

Abstract
Tumor-induced osteomalacia (TIO) is a rare paraneoplastic syndrome that results in renal phosphate wasting with hypophosphatemia. In most cases, the underlying cause of TIO is a small mesenchymal neoplasm that is often difficult to detect, resulting in delayed diagnosis. One such neoplasm is the phosphaturic mesenchymal tumor, mixed connective tissue variant (PMTMCT), an unusual entity with unique morphologic and biochemical features. The majority of these tumors are found at appendicular sites with only rare cases reported in the jaws. We describe a PMTMCT involving the mandible in a patient with a protracted history of osteomalacia. A review of the current literature is provided with emphasis on the clinical and histologic features, etiopathogenesis, and management of PMTMCT in the setting of TIO.
Osteomalacia is a metabolic bone disorder characterized by inadequate formation of mature bone due to defective mineralization of osteoid.1–3 Because the etiologic basis of this condition is diverse, successful management relies on recognizing the osteomalacia and identifying the underlying cause (Table I).1,2,4–7
Table I.
Etiologic factors associated with osteomalacia
|
FGF23 = fibroblast growth factor 23
PHEX = phosphate regulating gene with homologies to endopeptidases on the X-chromosome
DMP-1 = dentin matrix acidic phosphoprotein
Renal phosphate wasting with hypophosphatemia is an important mechanism for the development of osteomalacia. Both genetic and acquired causes of phosphate wasting have contributed greatly to our understanding of phosphate metabolism. Tumor-induced osteomalacia (TIO)—also known as oncogenic osteomalacia—is a rare, acquired form of osteomalacia resulting from renal phosphate wasting. Patients with TIO exhibit the clinical hallmarks of osteomalacia, including bone and muscle pain, severe muscle weakness, gait disturbances, and heightened susceptibility to fractures. 2,8 Long-term manifestations such as height loss, skeletal deformity and delayed growth, may also be observed.8 Laboratory analysis reveals a characteristic biochemical profile consisting of profound hypophosphatemia, hyperphosphaturia, and reduced or inappropriately normal 1,25-dihydroxyvitamin D (1,25-(OH)2D3) levels. Low renal tubular maximum for the reabsorption of phosphorus (TmP) and elevated serum alkaline phosphatase level may be observed as well. Serum calcium and parathyroid hormone levels are usually normal and the presence of significant hypocalcemia should prompt consideration of a different diagnosis such as vitamin D deficiency.
Because the underlying neoplasm in TIO may remain occult for months to years despite meticulous searches, the diagnosis of TIO is often established later in adulthood, typically the fourth and fifth decades,2,8,9 In many cases, the causative tumors may evade detection because they are small, slow-growing, and located in peculiar or atypical sites.1,2 Most frequently, the tumors are discovered in the soft tissues surrounding the appendicular skeleton.8,10,11 Head and neck involvement has been reported although gnathic localization is exceptionally rare.
Historically, the mostly mesenchymal tumors associated with TIO have comprised a heterogenous mix of soft tissue and bone neoplasms. However, the possibility that they may, for the most part, comprise a single distinct entity was suggested in the early 1970’s by Evans and Azzopardi12 as well as Olefsky et al13. This concept was further codified by Weidner and Santa Cruz7 who coined the term “phosphaturic mesenchymal tumor, mixed connective tissue variant” (PMTMCT) to describe 17 TIO-associated mesenchymal tumors, all showing similar histological features. More recently, a large study and detailed literature review by Folpe and colleagues further established that over 90% of TIO-associated mesenchymal tumors are in fact PMTMCTs.1 There is convincing evidence that most of these tumors produce fibroblast growth factor 23 (FGF23), a hormone that inhibits renal phosphate reabsorption and renal production of 1,25-(OH)2D3.14 Therefore, it may be challenging to distinguish TIO from other FGF23-mediated disorders such as autosomal dominant hypophosphatemic rickets, especially if the latter conditions manifest later in adulthood.15
We herein describe a patient with a prolonged history of osteomalacia who was discovered to have a PMTMCT involving her mandible.
CASE REPORT
A 42-year-old female was reportedly well until 9 years previously when she developed back and flank pain during pregnancy. Routine clinical laboratory assays performed at that time revealed hypophosphatemia in the setting of normal calcium and parathyroid hormone levels (Table II). Urine phosphate was in the low-normal range for a small individual, which was attributed to a low dietary phosphorus intake and her very low serum phosphate levels.16 An elevated alkaline phosphatase was also noted. DEXA scans were ordered and revealed low bone mineral density. A diagnosis of hypophosphatemic osteomalacia of unclear etiology was made and treatment with phosphorus (750mg four times daily) and vitamin D (ergocalciferol, 50,000 units twice weekly) was initiated.
Table II.
Laboratory values of current case
| Range | 9 years prior | Pre-operative (1 month) | Post-operative (11 days) | |
|---|---|---|---|---|
| Serum phosphate (mg/dl) | 2.5–4.1 | 1.4 | 2.4 | 3.2 |
| Urine phosphate (mg/24hr) | 800–1500 | 489 | 1815 | 874 |
| 1,25-(OH)2D3 (pg/dl) | 15–75 | 38 | 38 | 41 |
| Calcium (mg/dl) | 8.4–10.5 | 8.9 | 9.6 | 10.1 |
| Alkaline phosphatase (IU/L) | 44–147 | 159 | 59 | -- |
| Parathyroid hormone (pg/ml) | 10–65 | 44 | 31 | 47 |
| FGF23 (pg/ml) | 1–71 | -- | 192 | 98 |
| Treatment | none | phosphorus and ergocalciferol | none |
1,25-(OH)2D3 = 1,25-dihydroxyvitamin D
FGF23 = fibroblast growth factor 23
Further questioning revealed a negative family history of the heritable forms of osteomalacia. As TIO was considered a probable diagnosis, numerous exploratory investigations were undertaken over the following years in search for a causative tumor. During this time, she continued pharmacologic treatment with phosphorus and vitamin D with excellent clinical and biochemical response, including improvement of her bone mineral density by 35.3% at the hip, 31.5% at the 1/3 radius site, and 10.0% at the lumbar spine. Her evaluations included x-rays; bone scans; CT scans of the chest, abdomen, and pelvis; and whole body technetium-99M sestamibi scans, all of which were negative for tumors. An octreotide scan was also performed and showed a promising focus of increased uptake in the midline abdomen. A follow-up MRI suggested an area of arterial phase enhancement in the pancreatic head and an endoscopic ultrasound revealed a bilobar 11.0 × 6.0 mm hypoechoic lesion at the bifurcation of the celiac axis. The patient subsequently underwent open laparotomy and 2 small lymph nodes were excised. Microscopic examination of the nodes showed evidence of lipogranulomata, which have not been previously implicated in TIO. Furthermore, no change in her clinical status was observed post-operatively.
During the course of these evaluations, the patient revealed that a dental exam performed by her previous oral and maxillofacial surgeon had revealed a lesion in her right mandible. Radiographs retrieved from this assessment, performed 9 years prior, showed an entirely radiolucent lesion with distinct interradicular scalloping between the roots of #28-31 (Figure 1). According to the clinician’s records, the patient’s dentition was otherwise intact and the associated teeth were determined to be vital. The records indicated that the clinical and radiographic impression was that of a traumatic bone cyst and that a biopsy was performed which supported this diagnosis histologically. The patient reported that her mandibular lesion remained asymptomatic over the following years and that additional radiographs were not ordered.
Figure 1.
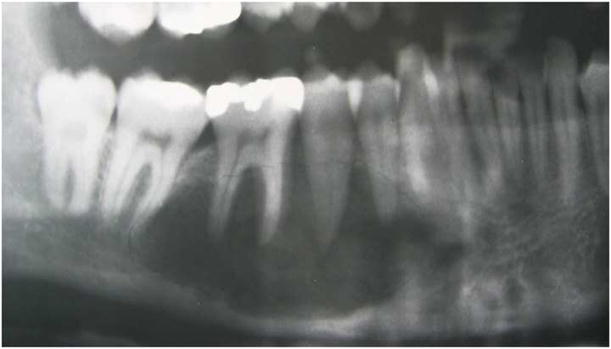
Panoramic radiograph taken 9 years prior to presentation showing a radiolucent lesion with interradicular scalloping.
Upon learning of the mandibular lesion, the patient’s endocrinologist immediately referred her to the Department of Oral and Maxillofacial Surgery at our institution for reevaluation. A new panoramic radiograph was obtained and demonstrated a multilocular lesion with predominantly corticated borders corresponding in the area of interest (Figure 2). The lesion remained confined to the #28-31 region but was now characterized by internal opacities and increased radiodensity towards the posterior aspect. The lamina dura of teeth #29 and 30 were effaced and the corresponding periodontal ligament spaces were irregularly widened. Scalloping of the inferior mandibular cortex was noted. Examination of a cone-beam computerized tomography (CBCT) scan (i-CAT Classic CBCT [Imaging Sciences, Hatfield, PA]) revealed a lesion of mixed density with well-demarcated borders anteriorly and less distinct borders posteriorly (Figures 3A and B). Significant expansion with associated cortical thinning was also detected. Internally, coarse opacities and variably-sized loculations were seen. A contrast-enhanced CT scan confirmed a lytic lesion with a focus of high density, but no abnormal enhancement, alleviating concerns of a high-flow lesion. The radiographic differential consisted of a calcifying epithelial odontogenic tumor, calcifying odontogenic cyst and its variants, ossifying fibroma, osteosarcoma, chondromyxoid fibroma, and chondrosarcoma. Given the clinical history of hypophosphatemia and hyperphosphaturia, a low-flow vascular lesion, central giant cell granuloma, and PMTMCT were also considered, all of which have been documented causes of TIO.
Figure 2.

Panoramic radiograph taken at presentation showing a multilocular lesion with internal opacities.
Figure 3.

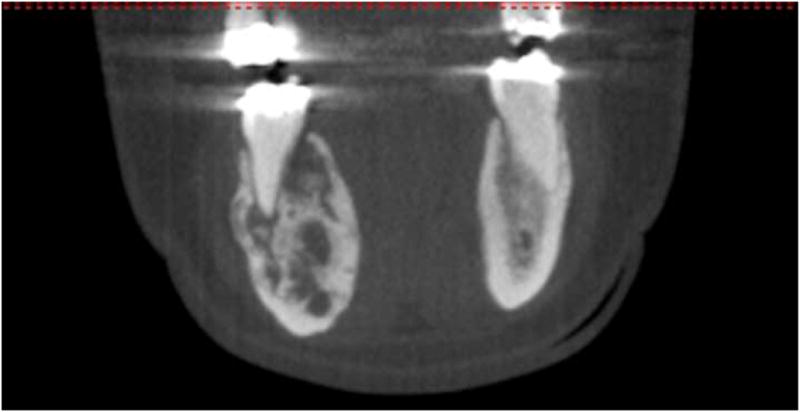
A mandibular cone-beam computerized tomography scan. Bucco-lingual expansion is evident. A, axial. B, coronal.
After a negative aspiration, the area was surgically explored and histologic examination of the biopsy specimen revealed a hypercellular tumor composed of a dense population of spindle-shaped cells. The cells had dark but uniform nuclei and were present in a fascicular pattern in focal areas (Figure 4A). Frequent deposits of acellular, “grungy”-type calcifications and ovoid fragments of viable bone were noted; scattered osteoclast-type multinucleated giant cells were also noted (Figure 4B). An immunohistochemical panel consisting of vimentin, pancytokeratin, desmin, S-100 and CD34 was performed. The tumor cells were strongly and diffusely positive with vimentin but exhibited no reactivity with the remaining markers. The microscopic features in concert with the patient’s clinical history supported a diagnosis of PMTMCT. Serum FGF23 concentrations were measured using an ELISA that detects only full length FGF23 (Intact FGF23 ELISA, Kainos Laboratories Tokyo, Japan). 17 Pre-operative FGF23 concentrations were 192 pg/ml (normal = 0–71 pg/ml), consistent with an FGF23-mediated cause of osteomalacia.
Figure 4.
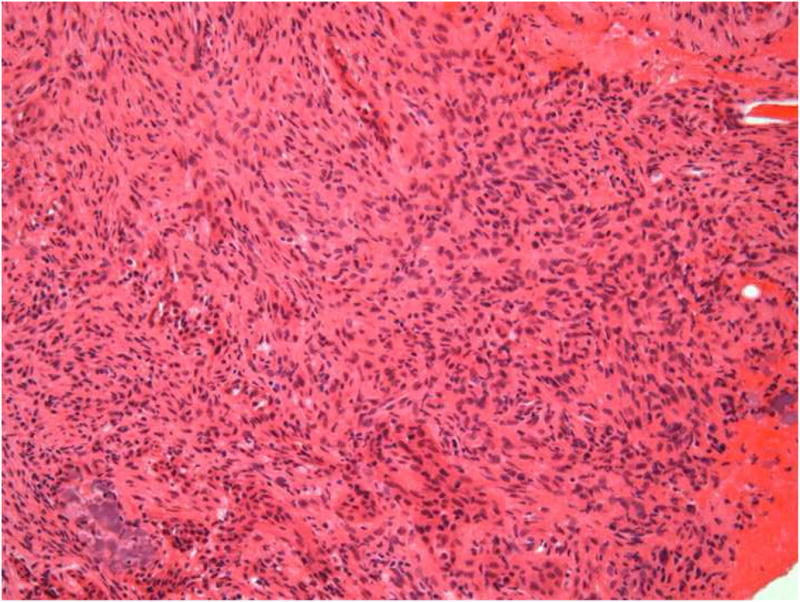
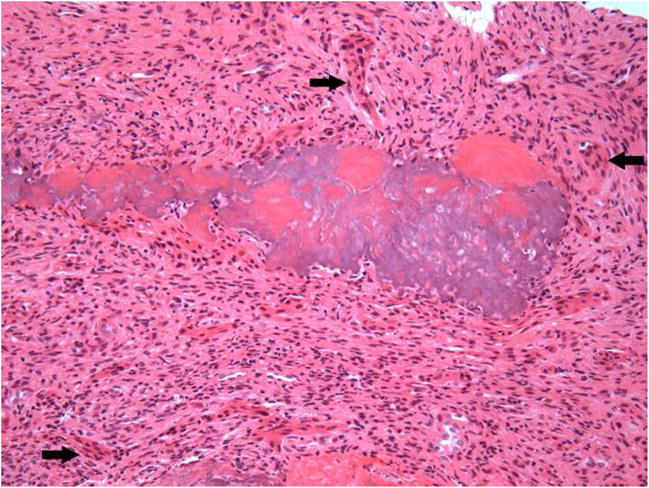

Microscopic images of lesion. A, A cellular spindle cell proliferation exhibiting a fascicular pattern (hematoxylin-eosin, original magnification X100). B, “Grungy”-type calcifications and osteoclast-like giant cells (arrows) (hematoxylin-eosin, original magnification X100). C, Positive staining with vimentin (original magnification X100).
Although a complete surgical excision of the lesion was planned, the tumor was found to be in direct contact with the inferior alveolar nerve intraoperatively. After great consideration, conservative curettage was performed to avoid sacrifice of the nerve although this limited the ability to completely resect the lesion. The histology of the curettage specimen was identical to that of the biopsy. Moreover, analysis of the excised tissue via reverse transcriptase polymerase chain reaction (RT-PCR) confirmed FGF23 expression (data not shown).
Following surgery, the patient experienced rapid normalization of her serum phosphate levels (Table II). Notably, there was a significant reduction of her plasma FGF23 level to 98 pg/ml 11 days after the surgery. A follow-up FGF23 measurement 4½ months post-surgery remained slightly elevated at 92 pg/ml. Although she was able to discontinue her medications for several months, she has since resumed supplementation with phosphorus and ergocalciferol, though at lower doses than her pre-surgical treatment. The patient continues to do well clinically but maintains a vigilant follow-up regimen due to the conservative nature of her surgery.
DISCUSSION
Clinical features and tumor detection
PMTMCTs are rare neoplasms that have often been described as “strange tumors that occur in strange places”.18 Approximately 53% of PMTMCTs occur in bone, 45% in soft tissue, and 3% in skin.6 Most arise within the extremities,1 where they may even be palpable. Craniofacial localization has been described in only 5% of cases, with the paranasal sinuses being the most favored sites in this region.1 Involvement of the mandible and mandibular soft tissues is exceptionally rare with only 4 clear cases reported in the English-language literature to date (Table III)1,7,9,19–26
Table III.
Reported cases of mandibular PMTs
| Reference | Age/Sex | Site | Initial diagnosis | Final diagnosis | Follow-up | Comments |
|---|---|---|---|---|---|---|
| Weidner and Santa Cruz (1987)7 | 27y/M | Mand | Osteo-sarcoma | PMT, ossifying fibroma-like | Recurred at 1.5 months; ANED, normal chemistry | Previously reported by Nomura et al19 as “osteosarcoma” |
| Yang et al (1997)20 | 31y/F | Peri-mand soft tissue | – | PMTMCT | ANED, normal chemistry | |
| Reyes-Múgica et al (2000)9 Folpe et al (2004)1 |
9y/F | L mand ramus | PMTMCT | PMTMCT | ANED, 5 years, normal chemistry | Also reported by Folpe et al1 |
| Dupond et al (2005)21 | 71y/M | L mand gingiva, #17, 18 | – | PMTMCT | Normal chemistries 15 days post-op |
y = year; M = male, F = female
Mand = mandible/mandibular; R = right, L = left
PMT = Phosphaturic mesenchymal tumor, PMTMCT = PMT, mixed connective tissue variant
ANED = alive, no evidence of disease; post-op = post-operatively
(Cases excluded from this table include mandibular tumors associated with OO but not reclassified as PMT22–24 or with insufficient data for revised analysis25; tumors associated with OO originating from the maxilla, paranasal sinus, and other oral soft tissue sites other than the mandibular tissues26; and cases associated with syndromes and/or disseminated carcinomas, as per Folpe et al 1.)
As expected, most PMTMCTs are discovered in middle-aged adults although cases have been documented in patients ranging from 3 to 73 years of age.1 No gender predilection has been noted. Clinically, patients with PMTMCT will often disclose a lengthy history of symptoms related to their osteomalacia. Diagnostic delays of months to years are common, with one case reportedly diagnosed 19 years after symptom onset.24
Affected patients exhibit the typical features of osteomalacia described earlier. However, the combination of hypophosphatemia, hyperphosphaturia, and inappropriately normal or low 1,25-(OH)2D3 should prompt consideration of an FGF23-mediated disorder including TIO. Once other causes of osteomalacia are excluded, a meticulous search for an underlying tumor is indicated. In addition to CT and magnetic resonance imaging (MRI), technicium Tc99 bone scintigraphy,27,28 positron emission tomography (PET) scanning,29 and f-18 FDG PET/CT scanning21 have been used to locate PMTs. The observation that PMTs may express somatostatin receptors (SSTRs) has also raised interest in indium In111-pentetreotide scintigraphy (octreotide scanning) as a method of detection.11,28,30 Unfortunately, octreotide scanning is not successful in many cases29 and was not helpful in the evaluation of our patient.
Histologic features and differential diagnosis
In the past, the histopathologic diagnoses associated with TIO have been varied and diverse. Weidner and Santa Cruz 7 were among the earliest to recognize the challenge in naming these tumors and suggested that most TIO-associated neoplasms could be classified as 4 entities based on recurring histologic patterns: 1) PMTMCT, 2) PMT, osteoblastoma-like, 3) PMT, non-ossifying fibroma-like, and 4) PMT, ossifying fibroma-like.2,7,31 It is now believed that the “osteoblastoma-like”, “non-ossifying fibroma-like” and “ossifying fibroma-like” variants represent bone-specific reaction patterns found within the spectrum of PMTMCTs.
The typical microscopic appearance of a PMTMCT is that of a highly vascular proliferation of spindled to stellate cells with low nuclear grade and low mitotic activity. The cells are embedded in a distinctive myxoid to chondromyxoid matrix that often contain foci of “grungy” or flocculent calcification. Other features that may be present include osteoclast-like giant cells, mature fat, chondroid or osteoid-like matrix, woven bone, and areas of microcystic change. Immunohistochemical stains for pancytokeratin, desmin, S-100, and CD34 are predominantly negative in the neoplastic cells.1 Table IV summarizes the histologic findings seen in PMTMCTs. 1,8
Table IV.
Histologic features of PMTMCT
| Cells |
| Spindled-shaped to stellate tumor cells |
| Low nuclear grade |
| Cellularity |
| Low to moderate |
| Mitotic activity |
| Absent to low (<1/10 high power field) |
| Matrix |
| Myxoid to chondromyxoid |
| ”Grungy” calcifications |
| Vasculature |
| Capillary |
| Thick-walled, hyalinized |
| Variable features |
| Osteoclast-like giant cells |
| Chondroblast-like cells |
| Mature adipocytes |
| Microcystic change |
| Fibrohistiocytic reaction |
| Aneurysmal bone cyst-like changes |
Folpe et al1 reviewed 29 tumors occurring in patients with known TIO and found that 21/29 (72.4%) exhibited the characteristic microscopic and immunohistochemical features of PMTMCT. It is interesting to note that three tumors in their series1 were diagnosed as malignant PMTMCTs based on atypical features such as high nuclear grade, hypercellularity, and mitotic activity exceeding 5 per high power field.
Entities that may be included in the histologic differential for PMTMCT are listed in Table V.1,2,6,9,32 With the exception of the mesenchymal malignancies, distinction between PMTMCT and these other tumors may be of academic interest as treatment (surgical resection with the goal of cure) remains the same in the setting of TIO. It is noteworthy that on rare occasions, TIO may develop in the setting of other conditions such as neurofibromatosis type-2; in these cases, the causative tumors may lack the morphologic features of PMTMCTs.1
Table V.
Histologic differential diagnosis of PMT
| Hemangiopericytoma |
| Giant cell tumors (giant cell reparative granuloma) |
| Spindle cell lipoma |
| Sclerosing hemangioma, hemangioma |
| Enchondroma |
| Osteoblastoma |
| Chondroblastoma |
| Mesenchymal chondrosarcoma |
| Osteosarcoma |
| Chondrosarcoma |
| Angiolipoma |
| Desmoplastic fibroma, fibromatosis |
| Myofibromatosis |
Pathophysiology and role of FGF23
TIO is a paraneoplastic syndrome caused by tumor-elaborated humoral factors known collectively as “phosphatonins”. Although multiple factors have been implicated, there is compelling evidence that FGF23 plays a significant role in this condition.
FGF23 belongs to the FGF19 subfamily and is produced primarily in the bone.33 Members of this subfamily circulate with hormonal activity. It has been previously demonstrated that FGF23 inhibits sodium-phosphate co-transport (via a decrease in NPT2A transporters) in the proximal tubular cells of the kidney in vitro.34,35 FGF23 has also been shown to inhibit 25-hydroxyvitamin D 1-alpha hydroxylase, thereby preventing conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. 36 Interestingly, investigators in this study found that mice injected with FGF23 showed reduced serum phosphorus levels, increased phosphate excretion, and features of osteomalacia.36 Likewise, patients with autosomal dominant hypophosphatemic rickets—a condition caused by FGF23 mutation resulting in excess FGF23—display a clinical phenotype similar to TIO.37
Most TIO-associated tumors in humans have been found to overexpress FGF23. Folpe et al1 confirmed 17/21 and 2/2 PMTMCTs in their series to be positive for FGF23 by immunohistochemistry and RT-PCR, respectively. Other investigators have also demonstrated FGF23 expression in PMTs by RT-PCR, 38 western blot,38–40 and in-situ hybridization studies.10 Lastly, several studies have shown that patients with TIO experience a decrease in post-operative FGF23 levels following successful tumor resection.14,39,41,42
Tumors associated with TIO can also produce other proteins, including secreted frizzled protein 4 (sFRP4),21 matrix extracellular phosphoglycoprotein (MEPE),21 and FGF743. Although FGF23 has the most clearly defined role in normal and abnormal phosphate metabolism, the function of these proteins and FGF23 in TIO requires further clarification.
Treatment and Prognosis
Complete resection of the causative tumor is the treatment of choice in TIO. Quite dramatic normalization of the biochemical abnormalities is typically observed within days to weeks following successful resection of the PMTMCT or TIO-associated tumor,1,2 while resolution of the osteomalacia may require months. It has been estimated that 90% of patients with TIO are cured with complete resection9,44 although early and delayed recurrences have been noted. In cases where anatomic restrictions or medical comorbidities prohibit complete resection, as in our patient, supplementation with phosphorus and calcitriol is often indicated if biochemical abnormalities persist.5 Periodic assessment of serum phosphate levels has been recommended as a means to monitor for recurrences, even in surgically “cured” TIO patients. Additionally, periodic serum FGF23 levels has been advocated as a tool to corroborate the success of surgical intervention and to help detect recurrences as well.45
In summary, we describe a case of PMTMCT involving the mandible in a patient with a 10 year history of osteomalacia. PMTMCTs involving the mandible are extraordinarily rare and our patient represents only the fifth case reported in the literature. Moreover, the radiographic presentation and previous diagnosis of traumatic bone cyst are intriguing aspects of this case. The diagnosis of TIO can be challenging but rewarding for both the clinician and patient. Successful diagnosis is dependent on a number of factors, including astute correlation between the clinical and biochemical facets of this condition, evaluating and excluding other causes of hypophosphatemia, performing a diligent search for the underlying tumor, and recognizing the histologic spectrum of TIO-associated tumors. Ultimately, early recognition remains of utmost importance to minimizing the debilitating consequences of long-term osteomalacia in affected patients.
Acknowledgments
We are grateful to Ms. Marianne Delabedia, RN, for her help with handling the specimens. We would also like to thank Dr. Rachelle Retoma for her assistance with images.
Funding: NIH (R01 AR42228)
Funding: Partial support NIH (NIDDK: DK32333)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: An analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30. doi: 10.1097/00000478-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Drezner MK. Tumor-induced osteomalacia. Rev Endocr Metab Disord. 2001;2:175–86. doi: 10.1023/a:1010006811394. [DOI] [PubMed] [Google Scholar]
- 3.Siris ES, Clemens TL, Dempster DW, Shane E, Segre GV, Lindsay R, et al. Tumor-induced osteomalacia. Kinetics of calcium, phosphorus, and vitamin D metabolism and characteristics of bone histomorphometry. Am J Med. 1987;82:307–12. doi: 10.1016/0002-9343(87)90075-1. [DOI] [PubMed] [Google Scholar]
- 4.Kaul M, Silverberg M, DiCarlo EF, Schneider R, Bass AR, Erkan D. Tumor-induced osteomalacia. Clin Rheumatol. 2007;26:1575–9. doi: 10.1007/s10067-006-0468-y. [DOI] [PubMed] [Google Scholar]
- 5.Schapira D, Izhak OB, Nachtigal A, Burstein A, Shalom RB, Shagrawi I, et al. Tumor-induced osteomalacia. Semin Arthritis Rheum. 1995;25(1):35–46. doi: 10.1016/s0049-0172(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 6.Ungari C, Rocchi G, Rinna C, Agreillo A, Lattanzi A, Pagnoni M. Hypophosphaturic mesenchymal tumor of the ethmoid associated with oncogenic osteomalacia. J Craniofac Surg. 2004;15(3):523–7. doi: 10.1097/00001665-200405000-00036. [DOI] [PubMed] [Google Scholar]
- 7.Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets. Cancer. 1987;59:1442–1454. doi: 10.1002/1097-0142(19870415)59:8<1442::aid-cncr2820590810>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 8.Reis-Filho JS, Paiva ME, Lopes JM. Pathologic Quiz Case: A 36-year-old woman with muscle pain and weakness. Arch Pathol Lab Med. 2002;126(10):1245–6. doi: 10.5858/2002-126-1245-PQCAYO. [DOI] [PubMed] [Google Scholar]
- 9.Reyes-Múgica M, Arnsmeier SL, Backeljauw PF, Persing J, Ellis B, Carpenter TO. Phosphaturic mesenchymal tumor-induced rickets. Pediatr Dev Pathol. 2000;3:61–9. doi: 10.1007/s100240050008. [DOI] [PubMed] [Google Scholar]
- 10.Larsson T, Zahradnik R, Lavigne J, Ljunggren Ö, Jüppner H, Jonsson K. Immunohistochemical detection of FGF-23 protein in tumors that cause oncogenic osteomalacia. Eur J Endocrinol. 2003;148:269–76. doi: 10.1530/eje.0.1480269. [DOI] [PubMed] [Google Scholar]
- 11.Rhee Y, Lee JD, Shin KH, Lee CH, Huh KB, Lim S-K. Oncogenic osteomalacia associated with mesenchymal tumor detected by indium-111 octreotide scintigraphy. Clin Endocrinol. 2001;54:551–4. doi: 10.1046/j.1365-2265.2001.01056.x. [DOI] [PubMed] [Google Scholar]
- 12.Evans DJ, Azzopardi JG. Distinctive tumours of bone and soft tissue causing acquired vitamin-D-resistant osteomalacia. Lancet. 1972;1:353–354. doi: 10.1016/s0140-6736(72)92844-9. [DOI] [PubMed] [Google Scholar]
- 13.Olefsky J, Kempson R, Jones H, Reaven G. “Tertiary” hyperparathyroidism and apparent “cure” of vitamin-D-resistant rickets after removal of an ossifying mesenchymal tumor of the pharynx. N Engl J Med. 1972;286:740–745. doi: 10.1056/NEJM197204062861402. [DOI] [PubMed] [Google Scholar]
- 14.Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, et al. Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J Clin Endocrinol Metab. 2006;91(6):2055–61. doi: 10.1210/jc.2005-2105. [DOI] [PubMed] [Google Scholar]
- 15.Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: Clinical characterization of a novel renal phosphate wasting disorder. J Clin Endocrinol Metab. 1997;82(2):674–681. doi: 10.1210/jcem.82.2.3765. [DOI] [PubMed] [Google Scholar]
- 16.Bijvoet OLM. Relation of plasma phosphate concentration to renal tubular resorption of phosphate. Clin Sci. 1969;37:23–36. [PubMed] [Google Scholar]
- 17.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87(11):4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 18.Weiss D, Bar RS, Weidner N, Wener N, Lee F. Oncogenic osteomalacia: strange tumours in strange places. Postgrad Med J. 1985;61:349–55. doi: 10.1136/pgmj.61.714.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nomura G, Koshino Y, Morimoto H, Kida H, Nomura S, Tamai K. Vitamin D resistant hypophosphatemic osteomalacia associated with osteosarcoma of the mandible: report of a case. Jpn J Med. 1982;21(1):35–9. doi: 10.2169/internalmedicine1962.21.35. [DOI] [PubMed] [Google Scholar]
- 20.Yang IM, Park YK, Hyun YJ, Kim DY, Woo JT, Kim SW, Kim JW, Kim YS, Choi YK. Oncogenic osteomalacia caused by a phosphaturic mesenchymal tumor of the oral cavity: a case report. Korean J Intern Med. 1997;12:89–95. doi: 10.3904/kjim.1997.12.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupond JL, Mahammedi H, Prié D, Collin F, Gil H, Blagosklonov O, et al. Oncogenic osteomalacia: Diagnostic importance of fibroblast growth factor 23 and F-18 fluorodeoxyglucose PET/CT SCAN for the diagnosis and follow-up in one case. Bone. 2005;36:375–8. doi: 10.1016/j.bone.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Ahn J-M, Kim H-K, Cha C-M, Kim J, Yim S-G, Kim HJ. Oncogenic osteomalacia: induced by tumor, cured by surgery. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:636–41. doi: 10.1016/j.tripleo.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 23.Kim YG, Choi YS, Lee SC, Ryu DM. mor-induced osteomalacia associatedTu with lesions in the oral and maxillofacial region: Report of two cases. J Oral Maxillofac Surg. 1996;54:1352–7. doi: 10.1016/s0278-2391(96)90497-8. [DOI] [PubMed] [Google Scholar]
- 24.Nitzan DW, Horowitz AT, Darmon D, Friedlaender MM, Rubinger D, Stein P, et al. Oncogenous osteomalacia: a case study. Bone Miner. 1989;6:191–7. doi: 10.1016/0169-6009(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 25.Avila NA, Skarulis M, Rubino DM, Doppman JL. Oncogenic Osteomalacia: Lesion detection by MR skeletal survey. AJR. 1996;167:343–5. doi: 10.2214/ajr.167.2.8686600. [DOI] [PubMed] [Google Scholar]
- 26.Yun K-I, Kim D-H, Pyo S-W. A phosphaturic mesenchymal tumor of the floor of the mouth with oncogenic osteomalacia. Report of a case. J Oral Maxillofac Surg. 2009;67:402–5. doi: 10.1016/j.joms.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Robertson A, Mansberg T, Mansberg V, Van der All H, Hooper M. Tumor-induced osteomalacia: A case of diagnostic dilemma. Clin Nucl Med. 2007;32(8):631–2. doi: 10.1097/RLU.0b013e3180a1ad1d. [DOI] [PubMed] [Google Scholar]
- 28.Jan de Beur SM. Tumor-induced osteomalacia. JAMA. 2005;294(10):1260–7. doi: 10.1001/jama.294.10.1260. [DOI] [PubMed] [Google Scholar]
- 29.Roarke MC, Nguyen BD. PET/CT localization of phosphaturic mesenchymal neoplasm causing tumor-induced osteomalacia. Clin Nucl Med. 1997;32(4):300–1. doi: 10.1097/01.rlu.0000257180.03964.51. [DOI] [PubMed] [Google Scholar]
- 30.Cheung FMF, Ma L, Wu WC, Siu TH, Choi PT, Tai YP. Oncogenic osteomalacia associated with an occult phosphaturic mesenchymal tumour: clinico-radiologico-pathological correlation with ultrastructural studies. Hong Kong Med J. 2006;12(4):319–21. [PubMed] [Google Scholar]
- 31.Shane E, Parisien M, Henderson JE, Dempster DW, Feldman F, Hardy MA, et al. Tumor-induced osteomalacia: clinical and basic studies. J Bone Miner Res. 1997;12:1502–11. doi: 10.1359/jbmr.1997.12.9.1502. [DOI] [PubMed] [Google Scholar]
- 32.Kumar R. Tumor-induced osteomalacia and the regulation of phosphate homeostasis. Bone. 2000;27(3):333–8. doi: 10.1016/s8756-3282(00)00334-3. [DOI] [PubMed] [Google Scholar]
- 33.Mirams M, Robinson BG, Mason RS, Nelson AE. Bone as a source of FGF23: regulation by phosphate? Bone. 2004;35:1192–1199. doi: 10.1016/j.bone.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Jonsson KB, Mannstadt M, Miyauchi A, Yang IM, Stein G, Ljunggren Ö, et al. Extracts from tumors causing oncogenic osteomalacia inhibit phosphate uptake in opossum kidney cells. J Endocrinol. 2001;169:613–20. doi: 10.1677/joe.0.1690613. [DOI] [PubMed] [Google Scholar]
- 35.Wilkins GE, Granleese S, Hegele RG, Holden J, Anderson DW, Bondy GP. Oncogenic osteomalacia: Evidence for a humoral phosphaturic factor. J Clin Endocrinol Metab. 1995;80(5):1628–34. doi: 10.1210/jcem.80.5.7745010. [DOI] [PubMed] [Google Scholar]
- 36.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis J. Bone Miner Res. 2004;19:429–35. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 37.ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–8. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 38.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001;98(11):6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koriyama N, Nishimoto K, Kodama T, Nakazaki M, Kurono Y, Yoshida H, et al. Oncogenic osteomalacia in a case with a maxillary sinus mesenchymal tumor. Am J Med Sci. 2006;332(3):142–7. doi: 10.1097/00000441-200609000-00010. [DOI] [PubMed] [Google Scholar]
- 40.White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, et al. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86:497–500. doi: 10.1210/jcem.86.2.7408. [DOI] [PubMed] [Google Scholar]
- 41.Paul S, Kurtz M, Mentzer SJ. Osteomalacia associated with fibroblast growth factor-23 secreting chest wall tumor. J Thorac Cardiovasc Surg. 2007;134(3):803–4. doi: 10.1016/j.jtcvs.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 42.Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, et al. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab. 2004;89:3979–82. doi: 10.1210/jc.2004-0406. [DOI] [PubMed] [Google Scholar]
- 43.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. FGF7 - an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Me tab. 2005;90(2):1012–1020. doi: 10.1210/jc.2004-0357. [DOI] [PubMed] [Google Scholar]
- 44.Ryan EA, Reiss E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am J Med. 1984;77:501–12. doi: 10.1016/0002-9343(84)90112-8. [DOI] [PubMed] [Google Scholar]
- 45.Seufert J, Ebert K, Muller J, Eulert J, Hendrich C, Werner E, et al. Octreotide therapy for tumor-induced osteomalacia. NEJM. 2001;345:1883–8. doi: 10.1056/NEJMoa010839. [DOI] [PubMed] [Google Scholar]