

Abstract
Engineering protein expression in vitro or in vivo is usually straightforward for single genes, but remains challenging for multiple genes because of the requirement of coordinated control. RNA and protein overexpression strategies often exploit T7 RNA polymerase and its natural TΦ Class I terminator. However, this terminator’s inefficiency and large size (100 bp) are problematic for multigene construction and expression. Here, we measure the effects of tandem copies of a small (18 bp) Class II T7 terminator from vesicular stomatitis virus on transcription in vitro and on translation in vitro and in vivo. We first test monomeric and dimeric gene constructs, then attempt extension to pentameric gene constructs. “BioBrick” versions of a pET vector and translation factor genes were constructed to facilitate cloning, and His-tags were incorporated to allow copurification of all protein products for relatively unbiased analysis and easy purification. Several results were surprising, including imbalanced expression of the pentameric constructs in vivo, illustrating the value of synthetic biology for investigating gene expression. However, these problems were solved rationally by changing the orders of the genes and by adding extra promoters to the upstream gene or by moving to a more predictable in vitro translation system. These successes were significant, given our initial unexpected results and that we are unaware of another example of coordinated overexpression of five proteins. Our modular, flexible, rational method should further empower synthetic biologists wishing to overexpress multiple proteins simultaneously.
Keywords: synthetic biology, gene expression, T7 RNA polymerase, transcription terminator, translation
Introduction
Synthetic biology can be defined as the complex engineering of in vivo- and in vitro-replicating systems (Forster and Church, 2007). Moving beyond traditional genetic engineering of overexpression of a single protein, synthetic biologists wish to express many genes simultaneously with coordinated control. Applications include microbial production of small-molecule drugs and biofuels (Pfleger et al., 2006), induction of pluripotent stem cells (Takahashi and Yamanaka, 2006), coexpression of subunits of protein complexes for biochemical or structural studies (Miranda et al., 2005) and our goals of synthesizing purified translation systems (Forster et al., 2001) and a minimal cell (Forster and Church, 2006). The protein expression system of choice varies with the protein and application: in vivo systems are preferred so far for yield and eukaryotic protein folding/post-translational modification, while in vitro systems are better for prokaryotic protein folding and proteins that are toxic, difficult to purify or contain unnatural amino acids.
Engineering protein expression in vivo is usually straightforward for single genes, but remains challenging for multiple genes. For example, successful, balanced, unamplified expression of three proteins in E. coli using a tricistronic mRNA transcribed by E. coli RNA polymerase required a combinatorial screen of intergenic sequences (Pfleger et al., 2006). For the goal of high overexpression of two genes in E. coli, bicistronic constructs under a single promoter for T7 RNA polymerase (T7RNAP) often gave low expression of the downstream gene (Kim et al., 2004; Rucker et al., 1997). This stimulated the use of two compatible plasmids (Johnston et al., 2000), which is limited by the number of compatible vectors, and the use of vectors containing two T7 promoters (Kim et al., 2004). The multicistronic and multipromoter approaches have been extended successfully to several genes per plasmid in vivo, although it is unclear if all proteins were highly expressed (Miranda et al., 2005; Watanabe et al., 2006). Less information is available for coexpression in vitro, where most multigene studies use a linear template for each gene (Ahn et al., 2008). Circular DNAs have the advantages of decreased degradation by exonucleases in crude in vitro translation systems (Lesley et al., 1991; Michel-Reydellet et al., 2005), compatibility with in vivo, and utility for building and replicating large synthetic genomes.
T7RNAP is the enzyme of choice for RNA synthesis in vitro and for mRNA synthesis for protein overexpression in vitro and in E. coli because of its efficiency and simplicity of function and control (Studier et al., 1990). However, controlling termination of transcription in multigene constructs is problematic. Like all RNA polymerases, it terminates inefficiently at its terminators (~70% Macdonald et al., 1994), so run through transcription into downstream genes is inevitable. This has led to use of tandem repeats (Baron and Barrett, 1997; Macdonald et al., 1993; Wirtz et al., 1998) of the standard TΦ terminator, a Class I terminator of 100 bp encoding at its 3′ end a stable stem–loop structure followed by a run of U residues (Macdonald et al., 1994). However, this strategy may be incompatible with constructing circular DNAs containing large numbers of genes because many repeats of a 200-bp direct repeat in close proximity in an extrachromosomal DNA would likely cause instability due to recombination. Shorter terminators are therefore desirable, but even minor truncations of TΦ reduce termination substantially (Macdonald et al., 1994). The testing of the much shorter Class II terminators (Lyakhov et al., 1998; Mead et al., 1986) in tandem was thus proposed (Forster and Church, 2007). For example, the vesicular stomatitis virus (vsv) Class II terminator is only 18 bp (bp; TATCTGTTAGTTTTTTTC) yet has a similar termination efficiency to TΦ in vitro (~70%; Lyakhov et al., 1998). Here, we construct 22 plasmids to test the utility of Class II terminators in protein expression in vitro and in vivo. We also test the modularity of this system for multigene expression using modular “BioBrick” plasmids (Shetty et al., 2008).
Materials and Methods
Constructs
E. coli translation initiation factors (IFs) IF1, IF2, and IF3 were His-tagged N-terminally and E. coli translation elongation factors EF-G and EF-Ts were tagged C-terminally using the sequence (CAC)6. Constructs 1 and 6 (Fig. 1A) are pAF3H and pAF1H in pET-derived vectors (Forster et al., 2001). The IF1 genes in Figures 1A and 2A were derived from pAF1H, while those in Figure 5A had the native nucleotide sequence (prepared by gene synthesis from synthetic oligonucleotides by Genscript, Piscataway, NJ). All constructs except 1 and 6 are flanked by pET-24a(+) (Novagen, Madison, WI) nucleotides 342 (BglII) to 3 (BspE1) and lack the 4 BioBrick sites EcoRI, XbaI, SpeI, and PstI (Figs. 1A, 2A, and 5A). All linker sequences were confirmed by sequencing of the constructs and are given in Figure S1.
Figure 1.

In vitro transcription of single gene constructs 1–9. A: Single gene constructs 1–9. Variable modules in these constructs are in gray. Percentages are mole % of total T7RNAP transcripts terminated at the above positions. Bracketed percentages are calculated termination efficiencies at the above terminators. T7pr, T7RNAP promoter; lacO, lac operator; RBS, ribosome binding site; TΦ and vsv, T7RNAP terminators. E, EcoRI; X, XbaI; S, SpeI; and P, PstI. B: In vitro transcription of BspEI linearized constructs 2–5 and 7–9. Transcripts were separated by 3.5% PAGE/urea gel and stained with toluidine blue. R, run off products; T, products terminating at terminators; M, marker sizes in nucleotides.
Figure 2.

In vitro transcription of dimeric gene constructs. A: Dimeric gene constructs. Variable modules in these constructs are in gray. Percentages are mole % of total T7RNAP transcripts terminated at the above positions. B: In vitro transcription of BamHI-linearized constructs 10–17. Transcripts were separated by 5% PAGE/urea gel and stained with toluidine blue. R, run off products; T, products terminating at terminators; M, marker sizes in nucleotides. The lower bands (<240 bases) for constructs 10–13 are run off products from the downstream T7 promoter and are equimolar (0.8–1.2×) compared with products from the upstream promoters.
Figure 5.

In vitro transcription of single and pentameric gene constructs. A: Single gene constructs 18–20 and pentameric gene constructs. All genes contain lacO/RBS and single vsv modules, but these modules are not depicted in pentameric gene constructs for simplicity. Variable modules in the constructs are in gray. B: In vitro transcription of PstI-linearized constructs. Transcripts were separated by 3.5% PAGE/urea gel and stained with toluidine blue. The products from the single gene constructs (8, 18, 3, 19, and 20) were used as markers. Note that only one of the run through markers was short enough to be resolved from the vsv-terminated marker (construct 8). Also, for constructs 21 and 22, only one of the run through products (EF-Ts followed by IF1) was short enough to be visible beneath the EF-G band (construct 22).
In Vitro Transcriptions With T7RNAP
Linearized plasmids (1 μg) were transcribed in 40 mM Tris–HCl, pH 8.0, 1 mM spermidine, 0.05 mg/mL BSA, 20 mM MgCl2, 5 mM DTT, 0.01% Triton-X100, 4 mM each NTP, 0.005 U/μL inorganic pyrophosphatase (Sigma, St. Louis, MO), 1 U/μL rRNasin ribonuclease inhibitor (Promega, Madison, WI) and 25 U T7 RNA polymerase (NEB) in 20 μL reaction at 37°C for 1 h (Milligan and Uhlenbeck, 1989). Bands on urea polyacrylamide gels stained with toluidine blue O (Sigma) were quantified by Quantity One software (Bio-Rad, Hercules, CA). Results were reproducible.
In Vitro Translations
In vitro combined transcription and translation was carried out using E. coli T7 S30 extract for circular DNA (Promega). Uncut plasmid (0.2 μg) and 35S-Met in a 5 μL reaction mix were incubated at 37°C for 1 h. Products were analyzed by SDS–PAGE (NuPAGE Novex 4–12% Bis–Tris gel running with MES–SDS buffer) and autoradiography. Results were reproducible.
In Vivo Translations
Transformed E. coli BL21 (λDE3) cells were grown in LB/kanamycin (50 μg/mL) to OD600 0.6–0.8, IPTG was added to 1 mM, and cells were harvested at 3–5 h. A portion of cells lysed with 8 M urea, 100 mM NaH2PO4, 10 mM Tris–HCl, pH 8.0, were purified using Ni-NTA agarose as described (Qiagen, Valencia, CA). Crude lysates and purified eluates were analyzed by SDS–PAGE and Coomassie Blue staining. Results were reproducible.
Results
Effect of Tandem Short Terminators on Termination In Vitro
In order to test the termination efficiency of a Class II terminator in tandem, we chose the most efficient such terminator (vsv; Lyakhov et al., 1998) and constructed plasmids containing 0–3 vsv terminators separated by 8 bp immediately downstream of E. coli IF3 or IF1 genes. The plasmids were linearized with BsaAI or BspEI (Fig. 1A) or BamHI (Fig. 2A), transcribed in vitro by T7 RNA polymerase, and the termination efficiencies assayed by urea gel electrophoresis (Figs. 1B and 2B).
Terminations at the single vsv terminators were 53–62% efficient, irrespective of the upstream gene (Figs. 1 and 2), as expected (Lyakhov et al., 1998). These termination efficiencies were similar to the 64–66% control efficiencies measured at TΦ terminators (results not shown; summarized in Fig. 1A). Addition of an adjacent vsv terminator did not increase the efficiencies at the upstream terminators (51–56%; Fig. 1), despite the possibility of another T7RNAP paused at the adjacent downstream terminator leading to queuing of T7RNAPs. Interestingly, terminations at the second and third vsv terminators were less efficient, ranging from 12% to 41% (Fig. 1). This shows that tandem vsv terminators function, but not independently. Nevertheless, the aggregate termination efficiencies of 65–75% achieved with 2–3 tandem small terminators (Figs. 1 and 2) were equal or better than the termination efficiencies of 64–66% achieved with the longer sequence of the single TΦ terminator (Fig. 1A).
Effect of Tandem Short Terminators on Combined Transcription and Translation In Vitro
The potential utility of a short terminator(s) for in vitro translation of circular DNA without needing linearization by restriction endonucleases or PCR was tested using a commercial, crude, E. coli translation system. First, the nine uncut monomeric constructs in Figure 1A were translated to measure the effects of terminators on upstream gene expression. Figure 3A shows that the predominant synthesized proteins were of the expected sizes. In the case of IF3, the relative yield of full-length protein decreased slightly with increasing numbers of vsv terminators. Decreasing yields were also seen in this E. coli crude translation system with shortening of the 3′-terminal stem–loop secondary structure of TΦ; the decreases were attributed to increased degradation by RNase (Ahn et al., 2008).
Figure 3.

In vitro translation of unlinearized constructs 1–17. Proteins synthesized from 35S-Met by a crude E. coli combined transcription/translation system were resolved by SDS–PAGE and detected by autoradiography. –, No DNA; 0, plasmid identical to construct 9 but lacking a T7 promoter. Marker sizes are given in kDa. A: Single gene constructs. B: Dimeric gene constructs.
Next, to further test the effect of vsv terminators on translation and to compare the relative protein yields from bicistrons versus adjacent monocistrons, the eight uncut dimeric gene constructs in Figure 2A were translated in vitro. In all cases, the downstream protein (IF1) theoretically can be produced from transcripts initiated at the promoter of the upstream gene (IF3). However, in the cases where the downstream gene lacked its own T7 promoter (constructs 14–17) as an additional source of transcripts, we predicted that expression of the downstream protein would be very sensitive to the presence and number of vsv terminators upstream. This was indeed the case (Fig. 3B): inhibition with increasing numbers of vsv terminators in approximate correlation with the increasing termination efficiencies between the genes was only pronounced for the bicistrons (constructs 14–17). This demonstrated that undesired in vitro translation of a downstream gene from the promoter of an upstream gene can be greatly reduced by tandem small T7 terminators. It also provided an example of efficient bicistronic expression in vitro in the absence of terminators between the genes (Fig. 3B, construct 14).
In summary, all of these crude in vitro translation yields correlated well with the relative numbers of open reading frames expected from the transcription initiation and termination results in the purified system (Figs. 1B and 2B).
Effect of Tandem Short Terminators on Translation In Vivo
Constructs 1–17 were transformed into E. coli BL21(DE3) cells for IPTG-induced overexpression in vivo. Over-expression of IF3 or IF1 was seen in all 17 induced cell lines (Fig. 4A), but expression in the uninduced cells was only seen in 4 of the 17 (Fig. 4A, constructs 1, 2, 6, 7). Better expression of IF1 was obtained with a single vsv terminator versus a single TΦ terminator. Surprisingly, 2 or 3 vsv terminators substantially inhibited overexpression for the single gene constructs of IF3. This demonstrates effects of tandem small terminators in vivo and indicates that 0–1 of the small vsv terminators may be optimal for expression in vivo. The IF3 constructs containing 2 and 3 small terminators may be suboptimal for expression because they favor synthesis of transcripts with unstable 3′-terminal secondary structures that may decay rapidly. The lack of a similar effect with IF1 indicates that upstream sequences may also play a role. Rapid and differential mRNA degradation can dominate control of gene expression in E. coli and is difficult to predict and measure (Carpousis, 2007; Deana and Belasco, 2005).
Figure 4.

In vivo translation of constructs 1–17. Transformed E. coli BL21(λDE3) cells were induced with IPTG and total proteins were analyzed by SDS–PAGE stained with Coomassie Blue. A: Single gene constructs. 0, control plasmid identical to construct 9 but lacking a T7 promoter. B: Dimeric gene constructs; M, marker sizes are given in kDa.
Also surprising was that the dimeric constructs with a deleted downstream promoter (constructs 14–17) gave similar protein yields from the downstream IF1 gene, irrespective of the number of vsv terminators immediately upstream (Fig. 4B). This suggests that, under conditions where both IF3 and IF1 proteins are overexpressed from a single promoter, run through transcription from the IF3 promoter into the IFI gene may be efficient enough to saturate IF1 translation in vivo, in contrast to in vitro (Fig. 3B, constructs 14–17).
In summary, the correlations between our purified transcriptions and in vitro translations of genes with different numbers and types of terminators do not extend to translation in vivo. This presumably reflects the complexity of control mechanisms in vivo. For example, deletion of the TΦ terminator affects the expression of some single genes in vivo but not others (Studier et al., 1990).
Overexpression of Five Proteins In Vitro and Vivo From Single Plasmids Containing Small Terminators
Given our goal of multigene overexpression from gene modules programmed for termination at their boundaries without long TΦ terminators, the assays above suggested that this might best be achieved using single, rather than tandem, vsv terminators between genes. In order to determine if this modular approach could be extended to more than two genes in tandem, we aimed to synthesize from a single plasmid 5 of the 6 His-tagged E. coli translation factors required for our simplified translation system (Forster et al., 2001; the sixth factor, EF-Tu, was omitted because it is required at much higher levels than the other five for reconstitution of translation). This should enable a simpler assembly of our system for peptidomimetic evolution (Forster et al., 2003). Anticipating that the least highly expressed of the five coexpressed proteins might be difficult to detect within the crude protein extract, we took advantage of copurification on Ni beads for relatively unbiased measurement of coexpression. The plasmid constructions were also simplified by using the modular BioBrick assembly strategy, which requires removal of all EcoRI, XbaI, SpeI, and PstI restriction sites from the plasmids. This was done by synthesizing the entire IF2, EF-G, and EF-Ts genes from synthetic oligodeoxyribonucleotides to facilitate necessary synonymous codon substitutions (Fig. 5A, constructs 18–20). Four standardized cloning steps from the five BioBrick genes gave construct 21 (Fig. 5A) which contains the five genes in an arbitrary order.
In vitro transcription of PstI-linearized constructs 18–21 verified that they could produce substantial amounts of all transcripts expected from terminations at all the vsv terminators (Fig. 5B). Combined in vitro transcription and translation of unlinearized pentameric gene construct 21 gave substantial amounts of all five protein products, as verified by copurification on Ni (Fig. 6). However, translation of construct 21 in vivo only overexpressed the four downstream gene products, not the upstream gene product IF1 (Fig. 7A). This result does not correlate with the relative numbers of transcripts terminated at the immediately downstream vsv terminators in vitro because the numbers of such transcripts from the IF1 and IF3 genes were calculated to be the same based on band densities (Fig. 5B, construct 21). Our working hypothesis was a combination of (i) IF1 staining less than the other proteins because it is much smaller, and (ii) the 5′ terminal gene is less transcribed than the other genes because it has only one promoter immediately upstream versus 2–5 promoters upstream for the other genes in construct 21, mindful that termination at vsv terminators is incomplete.
Figure 6.
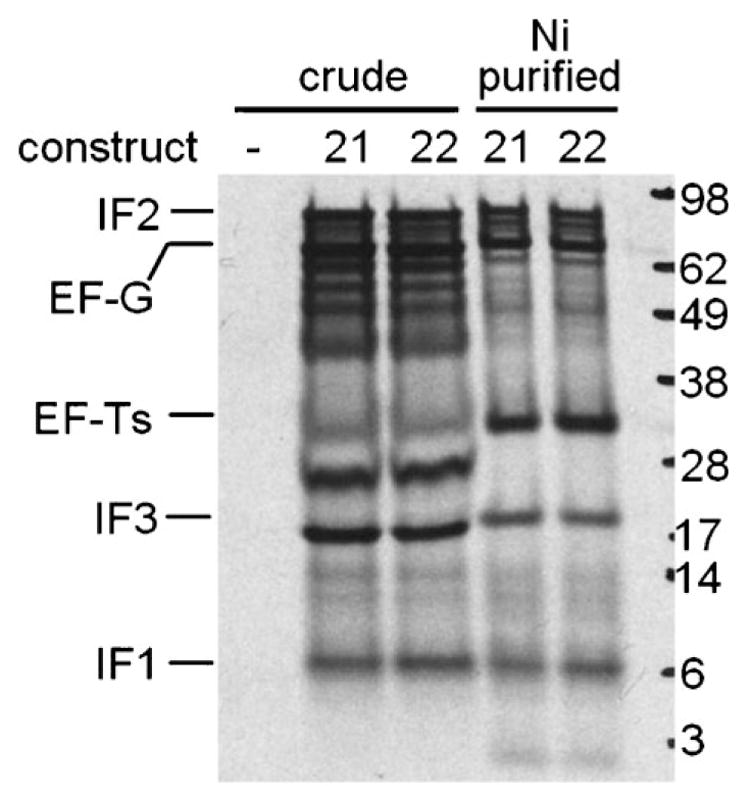
In vitro translation of unlinearized constructs 21 and 22. –, No DNA. Proteins synthesized from 35S-Met by a crude E. coli combined transcription/translation system were resolved by SDS–PAGE and detected by autoradiography. Marker sizes are given in kDa. Crude cell lysates and Ni-NTA purified eluates are shown.
Figure 7.

In vivo translation of pentameric constructs. Transformed E. coli BL21(λDE3) cells were induced with IPTG and total proteins were analyzed by SDS–PAGE stained with Coomassie Blue. A: Pentameric gene constructs 21 and 22. B: Pentameric gene constructs 23 and 24 (note with respect to the ip lanes, larger scale purifications under native conditions show reproducibly a single dominant protein band with the size of IF1). u, Uninduced; I, induced; c, crude cell lysates; p, Ni-NTA purified eluates; M, marker sizes are given in kDa.
Two Systematic Strategies for Adjusting the Relative In Vivo Expression of Five Proteins From Single Plasmids Containing Small Terminators
Our working hypothesis for the undetectable expression of IF1 in vivo was tested by changing the order of the genes to give the pentameric construct 22 (Fig. 5A). Indeed, IF1 expression was rescued when its gene was moved to the fourth position from the 5′ end, while IF3, the new 5′ terminal gene, became undetectable in vivo (Fig. 7A). Thus, undetectable expression from a gene in both of these pentameric constructs correlated with its 5′ terminal position, not with its size or identity. Nevertheless, the small size of IF1 also apparently affects the assay, as the IF1 band from 22 was lighter than the IF3 band from 21 (Fig. 7A). Altering the order of the genes is thus one strategy for systematically adjusting their relative levels of expression in vivo. Again, in vitro translation gave more equitable proportions of products than observed in vivo, with all five proteins being observed for construct 22 (Fig. 6).
Given that the 5′ terminal genes of the pentameric constructs 21 and 22 were apparently not overexpressed in vivo because they only have one promoter immediately upstream, we reasoned that expression might be increased by insertion of two additional upstream promoters into each construct. As predicted, these changes incorporated into pentameric gene constructs 23 and 24 gave detectable in vivo expression of all five proteins (Fig. 7B). As with constructs 21 and 22 (Fig. 7A), the small size of IF1 appears to affect the assay: the IF1 bands from 23 and 24 were lighter than those of IF3 (Fig. 7B). Also, IF1 may be more susceptible to proteolysis (Fig. 7B); it is known that overexpression in vivo of very small proteins can be challenging (Studier et al., 1990). Note that in vivo expression from all four pentameric constructs was leaky in the uninduced controls, effects that might be expected from increasing the number of plasmid-encoded lac operators 5–7× without altering the number of plasmid-encoded lac genes (still just one).
Discussion
The goal of this study was to develop new methods and principles to facilitate multigene expression with a focus on the potential utility of small T7 terminators. Some of the results were unexpected, such as decreased termination at the second vsv terminator of a tandem pair, minimal effects of TΦ terminator deletion on protein expression, inhibitory (Fig. 4A), and lack-of-inhibitory (Fig. 4B) effects of multiple vsv terminators on various protein expression experiments in vivo, and low protein expression in vivo of the upstream gene in pentameric gene constructs. This illustrates the value of synthetic biology for investigating basic cellular processes such as gene expression, even for “well understood” systems. Most of the unexpected results were associated with protein synthesis in vivo, where mRNA degradation can dominate control and is difficult to predict (Carpousis, 2007; Deana and Belasco, 2005).
Another value of synthetic biology illustrated here is that biological problems can be solved by rational engineering. The potential recombinational instability of many repeats of TΦ can be avoided by deleting TΦ or by substitution of smaller terminators. Inadequate expression in vivo of upstream genes in constructs containing several genes can be fixed by changing the orders of the genes and by adding extra promoters to the upstream gene or by moving to the in vitro translation system that we found to act more predictably. In addition to providing these new principles for multigene expression, we synthesized BioBrick vectors and inserts to facilitate plasmid construction and incorporated copurification of His-tagged proteins for easy analysis and purification. This new combination should provide the basis of a generalizable technology for overexpression of several or more proteins simultaneously in a modular, flexible manner.
Future applications beckon in synthetic biology for improved methods for multigene synthesis and expression. For example, our plans include coexpression of all 31 proteins of the PURE translation system (Shimizu et al., 2001) from 1 or 2 bacterial artificial chromosomes to dramatically decrease the number of individual fermentations and protein purifications required for construction of the system, thereby making it more scalable, economical, and useful. Also, together with the synthesis of a 15 kb operon containing all 21 genes for the small ribosomal subunit proteins of E. coli (Tian et al., 2004), this would be progress towards constructing our proposed self-replicating genome of 151 genes (Forster and Church, 2006). In these cases, the potential recombinational instability of many repeats of the promoters may need to be addressed. Different multigene expression goals will likely be solved in different ways, including use of different promoter sequences and more or less than one promoter per gene.
Supplementary Material
Acknowledgments
We thank George Church, Michael Jewett and Harris Wang for comments on the manuscript. A.C.F is supported by the National Institutes of Health and the American Cancer Society.
Contract grant sponsor: National Institutes of Health
Contract grant sponsor: American Cancer Society
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Ahn JH, Kang TJ, Kim DM. Tuning the expression level of recombinant proteins by modulating mRNA stability in a cell-free protein synthesis system. Biotechnol Bioeng. 2008;101:422–427. doi: 10.1002/bit.21884. [DOI] [PubMed] [Google Scholar]
- Baron MD, Barrett T. Rescue of rinderpest virus from cloned cDNA. J Virol. 1997;71:1265–1271. doi: 10.1128/jvi.71.2.1265-1271.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpousis AJ. The RNA degradosome of Escherichia coli: An mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- Deana A, Belasco JG. Lost in translation: The influence of ribosomes on bacterial mRNA decay. Genes Dev. 2005;19:2526–2533. doi: 10.1101/gad.1348805. [DOI] [PubMed] [Google Scholar]
- Forster AC, Church GM. Towards synthesis of a minimal cell. Mol Syst Biol. 2006;2:1–10. doi: 10.1038/msb4100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster AC, Church GM. Synthetic biology projects in vitro. Genome Res. 2007;17:1–6. doi: 10.1101/gr.5776007. [DOI] [PubMed] [Google Scholar]
- Forster AC, Weissbach H, Blacklow SC. A simplified reconstitution of mRNA-directed peptide synthesis: Activity of the epsilon enhancer and an unnatural amino acid. Anal Biochem. 2001;297:60–70. doi: 10.1006/abio.2001.5329. [DOI] [PubMed] [Google Scholar]
- Forster AC, Tan Z, Nalam MNL, Lin H, Qu H, Cornish VW, Blacklow SC. Programming peptidomimetic syntheses by translating genetic codes designed de novo. Proc Natl Acad Sci USA. 2003;100:6353–6357. doi: 10.1073/pnas.1132122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K, Clements A, Venkataramani RN, Trievel RC, Marmorstein R. Coexpression of proteins in bacteria using T7-based expression plasmids: Expression of heteromeric cell-cycle and transcriptional regulatory complexes. Protein Expr Purif. 2000;20:435–443. doi: 10.1006/prep.2000.1313. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Kim HE, Lee KH, Han W, Yi MJ, Jeong J, Oh BH. Two-promoter vector is highly efficient for overproduction of protein complexes. Protein Sci. 2004;13:1698–1703. doi: 10.1110/ps.04644504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley SA, Brow MA, Burgess RR. Use of in vitro protein synthesis from polymerase chain reaction-generated templates to study interaction of Escherichia coli transcription factors with core RNA polymerase and for epitope mapping of monoclonal antibodies. J Biol Chem. 1991;266:2632–2638. [PubMed] [Google Scholar]
- Lyakhov DL, He B, Zhang X, Studier FW, Dunn JJ, McAllister WT. Pausing and termination by bacteriophage T7 RNA polymerase. J Mol Biol. 1998;280:201–213. doi: 10.1006/jmbi.1998.1854. [DOI] [PubMed] [Google Scholar]
- Macdonald LE, Zhou Y, McAllister WT. Termination and slippage by bacteriophage T7 RNA polymerase. J Mol Biol. 1993;232:1030–1047. doi: 10.1006/jmbi.1993.1458. [DOI] [PubMed] [Google Scholar]
- Macdonald LE, Durbin RK, Dunn JJ, McAllister WT. Characterization of two types of termination signal for bacteriophage T7 RNA polymerase. J Mol Biol. 1994;238:145–158. doi: 10.1006/jmbi.1994.1277. [DOI] [PubMed] [Google Scholar]
- Mead DA, Szczesna-Skorupa E, Kemper B. Single-stranded DNA ‘blue’ T7 promoter plasmids: A versatile tandem promoter system for cloning and protein engineering. Protein Eng. 1986;1:67–74. doi: 10.1093/protein/1.1.67. [DOI] [PubMed] [Google Scholar]
- Michel-Reydellet N, Woodrow K, Swartz J. Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. J Mol Microbiol Biotechnol. 2005;9:26–34. doi: 10.1159/000088143. [DOI] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Miranda JJ, De Wulf P, Sorger PK, Harrison SC. The yeast DASH complex forms closed rings on microtubules. Nat Struct Mol Biol. 2005;12:138–143. doi: 10.1038/nsmb896. [DOI] [PubMed] [Google Scholar]
- Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol. 2006;24:1027–1032. doi: 10.1038/nbt1226. [DOI] [PubMed] [Google Scholar]
- Rucker P, Torti FM, Torti SV. Recombinant ferritin: Modulation of subunit stoichiometry in bacterial expression systems. Protein Eng. 1997;10:967–973. doi: 10.1093/protein/10.8.967. [DOI] [PubMed] [Google Scholar]
- Shetty RP, Endy D, Knight TF., Jr Engineering BioBrick vectors from BioBrick parts. J Biol Eng. 2008;2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tian J, Gong H, Sheng N, Zhou X, Gulari E, Gao X, Church G. Accurate multiplex gene synthesis from programmable DNA microchips. Nature. 2004;432:1050–1054. doi: 10.1038/nature03151. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Hotta K, Praseuth AP, Koketsu K, Migita A, Boddy CN, Wang CC, Oguri H, Oikawa H. Total biosynthesis of antitumor nonribosomal peptides in Escherichia coli. Nat Chem Biol. 2006;2:423–428. doi: 10.1038/nchembio803. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Hoek M, Cross GA. Regulated processive transcription of chromatin by T7 RNA polymerase in Trypanosoma brucei. Nucleic Acids Res. 1998;26:4626–4634. doi: 10.1093/nar/26.20.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.