism | 512 | 0.0115 | biopolymer modification | 46 | 0.057 | intracellular signaling cascade | 23 | 0.0196 |
Gene lists generated from ANOVAs (p<0.05) were subjected to GO-term enrichment analysis in DAVID. The GO database provides biological classification of gene function through functional categories relating to biological processes, molecular functions, or to cellular components. The number of genes associated with a particular GO term is shown. The p-values associated with annotation terms reflect the degree of enrichment bases on the threshold of EASE Score, a modified Fisher Exact P-Value, for gene-enrichment analysis.
Table 5.
Top Networks associated with different stages of illness as determined by Ingenuity Systems Pathways Analysis.
| Short-term DOI: | Long-term DOI: | ||||||
|---|---|---|---|---|---|---|---|
| Top Functions: | Score: | #: | Molecules in Network: | Top Functions: | Score: | #: | Molecules in Network: |
| Gene Expression, Cell Signaling, Connective Tissue Development and Function | 42 | 32 | ABCD2, ACSL1, AGXT, CBR1, CD55, CD79B, CEBPD, Creb, CYP51A1, DDAH1, DNA-directed RNA polymerase, DR1, ELF3, FOLH1, GPLD1, HIST2H4A, LIPA, MED12, MED14, MED17, MED21, MED23, MED30, NF1, OGT, POLR2E, POLR2L, PPARA, PRIM1, RBP7, RNA polymerase II, SLC2A3, SMYD3, SP1, TNFAIP3 | Inflammatory Disease, Cardiovascular Disease, Hematological Disease | 44 | 24 | Ap1, ARRB1, C1QB, C3AR1, CCL5, CLCF1, Creb, CREB3L4, DLL4, DMBT1, F3, HNRPAB, IL1, IL1F5, LDL, LTBR, Mmp, MMP2, NFIC, NFkB, Nuclear factor 1, Pdgf, PI3K, POU2F2, PTGER3, PTGS1, PTGS2, RHOH, SPP1, Tgf beta, TNFAIP8, TNFSF13, TNFSF18, Vegf, WDR5 |
| Cell Morphology, Muscular System Development and Function, Carbohydrate Metabolism | 35 | 29 | ADM, ANGPTL2, CA2, CA8, CA14, CARS, CPD, CREB1, DKK2, EDN1, HES1, HNRNPU, HRH3, IKZF1, LEF1, LTB4R2, Mek, NPY, PBEF1, PDGF BB, Pkg, PPP1R14A, PSMB4, SEC63, SFRS1, SLC1A1, SPP1, SRPK2, SYTL4, TPM4, VCAN, ZBTB16 | Organismal Injury and Abnormalities, Tissue Morphology, Gastrointestinal Disease | 41 | 23 | AFP, CAV3, CLOCK, E2f, ERK1/2, Fgf, FGF4, FGF10, Fgfr, FKBP5, G alphai, GRIN2B, GUCY2C, HCK, HIST2H4A, ITGB2, MT2A, MUC4, NCAPG, PFKFB1, Pka, PLCgamma, PP2A, Ras, SNTB2, SYK/ZAP, TACSTD2, TCR, TGFB1, TLR2, TLR10, TLX1, TSC22D3, TYRP1, VAV |
| Developmental Disorder, Neurological Disease, Organ Morphology | 29 | 26 | ADAM10, APLP2, APP, ARNT, C1q, CNTN1, DLX2, ENAH, GAD1, GLDN, KLK6, LDLRAP1, MAPK14, MHC Class I, MOG, MT1G, N-cor,Notch, NR0B2, NR4A2, NRCAM, OGDH, PGK1, PSEN1, RAPSN, Rar, REST, Rxr, SEC23A, Secretase gamma, TRIP11, Vegf, ZBTB33, ZNF335 | Connective Tissue Development and Function, Muscular System Development and Function | 36 | 21 | Akt, AR, CA12, CNKSR1, FPR1, FUS, GRB2, HIST3H3, Histone h3, Insulin, Jnk, KALRN, LBH, Mapk, MAPKAPK3, MAS1, Mek, MGST1, Nfat, OMD, P38 MAPK, PDGF BB, Pkc, PTPRE, Rac, RAC2, RNA polymerase II, SLC18A3, SNURF, SOX6, STAT5a/b, SUB1, TJP2, TPD52L1, UCP2 |
| Neurological Disease, Infectious Disease, Psychological Disorders | 27 | 25 | Actin, Alpha Actinin, ARL1, Calpain, CAPN3, CAPN7, Cbp/p300, Cpla2, DHX9, F Actin, GABAR-A, GABRA5, GABRB3, GABRG2, GABRP, GAL3ST1, IPP, LIMA1, MAG, MBP, MLPH, Pkc(s), Pld, PLD1, PLDN, PTPRD, Sox, SOX1, SOX10, SOX17, ST8SIA4, TMOD3, TWF1, UGT8, VIL1 | Gene Expression, Cell Death, Hematologic al Disease | 21 | 14 | ABCC5, ANKHD1, ARNT2, ATP12A, ATP1B1, C20ORF70, CAPZA2, DDX54, DEGS1, EGFR, FGF2, FN1, GJB3, GPD2, HCRTR2, KLK1B9, KLK1B22, LRRFIP1, MTCP1, NFKB1, NR3C1, PSMB2, PSMB8, PTPRK, RUFY1, SAPS2, SELENBP1, SEMA5B, SH2D3A, SLC17A1, TAL2, TYROBP, UGT2B15 |
| Intermediate-term DOI: | |||||||
| Top Functions: | Score: | #: | Molecules in Network: | Top Functions: | Score: | #: | Molecules in Network: |
| Cell-To-Cell Signaling and Interaction, Embryonic Development, Gene Expression | 47 | 28 | ALDH1A3, BCL3, CDKN1C, Cyclin E, DHFR, E2f, E2F7, EIF2AK1, FABP2, FGF5, FKHL18, HDAC1, HNRNPA1, HSPB1, IL8RB, LY96, NFAT complex, NFkB, NIBP, PAWR, PDGF BB, PIR, PLC gamma, PRTN3, Rb, SET, SMYD1, SP2, TAGLN, TFF1, TLR2, TLR7, TNFRSF12A, TNFSF18, TRIP6 | Nervous System Development and Function, Lipid Metabolism, Small Molecule Biochemistry | 27 | 19 | Akt, Ap1, CD2, CYBB, CYP3A7, DOCK10, Dynamin, EHD4, ETV5, HERC1, Hsp70, IGFBP5, IL1, Jnk, Mapk, MCAM, Mmp, NEU3, Nfat, NGFB, P38 MAPK, PAK2, PGLYRP1, PI3K, PIP5K3, PRKG1, Rac, RAC2, Ras, Rxr, SLC12A4, SOST, TCFL5, Tgf beta |
| Drug Metabolism, Molecular Transport, Small Molecule Biochemistry | 40 | 25 | ADCY6, ANXA5, Calpain, CAPN3, CDC42EP1, Cpla2, CUTL2, EDG2,EGF, ERK1/2, GAL3ST1, GUCY, GUCY1B2, KL, Mek1/2, NEK1, NPR1, OXT, PHLDA2, PITPNA, Pka, Pkc(s), PLA2G4B, PLC, PLCD1, PP2A, PPP2R3A, PTPRE, Rsk, RYR1, S100B, SLC6A2, TAC1, ZHX2, ZNF350 | ||||
The top Networks for each stage of illness are shown. All of the molecules that compose each network are listed; those shown in bold represent focus molecule that appeared as differentially expressed at p<0.05. The numbers of genes (#) is indicated. The score shown is based on a p-value calculation for enrichment using the right-tailed Fisher Exact Test.
Figure 2.

Venn diagram showing overlap of similar functions attributed to different stages of illness in schizophrenia. The functions/systems shown (A-G) represent a compilation of the results from Gene Ontology and Functional Clustering Analysis using DAVID, and Functional and Network analyses identified using Ingenuity Systems Pathways databases using the top differentially expressed genes at p<0.01 from each cohort. (See Tables 3 and 4 and Supplementary Table 5 for complete lists of annotations).
2.4 Real-time PCR validation of expression changes
To validate microarray findings, we selected a heterogeneous group of genes from these various functional categories for real-time PCR analysis on all subjects. These include protein phosphatase 1E (PPM1E), REST corepressor 3 (RCOR3), neuropeptide Y (NPY), solute carrier family 29, member 2 (SLC29A2), B and T lymphocyte associated (BTLA), F-box protein 31 (FBXO31), and sulfatase 1 (SULF1) for short-term DOI subjects, and Ras and Rab interactor 3 (RIN3), integrin beta 2 (ITGB2), lymphotoxin B receptor (LTBR), complement component 1, q subcomponent, B chain (C1QB) and early B-cell factor 1 (EBF1) for subjects with intermediate- and long-term DOI. Covariate analyses on the microarray expression values for these genes found no significant effects of pH, PMI, age, sex, suicide or drug dose on the expression levels for each of these genes (Table 6). Comparing the differential expression for the 12 genes determined by qPCR and microarray analyses revealed excellent correlation between the two methods (r = 0.84; p = 0.0006) (Figure 3A). In addition to validating expression differences for a subset of these genes in the same short-term DOI subject used for the microarray analysis, we also measured expression levels by qPCR in six additional schizophrenic subjects with DOI <7 yrs and their matched controls. Overall, significant decreases in expression were detected for PPM1E, RCOR3, NPY, SLC29A2, SULF1 and FBXO31 (Figure 3B).
Table 6.
Pearson's Product Moment correlation values (r values) and p-values of gene expression levels to demographic data.
| r values: | p-value: | |||||
|---|---|---|---|---|---|---|
| Gene: | pH: | PMI: | Age: | Drug Dose: | Sex: | Suicide: |
| NPY | -0.294 | 0.290 | -0.0004 | -0.065 | 0.876 | 0.643 |
| PPM1E | -0.013 | -0.048 | 0.188 | 0.217 | 0.053 | 0.809 |
| RCOR3 | -0.148 | 0.019 | -0.0004 | 0.048 | 0.214 | 0.442 |
| SLC29A2 | -0.268 | -0.178 | 0.374 | 0.0336 | 0.076 | ND |
| BTLA | -0.402 | -0.178 | -0.188 | 0.001 | 0.572 | ND |
| FBXO31 | 0.085 | -0.242 | -0.034 | 0.128 | 0.236 | 0.101 |
| SULF1 | 0.146 | -0.008 | -0.041 | 0.097 | 0.560 | 0.221 |
| LTBR | 0.021 | -0.523* | 0.062 | -0.364 | 0.605 | 0.647 |
| RIN3 | 0.143 | 0.282 | 0.066 | 0.092 | 0.285 | 0.520 |
| ITGB2 | 0.356 | -0.201 | 0.141 | -0.294 | 0.453 | 0.651 |
| C1QB | 0.291 | -0.057 | -0.083 | -0.019 | 0.346 | 0.782 |
| EBF | -0.372 | 0.058 | 0.432 | 0.020 | 0.960 | 0.103 |
Pearson Product Moment correlations were performed to assess the effects of pH, PMI, and drug dose (in chlorpromazine equivalents) on the microarray gene expression values. Student's t test (two-tailed, unpaired) was used to determine differences due to sex and suicide. N.D., not determined. Asterisk indicates significant correlation at P<0.05.
Figure 3.

Correlation between microarray expression values and real-time PCR results from control and schizophrenic subjects. A., Correlation analysis was performed between microarray and qPCR expression values for the 12 indicated genes. Differential (schizophrenic minus control) microarray expression average log2 ratios are plotted on the X axis, whereby the Y axis depicts the ΔΔCt (controls minus schizophrenic) from the real-time PCR experiments. Data for NPY, RCOR3, SLC29A1, BTLA, PPM1E, FBXO31 and SULF1 were generated from subjects with short-term DOI (n=8-12 schizophrenic subjects and matched controls) and RIN3, ITGB2, LTBR, C1QB and EBF1 from subjects with intermediate- and long-term DOI (Genes are denoted by Unigene gene symbols as defined in the text. The relative abundance of each gene expression was normalized by beta-2 microglobulin (B2M) in short-DOI subjects and by TUBB in intermediate- and long-DOI subjects. B. Expression levels of the indicated genes in schizophrenic subjects with short term illness only. Data are depicted as fold-change of the mean expression level ± SEM (n=10-12 schizophrenic subjects and matched controls). The relative abundance of each gene expression was normalized by B2M. Student's t tests were used to determine significant differences in gene expression levels. * denotes significantly different from control at p<0.05, two-tailed t test; + denotes significantly different from controls at p<0.05, one-tailed t test.
3. Discussion
In this study, we detected distinct gene expression profiles associated with different durations of illness in subjects with schizophrenia, which supports the hypothesis that the disease may have an evolving, progressive pathology. From comprehensive pathway analyses and database searches, we found that each stage was associated with dysfunctions in different biological systems, although some common biochemical themes underlying all stages were identified. The most striking findings from these studies were the high level of gene expression derangement observed early in the illness, and the contrasting expression profiles revealed between subjects with short and long duration illness.
Knowledge of gene expression patterns from subjects early in illness may be of particular importance as symptom profile and treatment responsiveness during the early stages of illness are thought to be the best predictors for disease outcome (Lieberman 2006). At the pathways analyses level, we found that early illness was associated with dysfunctions in diverse systems, including metal ion binding, vesicle-mediated transport, RNA processing, reproductive systems, nervous system development and carbohydrate metabolism. We validated expression differences by quantitative PCR analysis of genes associated with some of these systems, including PPM1E, RCOR3, NPY, SLC29A2, SULF1 and FBXO31, in an additional six schizophrenic subjects with duration of illness <7 years and matched controls, further implicating their involvement in short duration illness. We hypothesize that these gene expression changes may be more relevant to pathogenic mechanisms of the disease, as they may occur in closer temporal proximity to underlying pathological triggers.
Of further interest was the association of short-term illness with gene transcription dysfunction. Among our list of differentially expressed genes found in the brains of schizophrenic subjects ≤4 years of diagnosis, 28 were specifically annotated as encoding proteins with transcriptional regulatory activity. Alterations in the expression of these transcription factors may affect important developmental processes still happening in early life (throughout the 20s). Further, these transcription factors may drive other pathological or compensatory changes in gene expression happening later in illness. Significantly, one such gene (validated using qPCR analysis) was RCOR3, which is one of several proteins that function as co-repressors for the RE1 silencing transcription factor (REST) (Andres et al., 1999), a gene also found to be significantly altered in subjects with short-term illness (see Table 4; Supplementary Table 1). REST has been shown to mediate negative regulation of neuronal genes and play an important role in neuronal differentiation (Ballas et al., 2001; Greenway et al., 2007). It is thought that down-regulation of REST during neurogenesis is necessary for proper neuronal development (Ballas et al. 2001). Hence, altered expression of REST and RCOR3 into adolescence may be associated with abnormalities in synaptic pruning and connectivity that have been hypothesized to trigger the emergence of symptoms and pathological deficits of schizophrenia (Woo and Crowell, 2005).
Events occurring during the critical period following initial diagnosis of schizophrenia are thought to be critical for effective disease management and outcome (Lieberman et al. 2001; Lieberman et al., 2005). One reason that early intervention may be so effective could be due to the effect of antipsychotic drugs on gene expression activity. Antipsychotic drugs are known to alter the expression of many diverse genes, including transcription factors (Thomas 2006). Hence, early medication may help ameliorate widespread dysregulation of gene expression activity occurring early in illness, which may help prevent the cascade of downstream pathological outcomes.
Acknowledging the potential effects of antipsychotic drugs, our subjects with short-term DOI had histories of antipsychotic drug treatment for 2-4 years, while those of long duration may have been treated for decades. Hence, we cannot exclude that possibility that differences between cohorts of different disease duration are due to disparities in lifetime exposure to such drugs. However, as a preliminary argument against our findings being simply due to antipsychotic drug treatment, we did not find significant correlations between the expression levels of any validated gene and antipsychotic drug dose, reported in chlorpromazine equivalents, which considers lifetime exposure (see Table 6). There were also no significant differences among the average drug doses taken by the subjects in each cohort (Table 1). Moreover, we have identified changes in gene expression that replicate findings from a previous study that had ruled out an effect of drug treatment, as these expression changes were also found in the cortex of subjects who were medication-free at the time of death (Arion et al., 2007). As an alternative hypothesis to the idea that antipsychotic drug treatment is responsible for the changing molecular profiles at different stages of illness, we propose that the brain may undergo an adaptive response to pathology over the course of illness to achieve a new molecular homeostasis.
Another confound in our study was a higher rate of suicide in the short- and intermediate-term DOI subjects compared to the chronic subjects, none of whom died by suicide. Therefore we cannot rule the possibility that some changes in gene expression observed in the microarray datasets may have suicide as a confounding factor. However, we have shown that suicide is not a significant factor with regards to levels of mRNA for the genes validated using qPCR (Table 6), suggesting that suicide is not likely to be the cause of all the changes we identified using microarrays.
In contrast to early illness, chronic schizophrenia was found to be associated with quite different systems dysfunction. Top functional clustering categories and network analyses linked differential gene expression in our chronic subjects to immune function, stimulus response and inflammation. Related to these pathways, we validated the microarray expression results for ITGB2, LTBR, EBF and C1QB in subjects with long-term illness, as well as in a subset of subjects with intermediate-term illness. There is considerable evidence that CQ1B and other complement components are involved in CNS inflammatory and degenerative disorders (Johnson et al., 1992). As schizophrenia is not typically considered a neurodegenerative disorder, the increased expression of these genes may represent a sustained response to a progressive pathology that is occurring within the brains of schizophrenic patients, which, in its later stages, may cause some degenerative processes. Such a postulate would be consistent with the progressive brain structural deficits (i.e. gray matter shrinkage and ventricular enlargement) detected in chronic schizophrenic subjects. A confound in this part of our study is that the manifestation of immune- and inflammation-related expression changes detected in older individuals (aged 55-81) may reflect the impact of longer exposure to environmental factors and/or life stressors. However, a recent microarray study has also reported molecular evidence for altered immune function in schizophrenia, although different genes were highlighted (Arion et al. 2007). This defect was primarily found in a subset of subjects, which may be more related to our chronic subjects, although the durations of illness for these subjects was not specifically stated.
Genes related to several different systems previously implicated in the pathology of schizophrenia (i.e. GABAergic system, presynaptic machinery, myelination, ubiquitination) were also found to be differentially expressed in our study, highlighting the reliability of our findings. Interestingly, expression changes related to these systems were found most robustly in subjects with early illness. Additional genes we found to be altered in the subjects with short DOI, (increased expression of HSPB1 and decreased expression of EVI2, MAPK1, SCAMP1, SPOCK3, CDK5R1, PAFAH1B1), replicate findings from a recent microarray analysis that performed a rigorous assessment of differentially expressed genes in prefrontal cortex from schizophrenic subjects (Arion et al. 2007).
In summary, our findings provide evidence that patterns of gene expression in schizophrenia change with duration of illness. At the level of pathway analyses, we found that each stage was associated with dysfunction in distinct biological systems, although some common biochemical themes underlying all stages were identified. Further, we find that early stages of illness are associated with the highest level of gene expression derangement, which may result from persistent developmental insults, and that these may drive additional pathological and compensatory changes that occur throughout the course of disease. While this study has provided insight into the nature of such molecular changes that occur with disease progression in schizophrenia, further work should focus on understanding the mechanisms that control evolving gene expression, which may lead to future therapies that modify the course of disease.
4. Experimental Procedures
4.1 Subjects
Prior to commencement, approval for this study was obtained from the Ethics Committee of the Victorian Institute of Forensic Medicine and the North Western Mental Health Program Behavioral and Psychiatric Research and Ethics Committee. Psychiatric diagnoses of schizophrenia were made according to DSM-IV criteria (American Psychiatric Association, 1994) by consensus between two senior psychiatrists and a psychologist following extensive case history review using the Diagnostic Instrument for Brain Studies (DIBS) (Hill et al., 1996). As part of the DIBS protocol, it was noted when there was a Coroner's report of death by suicide. Subjects were carefully selected into three cohorts to represent different stages of illness. We obtained one cohort consisting of subjects very close to initial diagnosis (≤ 4 years duration of illness (DOI); n=8 patients, n=8 controls). A second cohort was comprised of chronic schizophrenics who had long-term DOI (> 28 years of illness; n=8 patients, n=8 controls). The third cohort consisted of subjects with intermediate ranges of DOI (7-18 years of illness; n=14 patients, n=14 controls). See Table 1 for subject summaries and Supplementary Table 3 for individual values and causes of death. DOI was calculated as the time from initial diagnosis to death. To minimize variation in the effects of antipsychotic drugs, all subjects chosen were treated with “typical” antipsychotic drugs. The final recorded dose of antipsychotic drug for each subject was converted to chlorpromazine equivalents, which incorporates lifetime exposure to antipsychotic medications (Bezchlibnyk-Butler and Jeffries 1999). In cases where death was witnessed, the time between death and autopsy was taken as the postmortem interval (PMI). Where death was not witnessed, tissue was taken only from individuals who had been seen alive up to 5 hours before being found dead; in these cases PMI was taken as the interval half way between the donors being found dead and last being seen alive. Importantly, in all cases, cadavers were refrigerated within 5 hours of being found and tissue was rapidly frozen to -70°C within 30 minutes of autopsy.
4.2 RNA Preparation and Microarray Analysis
Total RNA was extracted from the prefrontal cortex (Brodmann Area 46; 100 mg; left hemisphere) from all subjects as described previously (Desplats et al., 2006). RNA quantification was determined by spectrophotometer readings, and quality by Agilent Bioanalyzer scans. RNA integrity (RIN) numbers were not available at the time of RNA extraction, but Bioanalyzer traces of each RNA sample showed no evidence for degradation products. RNA samples were amplified, biotin-labeled and then hybridized to the Human Genome U133 Plus 2.0 array using standard Affymetrix protocol as described in Lockhart et al, 1996. Chips were scanned using the Affymetrix ScanArray 3000 using default settings and a target intensity of 250 for scaling. Present and Absent calls were determined with the Affymetrix algorithm (GCOS, Affymetrix, Santa Clara, CA). The quality controls of the hybridization for each array were assessed by the percent Present calls and GAPDH 3′/5′ ratios for each array. Hierarchical clustering methods were used (Eisen et al., 1998) to detect outliers in all samples, and from this analysis, one sample (short control 7) was omitted. Pearson's analyses found no significant correlations between the pH and PMI of each sample versus percent Present call and GAPDH 3′/5′ratios for each corresponding array using all 59 samples. Nonetheless, based on ANCOVA analysis on gene expression data, arrays with GAPDH 3′/5′ ratios of >2.0 from the intermediate DOI cohort were excluded (see below). This left 55 samples for unpaired statistical analyses and 50 samples for pairwise analyses.
RNA expression data from schizophrenic subjects and their matched controls were normalized using dChip (DNA-Chip Analyzer), a statistical model for the probe-level data which gives model-based estimates for gene expression indices (Li and Hung Wong 2001) and subjected to a batch adjustment normalization to allow for comparisons among all three cohorts (Johnson et al., 2007). Data were filtered to exclude those transcripts that were not present in at least one of the RNA samples, those with redundant probeset IDs (retaining those with the highest p-value for hybridization) and those showing the lowest variation; a list of 14,438 probesets remained for statistical testing. ANOVA analyses with error variance correction were performed using National Institutes of Aging (NIA) Array Tools (Sharov et al., 2005) on each cohort separately. To identify differentially expressed genes between schizophrenic and control subjects, we utilized both unpaired and pair-wise comparisons. For unpaired analyses, the geometric mean of the normalized expression values for all schizophrenic subjects in each cohort was compared against the geometric mean of the normalized expression values for all control subjects in each cohort. For the pair-wise analysis, the normalized expression value for each schizophrenic subject was compared against its matched control in a pair-wise manner, then, the results for all pairs in each cohort were averaged. Within the schizophrenic subjects only, a separate ANOVA was performed comparing expression levels of subjects with short-term DOI vs. those subjects with long-term DOI, and similarly in control subjects, the young vs. aged cohorts were compared. The resulting lists of differentially expressed genes (p-value <0.05) from both analyses were then filtered for an absolute log2 ratio >0.322 (i.e. 1.25 fold, increase or decrease).
In NIA Array Tools, the FDR was controlled according to Benjamini and Hochberg (Benjamini and Hochberg 1995) at a default setting of 0.1. Approximately 50 genes meeting these criteria were identified in our datasets from subjects with short DOI, with fewer genes in subjects with intermediate- and long-term DOI. It is difficult to compare these findings to the post-mortem microarray literature, as most previous microarray studies have not applied standard FDRs to their datasets. One drawback of such a multiple testing correction is that it assumes that each gene varies independently, which in the context of CNS gene expression, is often not the case (Sequeira and Turecki 2006). Therefore, to allow for a more inclusive understanding of potential pathological bases in schizophrenia, we have use a less stringent criteria, as discussed previously (Benes et al., 2006), which included genes with FDR≥0.1 for extended gene lists and pathways analysis. Nonetheless, as a surrogate test of our true false positive rate, we have performed large-scale real-time PCR analysis of 37 genes and find that ∼85% of the expression differences determined by the microarray analyses could be validated either in the same subjects used for the microarray analysis and/or in additional schizophrenic subjects with similar durations of illness. Data for 12 such genes are reported in the current manuscript.
4.3 Covariate analyses
The lists of differentially expressed genes from each cohort (at p<0.01) were subjected to a global ANCOVA according to (Goeman et al., 2004), to assess the effects of demographic (age and sex), sample (pH and PMI) and array (GAPDH 3′/5′ ratios) parameters on the variance of gene expression between controls and schizophrenic subjects within each cohort. A confounding effect of pH was found in subjects of the intermediate DOI cohort, however, after removing 4 subjects with high GAPDH 3′/5′ ratios, the effect of pH was no longer detected. This model is based on the empirical Bayesian generalized linear model. ANCOVA analysis using DOI as a continuous variable was not possible because control subjects do not have a DOI. For all genes validated by qPCR analysis, additional Pearson Product Moment correlations were performed to assess the effects of the continuous variables, age, pH and PMI on gene expression values in all subjects, and the effects of drug dose on expression in all subjects with schizophrenia. Before the Pearson's correlations were run, the datasets were tested for a Gaussian distribution using the Kolmogorov-Smirnov method, which confirmed that the expression data was normally distributed. Student's t test (two-tailed, unpaired) was used to determine differences in expression of these validated genes due to sex and suicide. All statistical analyses were performed using GraphPad Prism Software (GraphPad Inc, San Diego, CA).
4.4 Pathways Analyses
Pathways Analysis was performed using Ingenuity Pathways Analysis (IPA) program (www.ingenuity.com) and the Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/) to understand the potential biological relevance of differentially expressed genes at each stage of illness. The IPA Tools calculate a p-value using the right-tailed Fisher Exact Test, in order to determine statistically significant over-representation of functional analysis molecules in a given network of known interactions. The p-values associated with annotation terms from DAVID reflect the degree of enrichment bases on the threshold of EASE Score, a modified Fisher Exact P-value, for gene-enrichment analysis.
4.5 Real-Time PCR Analysis
Real-time PCR experiments were performed as described previously (Desplats et al. 2006), using specific primers for each sequence of interest and against four housekeeping genes: human β-2-microglobulin (B2M), beta-tubulin (TUBB), hypoxanthine guanine phosphoribosyl transferase (HPRT) and porphobilinogen deaminase (PBGD) (Supplementary Table 4). PCR reactions were performed on two independent sets of cDNA samples: those used for in the initial array experiments and on cDNA samples prepared from an extended cohort of subjects with short DOI (Supplementary Table 5). We first compared the expression of all four housekeeping genes in samples from control and schizophrenic subjects to assess variability in expression among all subjects and determine expression differences between control and schizophrenic subjects. In subjects with short DOI and their matched controls, B2M showed the least variation in threshold cycle (Ct) among all samples and showed no significant differences in expression between control and schizophrenic subjects, hence B2M was using as the internal control for subjects with short DOI and their controls. In subjects with long DOI and their matched controls, TUBB showed the least variation in Ct among all subjects and no significant differences in expression between control and schizophrenic subjects, hence TUBB was used for normalization. The amount of cDNA in each sample was calculated using SDS2.1 software by the comparative Ct method and expressed as 2exp(Ct). Significant differences in expression (p<0.05) were determined by Student's t tests (one- and two-tailed) (GraphPad Prism, San Diego, CA).
Supplementary Material
Acknowledgments
This study was funded by grants from the National Institutes of Health (NS44169 and MH069696 to E.A.T.). The authors wish to thank Kristi E. Kass and Lana Schaffer for excellent technical assistance.
Abbreviations
- DOI
duration of illness
- PPM1E
protein phosphatase 1E
- RCOR3
REST corepressor 3
- NPY
neuropeptide Y
- SLC29A2
solute carrier family 29, member 2
- BTLA
B and T lymphocyte associated
- FBXO31
F-box protein 31
- SULF1
sulfatase 1
- RIN3
Ras and Rab interactor 3
- ITGB2
integrin beta 2
- LTBR
lymphotoxin B receptor
- C1QB
complement component 1, q subcomponent, B chain
- EBF1
early B-cell factor 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci U S A. 1999;96:9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62:711–721. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston C, Jiang L, Sokolov BP. Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol Psychiatry. 2005;10:309–322. doi: 10.1038/sj.mp.4001565. [DOI] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Benes FM, Matzilevich D, Burke RE, Walsh J. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry. 2006;11:241–251. doi: 10.1038/sj.mp.4001758. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Bezchlibnyk-Butler K, Jeffries Je. Clinical Handbook of Psychotropic Drugs. Toronto: Hogrefe & Huber Publishers; 1999. [Google Scholar]
- Dean B, Keriakous D, Scarr E, Thomas EA. Gene expression profiling in Brodmann's area 46 from subjects with schizophrenia. Australian and New Zealand Journal of Psychiatry. 2007;41:308–320. doi: 10.1080/00048670701213245. [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Sakuma M, Maurizio AM, Relja M, Hoff AL. Cerebral ventricular change over the first 10 years after the onset of schizophrenia. Psychiatry Res. 2004;130:57–70. doi: 10.1016/j.pscychresns.2003.08.004. [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Szulc KU, Bertisch HC, Majcher M, Brown K. Understanding structural brain changes in schizophrenia. Dialogues Clin Neurosci. 2006;8:71–78. doi: 10.31887/DCNS.2006.8.1/ldelisi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSisto M, Harding CM, McCormick RV, Ashikaga T, Brooks GW. The Maine and Vermont three-decade studies of serious mental illness. II. Longitudinal course comparisons. Br J Psychiatry. 1995;167:338–342. doi: 10.1192/bjp.167.3.338. [DOI] [PubMed] [Google Scholar]
- Desplats PA, Kass KE, Gilmartin T, Stanwood GD, Woodward EL, Head SR, Sutcliffe JG, Thomas EA. Selective deficits in the expression of striatal-enriched mRNAs in Huntington's disease. J Neurochem. 2006;96:743–757. doi: 10.1111/j.1471-4159.2005.03588.x. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton WS, McGlashan TH. Natural history of schizophrenia subtypes. I. Longitudinal study of paranoid, hebephrenic, and undifferentiated schizophrenia. Arch Gen Psychiatry. 1991;48:969–977. doi: 10.1001/archpsyc.1991.01810350009002. [DOI] [PubMed] [Google Scholar]
- Friedman JI, Harvey PD, Coleman T, Moriarty PJ, Bowie C, Parrella M, White L, Adler D, Davis KL. Six-year follow-up study of cognitive and functional status across the lifespan in schizophrenia: a comparison with Alzheimer's disease and normal aging. Am J Psychiatry. 2001;158:1441–1448. doi: 10.1176/appi.ajp.158.9.1441. [DOI] [PubMed] [Google Scholar]
- Goeman JJ, van de Geer SA, de Kort F, van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Greenway DJ, Street M, Jeffries A, Buckley NJ. RE1 Silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells. 2007;25:354–363. doi: 10.1634/stemcells.2006-0207. [DOI] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutunian V, Katsel P, Dracheva S, Stewart DG, Davis KL. Variations in oligodendrocyte-related gene expression across multiple cortical regions: implications for the pathophysiology of schizophrenia. Int J Neuropsychopharmacol. 2007:1–9. doi: 10.1017/S1461145706007310. [DOI] [PubMed] [Google Scholar]
- Harvey PD, Bertisch H, Friedman JI, Marcus S, Parrella M, White L, Davis KL. The course of functional decline in geriatric patients with schizophrenia: cognitive-functional and clinical symptoms as determinants of change. Am J Geriatr Psychiatry. 2003;11:610–619. doi: 10.1176/appi.ajgp.11.6.610. [DOI] [PubMed] [Google Scholar]
- Harvey PD, Silverman JM, Mohs RC, Parrella M, White L, Powchik P, Davidson M, Davis KL. Cognitive decline in late-life schizophrenia: a longitudinal study of geriatric chronically hospitalized patients. Biol Psychiatry. 1999;45:32–40. doi: 10.1016/s0006-3223(98)00273-x. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2007 doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemby SE, Ginsberg SD, Brunk B, Arnold SE, Trojanowski JQ, Eberwine JH. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch Gen Psychiatry. 2002;59:631–640. doi: 10.1001/archpsyc.59.7.631. [DOI] [PubMed] [Google Scholar]
- Hill C, Keks NA, Roberts S, Opeskin K, Dean B, Copolov DL. Postmortem brain studies in schizophrenia: The problems of diagnosis. Am J Psychiatry. 1996;153:533–537. doi: 10.1176/ajp.153.4.533. [DOI] [PubMed] [Google Scholar]
- Ho BC, Andreasen NC, Nopoulos P, Arndt S, Magnotta V, Flaum M. Progressive structural brain abnormalities and their relationship to clinical outcome: a longitudinal magnetic resonance imaging study early in schizophrenia. Arch Gen Psychiatry. 2003;60:585–594. doi: 10.1001/archpsyc.60.6.585. [DOI] [PubMed] [Google Scholar]
- Huber G, Gross G, Schuttler R. A long-term follow-up study of schizophrenia: psychiatric course of illness and prognosis. Acta Psychiatr Scand. 1975;52:49–57. doi: 10.1111/j.1600-0447.1975.tb00022.x. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Kato T. Gene expression profiling in schizophrenia and related mental disorders. Neuroscientist. 2006;12:349–361. doi: 10.1177/1073858406287536. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Lampert-Etchells M, Pasinetti GM, Rozovsky I, Finch CE. Complement mRNA in the mammalian brain: responses to Alzheimer's disease and experimental brain lesioning. Neurobiol Aging. 1992;13:641–648. doi: 10.1016/0197-4580(92)90086-d. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Katsel P, Davis KL, Haroutunian V. Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions in schizophrenia: a gene ontology study. Schizophr Res. 2005;79:157–173. doi: 10.1016/j.schres.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Katsel PL, Davis KL, Haroutunian V. Large-scale microarray studies of gene expression in multiple regions of the brain in schizophrenia and Alzheimer's disease. Int Rev Neurobiol. 2005;63:41–82. doi: 10.1016/S0074-7742(05)63003-6. [DOI] [PubMed] [Google Scholar]
- Knable MB, Torrey EF, Webster MJ, Bartko JJ. Multivariate analysis of prefrontal cortical data from the Stanley Foundation Neuropathology Consortium. Brain Res Bull. 2001;55:651–659. doi: 10.1016/s0361-9230(01)00521-4. [DOI] [PubMed] [Google Scholar]
- Kraepelin E. Dementia praecox and paraphrenia. In: Kreiger RE, editor. Melbourne. 1971. original 1919. [Google Scholar]
- Kurtz MM. Neurocognitive impairment across the lifespan in schizophrenia: an update. Schizophr Res. 2005;74:15–26. doi: 10.1016/j.schres.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Kurtz MM, Seltzer JC, Ferrand JL, Wexler BE. Neurocognitive function in schizophrenia at a 10-year follow-up: a preliminary investigation. CNS Spectr. 2005;10:277–280. doi: 10.1017/s1092852900022598. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA. Is schizophrenia a neurodegenerative disorder? A clinical and neurobiological perspective. Biol Psychiatry. 1999;46:729–739. doi: 10.1016/s0006-3223(99)00147-x. [DOI] [PubMed] [Google Scholar]
- Lieberman JA. Neurobiology and the natural history of schizophrenia. J Clin Psychiatry. 2006;67:e14. [PubMed] [Google Scholar]
- Lieberman JA, Perkins D, Belger A, Chakos M, Jarskog F, Boteva K, Gilmore J. The early stages of schizophrenia: speculations on pathogenesis, pathophysiology, and therapeutic approaches. Biol Psychiatry. 2001;50:884–897. doi: 10.1016/s0006-3223(01)01303-8. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Mathalon DH, Sullivan EV, Lim KO, Pfefferbaum A. Progressive brain volume changes and the clinical course of schizophrenia in men: a longitudinal magnetic resonance imaging study. Arch Gen Psychiatry. 2001;58:148–157. doi: 10.1001/archpsyc.58.2.148. [DOI] [PubMed] [Google Scholar]
- McGlashan TH. A selective review of recent North American long-term followup studies of schizophrenia. Schizophr Bull. 1988;14:515–542. doi: 10.1093/schbul/14.4.515. [DOI] [PubMed] [Google Scholar]
- McGlashan TH. The profiles of clinical deterioration in schizophrenia. J Psychiatr Res. 1998;32:133–141. doi: 10.1016/s0022-3956(97)00015-0. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22:2718–2729. doi: 10.1523/JNEUROSCI.22-07-02718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Levitt P, Lewis DA. Critical appraisal of DNA microarrays in psychiatric genomics. Biol Psychiatry. 2006;60:163–176. doi: 10.1016/j.biopsych.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Mori T, Ohnishi T, Hashimoto R, Nemoto K, Moriguchi Y, Noguchi H, Nakabayashi T, Hori H, Harada S, Saitoh O, Matsuda H, Kunugi H. Progressive changes of white matter integrity in schizophrenia revealed by diffusion tensor imaging. Psychiatry Res. 2007;154:133–145. doi: 10.1016/j.pscychresns.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Saijo T, Abe T, Someya Y, Sassa T, Sudo Y, Suhara T, Shuno T, Asai K, Okubo Y. Ten year progressive ventricular enlargement in schizophrenia: an MRI morphometrical study. Psychiatry Clin Neurosci. 2001;55:41–47. doi: 10.1046/j.1440-1819.2001.00783.x. [DOI] [PubMed] [Google Scholar]
- Sequeira A, Turecki G. Genome wide gene expression studies in mood disorders. Omics. 2006;10:444–454. doi: 10.1089/omi.2006.10.444. [DOI] [PubMed] [Google Scholar]
- Sharov AA, Dudekula DB, Ko MS. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics. 2005;21:2548–2549. doi: 10.1093/bioinformatics/bti343. [DOI] [PubMed] [Google Scholar]
- Sporn AL, Greenstein DK, Gogtay N, Jeffries NO, Lenane M, Gochman P, Clasen LS, Blumenthal J, Giedd JN, Rapoport JL. Progressive brain volume loss during adolescence in childhood-onset schizophrenia. Am J Psychiatry. 2003;160:2181–2189. doi: 10.1176/appi.ajp.160.12.2181. [DOI] [PubMed] [Google Scholar]
- Steinmeyer EM, Marneros A, Deister A, Rohde A, Junemann H. Long-term outcome of schizoaffective and schizophrenic disorders: a comparative study. II. Causal-analytical investigations. Eur Arch Psychiatry Neurol Sci. 1989;238:126–134. doi: 10.1007/BF00450999. [DOI] [PubMed] [Google Scholar]
- Stephens JH, Richard P, McHugh PR. Long-term follow-up of patients hospitalized for schizophrenia, 1913 to 1940. J Nerv Ment Dis. 1997;185:715–721. doi: 10.1097/00005053-199712000-00001. [DOI] [PubMed] [Google Scholar]
- Sugai T, Kawamura M, Iritani S, Araki K, Makifuchi T, Imai C, Nakamura R, Kakita A, Takahashi H, Nawa H. Prefrontal abnormality of schizophrenia revealed by DNA microarray: impact on glial and neurotrophic gene expression. Ann N Y Acad Sci. 2004;1025:84–91. doi: 10.1196/annals.1316.011. [DOI] [PubMed] [Google Scholar]
- Thomas EA. Molecular profiling of antipsychotic drug function: convergent mechanisms in the pathology and treatment of psychiatric disorders. Mol Neurobiol. 2006;34:109–128. doi: 10.1385/MN:34:2:109. [DOI] [PubMed] [Google Scholar]
- Tkachev D, Mimmack ML, Huffaker SJ, Ryan M, Bahn S. Further evidence for altered myelin biosynthesis and glutamatergic dysfunction in schizophrenia. Int J Neuropsychopharmacol. 2007:1–7. doi: 10.1017/S1461145706007334. [DOI] [PubMed] [Google Scholar]
- Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, Bahn S. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362:798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- van Haren NE, Hulshoff Pol HE, Schnack HG, Cahn W, Mandl RC, Collins DL, Evans AC, Kahn RS. Focal gray matter changes in schizophrenia across the course of the illness: a 5-year follow-up study. Neuropsychopharmacology. 2007;32:2057–2066. doi: 10.1038/sj.npp.1301347. [DOI] [PubMed] [Google Scholar]
- van Haren NE, Pol HE, Schnack HG, Cahn W, Brans R, Carati I, Rais M, Kahn RS. Progressive Brain Volume Loss in Schizophrenia Over the Course of the Illness: Evidence of Maturational Abnormalities in Early Adulthood. Biol Psychiatry. 2008;63:106–113. doi: 10.1016/j.biopsych.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Barrett T, Cheadle C, Sokolov BP, Wood WH, 3rd, Donovan DM, Webster M, Freed WJ, Becker KG. Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res Bull. 2001;55:641–650. doi: 10.1016/s0361-9230(01)00522-6. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Crook JM, Hyde TM, Kleinman JE, Weinberger DR, Becker KG, Freed WJ. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr Res. 2002;58:11–20. doi: 10.1016/s0920-9964(01)00377-2. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ. Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:571–578. doi: 10.1038/sj.mp.4001158. [DOI] [PubMed] [Google Scholar]
- Whitford TJ, Grieve SM, Farrow TF, Gomes L, Brennan J, Harris AW, Gordon E, Williams LM. Volumetric white matter abnormalities in first-episode schizophrenia: a longitudinal, tensor-based morphometry study. Am J Psychiatry. 2007;164:1082–1089. doi: 10.1176/ajp.2007.164.7.1082. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Velakoulis D, Smith DJ, Bond D, Stuart GW, McGorry PD, Brewer WJ, Bridle N, Eritaia J, Desmond P, Singh B, Copolov D, Pantelis C. A longitudinal study of hippocampal volume in first episode psychosis and chronic schizophrenia. Schizophr Res. 2001;52:37–46. doi: 10.1016/s0920-9964(01)00175-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.