

Abstract
Plasmodium falciparum is a purine auxotroph. The transport of purine nucleosides and nucleobases from the host erythrocyte to the parasite cytoplasm is essential to support parasite growth. P. falciparum Equilibrative Nucleoside Transporter 1 (PfENT1) is a major route for purine transport across the parasite plasma membrane. Malarial parasites are sensitive to inhibitors of purine salvage pathway enzymes. The immucillin class of purine nucleoside phosphorylase inhibitors and the adenosine analog, tubercidin, block growth of P. falciparum under in vitro culture conditions. We sought to determine whether these inhibitors utilize PfENT1 to gain access to the parasite cytosol. There is considerable controversy in the literature regarding the Km and/or Ki for purine transport by PfENT1 in the Xenopus oocyte expression system. We show that oocytes metabolize adenosine but not hypoxanthine. For adenosine, metabolism is the rate limiting step in oocyte uptake assays, making hypoxanthine the preferred substrate for PfENT1 transport studies in oocytes. We demonstrate that the Ki for PfENT1 transport of hypoxanthine and adenosine is in the 300–700 μM range. Effects of substrate metabolism on uptake studies may explain conflicting results in the literature regarding the PfENT1 adenosine transport Km. PfENT1 transports the tubercidin class of compounds. None of the immucillin compounds tested inhibited PfENT1 transport of [3H]hypoxanthine or [3H]adenosine. Although nucleobases are transported, modifications of the ribose ring in corresponding nucleoside analogs affects substrate recognition by PfENT1. These results provide new insights into PfENT1 and the mechanism by which purine salvage pathway inhibitors are transported into the parasite cytoplasm.
Keywords: Malaria, nucleoside, purine, transport, tubercidin, immucillin
1. Introduction
Malaria, caused by infection with parasites of the Plasmodium genus, remains a devastating global health problem. It accounts for 300–500 million clinical cases and 1 – 2 million deaths each year. Due to the increasing emergence of resistance to current antimalarial drugs, efforts to establish new drug targets within the parasite have become increasingly important. The essential role of DNA synthesis during malaria’s 48 hour intra-erythrocytic growth phase suggests that purine metabolic pathways may represent promising targets for the development of new anti-malarial drugs. Like many protozoan parasites, Plasmodia are purine auxotrophs incapable of synthesizing purines de novo [1, 2]. While the parasite’s dependence upon an external purine source has been known for nearly two decades [3], recent studies have begun to elucidate the molecular details involved in purine transport and metabolism.
The intra-erythrocytic malarial parasite transports purine nucleosides and nucleobases from the erythrocyte cytoplasm into the parasite cytosol via the PfENT1 equilibrative nucleoside transporter [4–6]. In the parasite cytoplasm, purine nucleosides and nucleobases are metabolized to generate nucleotides needed for nucleic acid synthesis, ATP generation, and intracellular signaling. However, the set of purine metabolic enzymes within the malarial parasite is more limited than those found in most mammalian cells. Plasmodium parasites do not contain a gene for adenosine kinase (AK) and thus cannot directly convert adenosine to AMP [7, 8]. For this reason, adenosine that is transported into the parasite cytosol is converted to hypoxanthine via the successive action of adenosine deaminase (PfADA) and purine nucleoside phosphorylase (PfPNP). Hypoxanthine is then utilized by hypoxanthine-guanine-xanthine phosphoribosyltransferase (PfHGXPRT) to generate inosine 5′-monophosphate (IMP) [9]. IMP is the branch-point for synthesis of all other parasite purine nucleotides. The majority of purines salvaged by P. falciparum are metabolized through this pathway.
During malaria infection in humans, plasma purines provide a source of purines that the parasites can use. The concentrations of various purines in human plasma is in the range of 0.4 to 6 μM [10]. During growth under in vitro culture conditions P. falciparum can proliferate in media containing a single purine source (hypoxanthine, adenine, guanine, xanthine, inosine, adenosine or guanosine) at a concentration greater than ~2 to 5 μM [11]. Parasite growth, however, with just guanine, guanosine or xanthine as the sole purine source is less robust than with the other purines and they are toxic at concentrations >50 μM [11]. Thus, during malaria infection the total plasma purine concentration available to the intraerythrocytic parasites is ~10 to 30 μM [10]. Pfnt1 knockout parasites can survive in culture but only in media supplemented with supraphysiological purine concentrations [6, 11].
Several purine salvage pathway inhibitors have antimalarial activity under in vitro culture conditions. The immucillin family of nucleoside analogs inhibit PNP. Immucillins inhibit both the erythrocytic and malarial PNP enzymes [12, 13]. The inhibition constant for immucillinH inhibition of PfPNP is 29 nM [14]. Immucillins inhibit in vitro P. falciparum growth in cultures containing hypoxanthine at a concentration higher than that found in human plasma [15]. In the presence of 10 μM hypoxanthine, 10 μM immucillinH completely inhibited parasite growth [15]. Tubercidin, an adenosine analog, is a substrate for adenosine kinase (AK). It is phosphorylated by AK and can act as a competitive inhibitor of AK phosphorylation of adenosine. Tubercidin also blocks parasite growth [16], although P. falciparum lacks AK activity. It is unclear whether tubercidin compounds exert their effect solely upon erythrocyte AK or interact with an as yet unidentified target within the parasite. The transport pathway(s) by which these purine salvage pathway inhibitors enter into the parasite is unknown.
DNA sequence analysis suggests that the P. falciparum genome encodes four putative nucleoside transporters [17], however only PfENT1 has been characterized [4, 5, 18, 19]. PfENT1 is an equilibrative nucleoside transporter localized to the parasite plasma membrane [20] that transports both nucleosides and nucleobases. However, disparate values have been reported for the transport Km or Ki of PfENT1 for various physiologic substrates [4, 5, 18, 19, 21]. To date, nearly all functional studies of PfENT1 have been performed using radioactive substrate uptake in the Xenopus laevis oocyte heterologous expression system. Metabolism of transported substrates in the oocyte cytoplasm to non-transportable products can have significant effects on the interpretation of radiolabeled substrate uptake experiments, especially if the metabolic enzyme is the rate-limiting step under the “uptake” assay conditions. Previous studies of PfENT1 mediated transport in Xenopus oocytes have not accounted for the potential effects of substrate metabolism in the oocyte cytoplasm in the interpretation of their experimental results. In order to study the transport of purine salvage pathway inhibitors that could alter the metabolism of transport substrates in the Xenopus oocyte expression system it was essential to characterize the effects of purine metabolism on the apparent transport properties of PfENT1 in the Xenopus oocyte system. We expressed PfENT1 in oocytes and investigated the metabolism of transported substrates, the transport of immucillin and tubercidin derivatives and the transport of purine nucleoside and nucleobase analogs to define the structural determinants of substrate specificity. The results help to resolve conflicting data in the literature, define the transport pathway for the tubercidins and demonstrate that immucillin uptake is mediated by a transport pathway other than PfENT1.
2. Materials and methods
2.1. Materials
Oligonucleotides to clone PfENT1 were synthesized by Sigma Genosys Biotechnologies, Inc (The Woodlands, TX). Restriction enzymes were purchased from New England Biolabs, Inc (Beverly, MA). [3H]Adenosine was purchased from Amersham Biosciences and [3H]hypoxanthine was purchased from Perkin Elmer NEN Radiochemicals (Waltham, MA). [3H]Tubercidin was purchased from Moravek Biochemicals and Radiochemicals (Brea, CA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
2.2. Chemistry: Synthesis of MT-tubercidin and other reagents
5′-methylthiotubercidin (MT-tubercidin, 10) was synthesized as described in detail in the Supplementary Information. [3H]Immucillin-H was synthesized as described previously [22]. Adenosine kinase inhibitor 1 (4-Chloro-5-iodo-7-(5-amino-5-deoxy-β-d-ribofuranosyl)pyrrolo[2,3-d]pyrimidine) was made as previously described by Ugarkar and colleagues [23]. 9-Deazahypoxanthine was synthesized as described previously [24].
2.3. Cloning and Expression of PfENT1 in X. laevis oocytes
The coding region of the PfENT1 gene was PCR amplified from genomic DNA preparations of P. falciparum 3D7 strain parasites, as described previously [18, 19]. The sense primer contained a BamH1 site prior to the initiating Met (5′CGAGGATCCATGAGTACCGGTAAAGAGTC3′), while the anti-sense primer contained an EcoR1 site downstream from the termination codon (5′CGAGAATTCTTATTGTGTTACATCGATGGGTGG3′). The ~1.3 kb PfENT1 PCR product was cloned into a pXOON expression vector [25] and the fidelity of the complete PfENT1 DNA sequence was verified by DNA sequencing. The pXOON plasmid was linearized with NheI and capped mRNA was prepared using T7 RNA polymerase (mMessage mMachine, Ambion, Austin, TX) and dissolved in diethylpyrocarbonate (DEPC) treated water.
Defolliculated X. laevis oocytes were prepared as described previously [26]. Oocytes were stored at 16 °C in SOS medium (in mM): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 HEPES, pH 7.5 with 100 IU/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B (Invitrogen, Carlsbad, CA) and 5% horse serum (Sigma-Aldrich). 24 hours post-isolation, oocytes were injected either with 23 nl of 1 ng/nl PfENT1 capped mRNA or DEPC treated water. Nucleoside/nucleobase uptake assays were performed 3 or 4 days post-injection.
2.4. Time course assays
PfENT1 mediated uptake of [8-3H]adenosine (23 Ci/mmol), [2, 8-3H]hypoxanthine (30 Ci/mmol), [5′-3H]Immucillin-H (0.45 Ci/mmol) or [8-3H]tubercidin (2 Ci/mmol) was determined in E1 buffer (in mM) (140 NaCl, 2.8 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, pH 7.4). For uptake of [5′-3H]Immucillin-H over a range of pH values (pH 5 – pH 8), uptake was determined in E2 buffer (in mM) (135 NaCl, 2.8 KCl, 2 MgCl2, 1 CaCl2, 7.5 HEPES, 7.5 citric acid). Three to five PfENT1 expressing oocytes were added to buffer containing a 1.5 μM concentration of the radioactive nucleoside to be tested and incubated at room temperature for 5, 15, 30, 60, or 120 min. Assays were terminated by five washes with ice cold E1 buffer, followed by solubilization of the oocyte with 200 μl 5% SDS. Uptake of radiolabeled nucleoside or nucleobase was determined with a Wallac WinSpectral 1414 Liquid Scintillation counter. All data points represent the average of at least ten individual oocytes derived from three separate oocyte isolations.
To account for non-PfENT1 mediated uptake, all time course experiments were performed in parallel with DEPC-treated water injected oocytes or uninjected oocytes. For all of the substrates tested, no significant difference was observed between DEPC injected or uninjected oocytes. For this reason, these data sets were pooled to represent background uptake values.
2.5. Analysis of purine metabolism by high-performance liquid chromatography (HPLC)
Purine metabolites were extracted from PfENT1 expressing oocytes incubated for 10 min with [8-3H]adenosine, [2, 8-3H]hypoxanthine or [8-3H]tubercidin. Five oocytes per condition were homogenized in 50 μl of water. Proteins and nucleic acids were removed by perchloric acid treatment. Briefly, samples were treated with 0.5 M HClO4 at 1:6 (v/v), vigorously mixed and incubated for 20 min at 4 °C. Samples were neutralized with 5 M KOH for 20 min at 4 °C and filtered through a YM10 Centricon spin column (MW retention = 10000; Amicon). Aliquots of each extract were monitored for radioactivity with a 1414 WinSpectral scintillation counter (Wallac, Gaithersburg, MD). All samples were analyzed in a reverse-phase (Luna C18(2), 150 × 4.6 mm, 3 μm, Phenomenex, Torrance, CA) ion-pair HPLC system. The mobile phases were 8 mM tetrabutylammonium bisulfate (Fluka) and 100 mM KH2PO4 with the pH adjusted to 6.0 with KOH (solution A) and 30% acetonitrile containing 8 mM tetrabutylammonium bisulfate and 100 mM KH2PO4 (pH 6) as solution B. The HPLC gradient was from 0% to 100% solution B in 20 min. The eluant was monitored at 254 nm and the flow rate was 1 ml/min. Fractions were collected every minute for 30 minutes and subjected to liquid scintillation counting. Radiolabeled peaks were identified based on UV detection of nucleoside internal standards: adenine, adenosine, AMP, ADP, ATP, guanine, guanosine, GMP, GDP, GTP, hypoxanthine, tubercidin (Sigma-Aldrich), tubercidin 5′-mono, -di and triphosphate (Axxora, LLC, San Diego, CA)..
2.6. Purine nucleotide phosphorylase activity assay
Activity of PNP in PfENT1 expressing and control oocytes was measured as previously described except that the oocyte homogenate was prepared without heparin [27]. Two oocytes were suspended in either PBS or PBS + 0.3% Triton X-100 (v/v) and manually homogenized. The homogenate was spun for 10 min at 16000 ×g at 4 °C. PNP activity was measured by a xanthine oxidase coupled assay in which inosine phosphorolysis is followed by hypoxanthine oxidation to uric acid producing an increase in UV absorbance at 293 nm. Up to 50 μl of supernatant was added to PNP reaction buffer (50 mM KPO4 pH 7.4, 1 mM inosine, 1 mM DTT and 20 mU xanthine oxidase) and reaction progress was monitored on a Cary 100 UV/Vis spectrophotometer (Varian Inc., Piscataway, NJ). Human red blood cells lysed in PBS + 0.3% Triton X-100 were used as a positive control.
2.7. Uptake competition assays
In order to measure the transport inhibition constant, Ki, of non-radiolabeled substrates via PfENT1, competition against the uptake of either [8-3H]adenosine (23 Ci/mmol) or [2,8-3H]hypoxanthine (30 Ci/mmol) was assayed. PfENT1 injected oocytes were incubated for 10 minutes in E1 buffer containing a fixed concentration of 100 nM radioactive adenosine or hypoxanthine and the competitor to be tested over a concentration range of ~2 μM to 8 mM. Following uptake, oocytes were washed and solubilized in SDS as described above for the time course assays. In order to normalize the competition data, uptake by PfENT1 injected oocytes in the absence of competitor was measured to determine 100% uptake. 0% uptake was determined by examining uptake by uninjected oocytes incubated in the presence of the lowest competitor concentration being tested.
9-Deazahypoxanthine and all immucillin compounds tested were prepared as 10 mM stock solutions in E1 buffer and serially diluted to final working concentrations. Tubercidin and 5′-iodotubercidin were dissolved in DMSO at 10 mM and 20 mM respectively. MT-tubercidin (10) was dissolved in a 50% DMSO/50% E1 buffer solution at a 10 mM concentration. 1 was dissolved in E1 buffer at a 4 mM final concentration. For all experiments in which DMSO was present in the stock solution, the equivalent concentration of DMSO was added for each competitor concentration tested.
2.8. Data analysis
Curve-fitting and statistical analysis was performed with Prism version 4.0 software (GraphPad Software, La Jolla, CA). For competition studies, two oocytes for each concentration of competitor were assayed and then averaged to yield each point on the competition curve. A one site fit (or two site fit for Iodotubercidin and 1 vs. [3H]adenosine) nonlinear regression analysis was performed to generate IC50 values; Ki values were determined by using the Cheng-Prusoff equation [28]. The Ki values from at least 3 independent assays were averaged to generate Ki and SEM values.
3. Results
3.1. PfENT1 mediated transport of natural purine substrates
Substrate transport by PfENT1 was measured by tritiated substrate uptake into oocytes expressing PfENT1. Fig. 1 shows a time course for the uptake of either 1.5 μM adenosine or hypoxanthine into PfENT1 expressing oocytes. While the uptake of both adenosine and hypoxanthine proceeds linearly for the first 30 min (Fig. 1, inset), the rate of adenosine uptake is nearly 4 times faster than the rate of hypoxanthine uptake. Similar observations were reported elsewhere [19] but no explanation has been given for the difference in the rates of uptake of the two substrates. Uptake over 120 minutes, a longer time period than previously examined, established that transport of adenosine continued at a constant rate while the transport of hypoxanthine slowed, approaching a plateau at 2.67 ± 0.36 pmol of hypoxanthine uptake per oocyte (Fig. 1B, dotted line).
Fig. 1.

Time course for uptake of adenosine or hypoxanthine by PfENT1 expressing X. laevis oocytes. (A) The initial thirty minute uptake time course showing uptake of 1.5 μM [8-3H] adenosine (■) or [2,8-3H] hypoxanthine (▲) by PfENT1-expressing (solid symbols) and control water-injected (open symbols) oocytes to illustrate the linear nature of uptake of at these initial time points. (B) PfENT1 mediated uptake of 1.5 μM [8-3H] adenosine (■) or [2,8-3H] hypoxanthine (▲) over a two hour time course. While uptake of adenosine is linear throughout the time course, uptake of hypoxanthine approaches a plateau (dashed line), predicted by nonlinear regression analysis. Transport was determined by assessing uptake in PfENT1-injected oocytes and subtracting uptake by water injected oocytes. Each data point represents nucleoside or nucleobase uptake by at least 10 PfENT1 and 10 water injected oocytes, derived from at least three separate oocyte isolations.
PfENT1 is an equilibrative nucleoside transporter and an explanation for the plateau in hypoxanthine uptake is that the oocyte’s internal hypoxanthine concentration has reached equilibrium with the external 1.5 μM concentration. The diameter of a Xenopus laevis oocyte is ~1.2 mm [29] which gives a calculated internal volume of 0.9 μl similar to that measured experimentally [30]. Equilibration of a 1.5 μM external solution should occur at about 1.35 pmol of uptake per oocyte ([internal volume]×[external hypoxanthine concentration]). The 2.67 pmol per oocyte plateau observed for the hypoxanthine time course data is about twice the expected value. Hypoxanthine is hydrophobic; its non-specific binding to the external surface of the oocyte is greater than that of adenosine (Fig. 1A). It is likely that there is non-specific binding to membranes or proteins in the oocyte cytoplasm leading to the plateau uptake level that is greater than predicted by simple equilibration. As we show below, hypoxanthine is not metabolized within the oocyte, thus metabolism does not explain the increased level.
In contrast to hypoxanthine, PfENT1 mediated adenosine transport continued linearly throughout the entire 120 min duration of the experiment and did not reach a plateau value. Adenosine uptake at 60 min reached nearly 7 pmol of uptake per oocyte, well beyond the plateau range predicted for simple equilibration. This suggests that adenosine is either metabolized within the oocyte to a form that is impermeable through PfENT1, or sequestered from the oocyte cytoplasm in a subcellular compartment by a concentrative transporter or tightly protein bound. In any of these cases, the adenosine concentration within the oocyte cytoplasm remains low, permitting continuous adenosine uptake at a rate limited by the activity of the metabolic enzyme or sequestration process, not by the transporter.
3.2. Purine metabolism in Xenopus laevis oocytes
The metabolic fate of radiolabeled adenosine and hypoxanthine within the oocyte was analyzed by HPLC analysis of metabolites (Fig. 2). Oocytes expressing PfENT1 were incubated in the presence of [3H]adenosine or [3H]hypoxanthine for 10 minutes and then washed to remove excess radiolabeled purine. HPLC analysis of oocyte metabolites showed that 93% of the transported [3H]adenosine was metabolized to AMP, ADP, and ATP, presumably by X. laevis adenosine kinase activity. In contrast, hypoxanthine was not metabolized within the oocyte, suggesting that X. laevis oocytes contain minimal or no hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRT) activity and hypoxanthine is not a substrate for adenosine kinase.
Fig. 2.
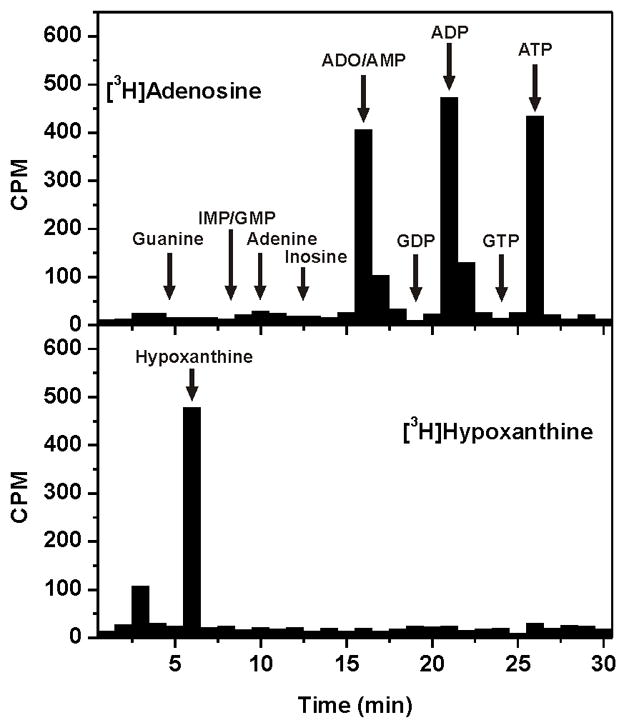
Xenopus laevis oocyte purine metabolism. Oocytes expressing PfENT1 were metabolically labeled for 10 minutes with 1.5 μM [8-3H] adenosine (top panel) or [2,8-3H] hypoxanthine (bottom panel) and analyzed by HPLC. Fractions were collected and analyzed for the presence of radioactivity. The retention times for adenine, adenosine (ADO), adenosine 5′-mono- (AMP), di- (ADP) and tri-phosphate (ATP), guanine, guanosine 5′-mono (GMP), di- (GDP) and tri-phosphate (GTP), hypoxanthine, inosine, and inosine 5′-monophosphate (IMP) are indicated.
Because transported hypoxanthine is not metabolized inside the oocyte, its concentration within the oocyte can rise and equilibrate with the external concentration, leading to the observed plateau. By contrast, adenosine metabolism maintains the internal adenosine concentration at a low level, thereby maintaining the transport gradient across the oocyte membrane and sustaining a steady rate of adenosine influx determined by the AK activity. We have recently shown that AMP is not transported by PfENT1 [7], thus the phosphorylated products of X. laevis adenosine metabolism are trapped within the oocyte and are not substrates for transport by PfENT1.
3.3. Consequences of purine metabolism on PfENT1 transport studies in oocytes
There is significant debate in the PfENT1 literature regarding its Km for purine substrates including adenosine and hypoxanthine [4, 5, 18, 19, 21]. Purine metabolism within the oocyte may account for some of these differences, because the Km of oocyte metabolic enzymes for the transported substrates may differ from the Km of PfENT1 for those substrates. Depending on the uptake assay conditions the transporter or the metabolic enzymes may be the rate limiting step.
The experiments in Figure 3 explicitly demonstrate the effect of oocyte metabolism on PfENT1 transport competition studies. We examined the competition of an ascending concentration of unlabeled hypoxanthine on the uptake of either 100 nM [3H]hypoxanthine or 100 nM [3H]adenosine (Fig. 3A). We present the results as inhibition curves to allow comparison when the cold competitor, in this case hypoxanthine, is the either the same or different than the tritiated substrate. As one would expect for competition at a single site, i.e. the transporter, unlabeled hypoxanthine competed with the uptake of both radiolabeled substrates with nearly identical inhibition constants. The Ki for unlabeled hypoxanthine vs [3H]hypoxanthine was 300 ± 32 μM and was 479 ± 87 μM vs [3H]adenosine (Table 1). In contrast, performing a similar experiment with an ascending concentration series of unlabeled adenosine yielded a different result (Fig. 3B). Competition of unlabeled adenosine with 100 nM [3H]hypoxanthine occurred with a Ki of 655 ± 60 μM whereas competition of unlabeled adenosine with 100 nM [3H]adenosine occurred with a Ki of 48 ± 15 μM, 10-fold lower than when hypoxanthine is the labeled substrate (Table 1). This implies that cold adenosine competes with labeled hypoxanthine at one site, presumably PfENT1, and with labeled adenosine at a different site, presumably adenosine kinase.
Fig. 3.

PfENT1 substrate affinity measurements. Competition with 10 minutes of uptake of either 100 nM [8-3H]adenosine (■) or 100 nM [2,8-3H]hypoxanthine (▲) was measured for a range of concentrations of either hypoxanthine (A) or adenosine (B). Competition data was normalized by determining uptake in PfENT1 expressing (100%) and uninjected (0%) oocytes in the absence of competitor.
Table 1.
PfENT1 substrate inhibition constants determined by uptake competition studies with the indicated radiolabeled substrate.a
| Competition vs. [3H]hypoxanthine | Competition vs. [3H]adenosine | |||
|---|---|---|---|---|
| Ki (μM) | n | Ki (μM) | n | |
| Adenosine | 650 ± 60 | 3 | 48 ± 15 | 4 |
| Hypoxanthine | 300 ± 32 | 4 | 480 ± 87 | 6 |
| 9-Deaza-hypoxanthine | 710 ± 410 | 3 | 940 ± 270 | 5 |
| Tubercidin | 420 ± 110 | 4 | 590 ± 100 | 5 |
| Iodotubercidin | 54 ± 8 | 3 | 66 ± 15, 0.048 ± 0.026b | 3 |
| 1 | 3,400 ± 830 | 3 | 1200 ± 300, 0.15 ± 0.01b | 3 |
| MT-Tubercidin (10) | 1,100 ± 130 | 3 | 1,600 ± 140 | 5 |
Given are the mean ± SEM.
Denotes two site fit.
Oocyte metabolism of adenosine provides an explanation for the seemingly paradoxical result that the inhibition constant of PfENT1 for adenosine is 10-fold different depending on whether the labeled substrate is hypoxanthine or adenosine. Because hypoxanthine is not metabolized, competition of unlabeled hypoxanthine with the uptake of either radiolabeled substrate (Fig. 3A) occurs at only one site, the PfENT1 transport pathway. Similarly, uptake of [3H]hypoxanthine is unaffected by metabolism and thus the competition of unlabeled adenosine with the uptake of [3H]hypoxanthine (Fig. 3B, triangles) can only occur at PfENT1. In contrast, adenosine is metabolized inside the oocyte and thus the competition of unlabeled adenosine with [3H]adenosine uptake (Fig. 3B, squares) can occur at two sites, transport via PfENT1 and metabolism within the oocyte. The measured Ki will depend on which process, transport or metabolism, is rate limiting. The observed 10-fold difference in the inhibition constant of PfENT1 for adenosine depending on the labeled substrate in Fig. 3B, implies that the lower adenosine Ki being measured in competition with labeled adenosine reflects the Ki of adenosine metabolism not transport via PfENT1. This implies that metabolism is the rate limiting step when measuring adenosine uptake vs labeled adenosine.
The capacity of oocyte purine metabolism to alter the measurement of Km or Ki in PfENT1 mediated transport studies is an important consideration for ongoing studies of this transporter in oocytes. The remainder of this study is directed toward examining PfENT1 mediated transport of parasiticidal nucleoside-analog inhibitors of purine metabolism. In order to avoid the complications involved in differentiating between competition at the PfENT1 transporter and blockade of metabolic enzymes by these nucleoside-analog inhibitors, all competition assays were performed using [3H]hypoxanthine, a PfENT1 substrate that we have shown is not metabolized inside the oocyte.
3.4. Tubercidin based compounds are transported by PfENT1
The tubercidins (7-deaza-adenosine derivatives) are adenosine analogs that compete with adenosine for metabolism by adenosine kinase. They are reported to have antimalarial activity under in vitro culture conditions [16]. However, P. falciparum lacks adenosine kinase activity [7] and its genome lacks an adenosine kinase gene homologue [8]. Thus, it is not clear whether the tubercidin compounds exert their effect within the parasite or in the erythrocyte cytoplasm. In order to determine whether tubercidin analogs gain access to the parasite through PfENT1 mediated transport, we measured the time course of [3H]tubercidin uptake into PfENT1-injected oocytes (Fig. 4A). Tubercidin uptake was linear over a 60 minute period. Interestingly, tubercidin accumulation reached a level that surpassed the plateau expected for equilibration with external tubercidin. As we found for adenosine (Fig. 2), HPLC analysis of oocytes metabolically labeled with [3H]tubercidin showed that tubercidin was metabolized within the oocyte to mono-, di- and tri-phosphate derivatives (TuMP, TuDP and TuTP) (Fig. 4B). Although tubercidins are referred to as inhibitors of adenosine kinase they are also substrates and are phosphorylated by it [31]. A variety of tubercidin analogs have been synthesized to test for their affinity against adenosine kinase and/or to decrease toxicity to mammalian cells (Fig. 5A). To assess the structure-activity relationship of tubercidin derivatives for PfENT1 the transport inhibition constants of several tubercidin derivatives were determined by competition for [3H]hypoxanthine uptake.
Fig. 4.
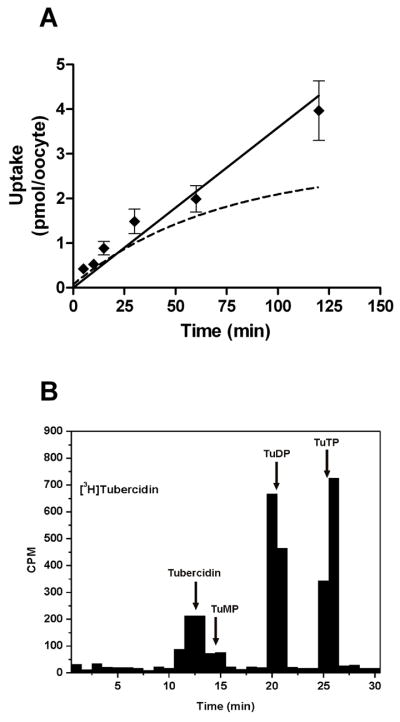
PfENT1 transports tubercidin compounds. A. A time course for the uptake of 1.5 μM [8-3H]tubercidin. For comparison, the time course of uptake of 1.5 uM [2,8-3H]hypoxanthine (dashed line) described in Fig. 1 is reproduced here. B. Oocytes expressing PfNT1 were metabolically labeled for 10 minutes with 1.5 μM [8-3H]tubercidin and analyzed by HPLC. Fractions were collected and analyzed for the presence of radioactivity. The retention times for Tubercidin, Tubercidin 5′-mono- (TuMP), di- (TuDP) and tri-phosphate (TuTP) are indicated.
Fig. 5.

PfENT1 specificity for tubercidin compounds. The structures of all of the tubercidin compounds tested in this study – Tubercidin, MT-Tubercidin (10), Iodotubercidin, and 1 – are shown in (A). Competition of a range of concentrations of each of these compounds with 10 minutes of uptake of 100 nM [2,8-3H]hypoxanthine is shown in Panels B-E. Competition data was normalized by determining uptake in PfENT1 expressing (100%) and uninjected (0%) oocytes in the absence of competitor. For comparison, competition of a range of hypoxanthine concentrations with 10 minutes of uptake of 100 nM [2,8-3H]hypoxanthine is reproduced in this figure (dashed line).
Tubercidin derivatives competed with [3H]hypoxanthine uptake, albeit with different inhibition constants (Fig. 5, Table 1). The tubercidin Ki vs [3H]hypoxanthine was similar to the adenosine Ki vs [3H]hypoxanthine, suggesting that the N7 position in the purine ring is not a determinant of purine recognition for PfENT1. The iodotubercidin Ki for PfENT1 transport was ten-fold lower than the Ki’s for tubercidin and adenosine. By contrast, the modifications present on MT-tubercidin and compound 1 increased the transport Ki’s of these compounds compared to tubercidin by 2.5 fold and 8 fold respectively.
Having shown that PfENT1 is competitively inhibited by the tubercidin derivatives, we tested the hypothesis that intra-oocyte adenosine kinase activity was responsible for complicating PfENT1 competition studies performed using [3H]adenosine. By comparing the competition of iodotubercidin with the uptake of either [3H]hypoxanthine or [3H]adenosine (Fig. 6), we found an explicit demonstration of the effect of adenosine kinase activity upon PfENT1 uptake studies. Uptake data for the competition of iodotubercidin with [3H]hypoxanthine was best fit by a one site competition equation with a Ki for iodotubercidin of 54 μM (Table 1). In contrast, uptake data for iodotubercidin competition with [3H]adenosine was best fit by a two-site competition equation with Ki’s of 66 μM and 0.048 μM (Table 1). With both labeled substrates we found one Ki value for iodotubercidin of ~60 μM, which we attribute to competition of iodotubercidin with the labeled substrate at the PfENT1 transporter. In the competition of iodotubercidin (or 1, see Table 1) with [3H]adenosine a second site with 1000-fold higher apparent affinity was found. This second site most likely represents competition between iodotubercidin and [3H]adenosine at the adenosine kinase enzyme. These results further demonstrate the complications inherent in the interplay of transport and metabolism in the study of purine transport in oocytes.
Fig. 6.

Iodotubercidin competition with metabolized and unmetabolized PfENT1 substrates. Competition of a range of concentrations of iodotubercidin with 10 minutes of uptake of either 100 nM [8-3H]adenosine (■) or 100 nM [2,8-3H]hypoxanthine (▲). Competition data was normalized by determining uptake in PfENT1 expressing (100%) and uninjected (0%) oocytes in the absence of iodotubercidin. Non-linear regression analysis of hypoxanthine competition data (one-site fit) and adenosine competition data (two site fit) are shown.
3.5. Immucillin compounds are not transported by PfENT1
Immucillins inhibit both human and malarial purine nucleoside phosphorylase (PNP) with sub-nanomolar affinity and inhibit parasite growth during in vitro culture in media containing hypoxanthine [12, 13, 15]. Additionally, immucillin treatment of intact erythrocyte-free malarial parasites blocks malarial PNP [5]. This shows that immucillins gain access to the parasite cytosol. Because immucillins are purine-analogues, we hypothesized that these compounds would enter the parasite through PfENT1. To address this question, we examined the ability of PfENT1 expressing oocytes to transport [3H]Immucillin-H (ImmH).
The time course for accumulation of 1.5 μM [3H]ImmH by PfENT1-expressing oocytes is shown in Fig. 7A. ImmH uptake differed from hypoxanthine uptake, exhibiting an initial uptake rate approximately 10-fold slower than the observed initial rate of uptake for 1.5 μM hypoxanthine. Additionally, ImmH uptake reached a plateau after 30–60 minutes of uptake at an amount that is ~25% of the level expected for equilibration with external ImmH (Fig. 7A). The ImmH accumulation, however, was specific for PfENT1 expressing oocytes and was not seen in water injected or uninjected oocytes (Fig. 7A, open circles). To rule out the possibility that uptake of [3H]ImmH was due to increased endocytic trafficking resulting in pinocytotic uptake of fluid phase [3H]ImmH by PfENT1 expressing oocytes, we measured uptake of 1.5 μM [3H]glucose by PfENT1 expressing and control oocytes. There was no [3H]glucose uptake during a 60 min experiment (data not shown) ruling out this possibility. Furthermore, to rule out the possibility that [3H]ImmH was binding to PNP in the oocytes, we assayed the PNP activity in PfENT1 expressing and control oocytes. No PNP activity was detected in either PfENT1 or control oocytes (data not shown). We conclude that the slow rate of [3H]ImmH uptake and the failure to reach equilibrium at an amount of uptake consistent with the external concentration suggests that ImmH was not transported into the oocyte by PfENT1 but rather might be binding to the PfENT1 protein at a site distinct from the transport pathway.
Fig. 7.

PfENT1 does not exhibit specificity for immucillin compounds. A. A time course for the uptake of 1.5 μM [5′-3H] Immucillin H by PfENT1 expressing (●) and DEPC water injected oocytes (○). The chemical structure of Immucillin H is shown. B. Competition of 2.5 mM or 10 mM Immucillin H (black bars), Immucillin A (grey bars), and Immucillin G (white bars) with 10 minutes of uptake of 100 nM [2,8-3H]hypoxanthine. Competition of 2.5 mM hypoxanthine (hashed bar) with 10 minutes of uptake of 100 nM [2,8-3H]hypoxanthine is shown. C. The structure of 9-deaza-hypoxanthine is shown. A range of 9-deaza-hypoxanthine concentrations (●) competes with 10 minutes of uptake of 100 nM [2,8-3H]hypoxanthine. Competition of a range of hypoxanthine concentrations with 10 minutes of uptake of 100 nM [2,8-3H]hypoxanthine was shown in Fig. 3A and is reproduced in this figure (dashed line) for purposes of comparison. D. PfNT1 mediated uptake of 1.5 μM [5′-3H]Immucillin H over a range of buffer pH values. For each pH, 10 minutes of ImmH uptake by PfNT1 expressing oocytes was measured and uptake by DEPC water injected oocytes is subtracted to account for background.
To examine further whether PfENT1 transports immucillin compounds, we examined whether high concentrations of unlabeled immucillins would compete with [3H]hypoxanthine uptake. At both 2.5 and 10 mM concentrations, ImmH, Immucillin-A, and Immucillin-G failed to compete with the uptake of 100 nM [3H]hypoxanthine, whereas 2 mM hypoxanthine blocked ~90% of [3H]hypoxanthine uptake (Fig. 7B). These concentrations of immucillins also failed to compete with the uptake of 100 nM [3H]adenosine (data not shown).
Immucillins are nucleoside analogs that contain modifications in both the purine and ribose rings. These compounds are 9-deaza purines and contain a cationic amino group in place of the ribose ring oxygen. In order to determine which of these modifications was responsible for abrogating PfENT1 transport, we examined PfENT1’s ability to transport 9-deazahypoxanthine (9DHx). 9DHx is the nucleobase present in ImmH, but lacks a sugar moiety. As shown in Fig. 7C and Table 1, 9DHx competed with [3H]hypoxanthine uptake with similar Ki to that of hypoxanthine. This implies that the nitrogen at the 9 position of the purine ring is not an important determinant of PfENT1 substrate recognition, because its removal did not affect apparent transport affinity. This suggests that the lack of immucillin transport by PfENT1 is not due to the modification within the purine ring but rather the replacement of the natural ribose sugar with an iminoribitol moiety.
While the ribose sugar of natural nucleosides is uncharged, the iminoribitol ring within immucillins contains an imino group with a pKa of 6.9 [32]. To examine whether the charge character at the iminoribitol nitrogen is responsible for blocking PfENT1 transport of immucillins, we examined uptake of 1.5 μM [3H]ImmH into PfENT1 expressing oocytes over a range of pH values, from pH 5 to pH 8. If PfENT1 recognition is affected by the charge at the iminoribitol nitrogen, then the rate of ImmH uptake should be pH dependent, because ImmH will be cationic at pH 5 and neutral at pH 8. Previous studies have shown that the rate of PfENT1 transport of adenosine is unaltered over this pH range [19]. We found that ImmH accumulation was unchanged by changes in extracellular pH (Fig. 7D). Thus, the ionization state of the iminoribitol nitrogen is not responsible for abrogating PfENT1 recognition of immucillins. The difference in hydrogen bonding properties of the iminoribitol and ribose sugars therefore affects transport. The ribose oxygen acts as a hydrogen bond acceptor, while the iminoribitol nitrogen is a hydrogen bond donor. Alternatively, ribosyl geometric constraints within the PfENT1 purine recognition site could be affected by the iminoribitol modification of the natural purine sugar. We consider this possibility to be less likely because ribosyl and iminoribitol groups are flexible with low energy barriers for conformational changes.
4. Discussion
We sought to examine PfENT1’s ability to transport purine salvage pathway enzyme inhibitors in a heterologous Xenopus oocyte expression system. We found that oocyte mediated purine metabolism had a significant confounding effect upon PfENT1 transport studies. We showed that adenosine is phosphorylated to AMP, ADP, and ATP in the oocyte cytoplasm, presumably via adenosine kinase and adenylate kinase. This raises an important issue for the use of radiolabeled substrate uptake experiments for the determination of the transport parameters of PfENT1 because the measured parameters will depend on whether transport or metabolism is the rate limiting step in the uptake process. For radiolabeled adenosine intra-oocyte adenosine metabolism affects the initial rate of PfENT1-mediated adenosine transport, thus metabolism alters measurement of the adenosine Km when [3H]adenosine is used as the labeled substrate. Unlike adenosine, hypoxanthine is not metabolized within the oocyte during the time course of our experiments. Both adenosine and hypoxanthine competed with [3H]hypoxanthine uptake with similar Ki values 655 μM and 300 μM (Fig. 3A), suggesting that PfENT1 exhibits similar affinities for these two substrates. To avoid the confounding effects of oocyte purine metabolism in our study, we used [3H]hypoxanthine as the transported substrate for uptake assays with the purine salvage pathway inhibitors. The tubercidin class of purine metabolism inhibitors was transported by PfENT1. In contrast, immucillins did not compete with PfENT1 mediated uptake of either [3H]hypoxanthine or [3H]adenosine, suggesting that they are not transported by PfENT1.
The PfENT1 transportable purine substrates present in erythrocytes includes, in order of increasing concentration, adenine (0.4 μM), adenosine (0.9 μM), inosine (2.5 μM), xanthine (3.6 μM) and hypoxanthine (13 μM) [10, 33]. The concentration of hypoxanthine in the erythrocyte is about twice the sum of the concentrations of the other PfENT1 transportable purine substrates. It should be noted that in P. falciparum infected erythrocytes the concentrations of these purines might be different due to the presence of the new permeability pathway in the erythrocyte plasma membrane [34, 35].
Three groups have studied PfENT1 transport in Xenopus oocytes using similar transport assays to those used in the current work, but have obtained disparate results. These differences may be due in part to effects of intra-oocyte purine metabolism. Parker et al. reported that PfENT1 exhibits nearly identical transport Km’s for adenosine (320 μM), hypoxanthine (410 μM), and adenine (320 μM) [19], similar to the Ki measured in the present study. Downie et al. reported that the PfENT1 adenosine Km was 1.86 mM [5]. In contrast, Carter and coworkers reported that PfENT1 exhibits a much lower Km for adenosine (13 μM) than the other groups but found that the Km for inosine (253 μM) was similar to that reported for adenosine by the other groups [18]. It is important to note that the highest adenosine concentration tested in the Carter study was ~110 μM. This concentration is not sufficiently high enough to detect the transport Km of PfENT1 for adenosine demonstrated in the present study or reported by Parker et al. (2000) or Downie et al. (2006). The 13 μM adenosine Km reported by Carter et al. is more likely to be due to mixed effects of competition with adenosine kinase, the primary metabolic enzyme for adenosine in the oocyte, not PfENT1 or the result of a one-site fit to the combination of the affinities of both adenosine kinase and PfENT1. This is consistent with the metabolism of adenosine, not transport, being the rate limiting factor in the labeled adenosine uptake experiments. In addition, Carter et al. found no evidence of nucleobase transport by PfENT1 [18]. Under the conditions that Carter et al. performed their adenosine uptake assays they were largely assaying the rate of adenosine kinase action not transport activity, i.e. metabolism rather than transport was rate limiting, thus, their failure to detect PfENT1 mediated nucleobase transport may reflect the fact that hypoxanthine is not a substrate for adenosine kinase and therefore does not compete with adenosine for phosphorylation by adenosine kinase, the rate limiting step in adenosine uptake experiments. Carter et al. did not use a sufficiently high concentrations of either adenosine or hypoxanthine to detect competition for PfENT1 mediated transport given the transport Ki’s measured in the present work.
In the studies of Parker et al., oocytes were treated with 2-deoxycoformycin to block oocyte adenosine deaminase while Carter et al. did not. Although 2-deoxycoformycin inhibits adenosine deaminase activity, we found that the major path for intra-oocyte adenosine metabolism is adenosine kinase because no inosine, hypoxanthine or guanosine derivatives were detected by HPLC (Fig. 2). Thus, both studies permit the conversion of transported adenosine to AMP, ADP and ATP. All of their results with adenosine most likely report on the combination of PfENT1 transport and adenosine metabolism by adenosine kinase.
Transport experiments with [3H]ImmH revealed that [3H]ImmH accumulation was ~4 fold less than the amount expected for equilibration with extracellular ImmH. Furthermore, the immucillins did not compete with [3H]hypoxanthine or [3H]adenosine uptake, even at 10 mM concentrations. Regardless of the explanation for the [3H]ImmH uptake, the results indicate that PfENT1 does not transport immucillins. We suggest that the apparent uptake of [3H]ImmH might be due to high affinity binding to the surface of PfENT1 expressing oocytes. The interaction was sufficiently robust so that ImmH remained bound during the 5 min wash period at 4 °C. Further work will be needed to understand the nature of the interaction.
Immucillins inhibit both the human and malarial PNP enzymes [12, 13]. Blockade of PNP activity is presumably the mechanism by which immucillins exert their antimalarial effect, because immucillin-induced parasite death, can be rescued by supplementation with supraphysiological levels of hypoxanthine, the downstream product of PNP [15]. Additionally, an immucillin analog specific for the malarial PNP enzyme (MT-ImmH) is lethal to malaria parasites during in vitro culture [36], suggesting that blockade of PfPNP is necessary for the antimalarial activity if immucillins. As PfENT1 does not transport immucillins, an alternative immucillin transport pathway into the parasite remains to be identified.
Malaria genome database studies have identified three putative nucleoside transporters besides PfENT1 [17], but their substrate specificities and localization within the parasite have not been determined. A recent study by Quashie et al. (2008) reported four distinct purine transport activities in malaria parasites and attempted to equate them with the four putative purine transporters identified in the genomic studies [21]. As has been noted elsewhere [37, 38], Quashie et al. failed to prove that transport and not metabolism was the rate limiting process in their uptake studies in intact parasites. Nor did they exclude effects of intra-parasite purine metabolism on their uptake competition studies. It is critical to determine whether in the parasite, transport or metabolism is rate limiting before one attributes the results of substrate competition experiments to the transporter: Because, as we have demonstrated here, intra-oocyte purine metabolism can have a significant impact on the measured Km or Ki due to competition at a cytoplasmic metabolic enzyme, in our case adenosine kinase. While several papers have reported activities of purine salvage pathway enzymes from parasite lysates [9, 39], the activity measurements were performed with totally non-physiological substrate concentrations meaning that the reported enzyme activities must be corrected for substrate concentrations found under in vivo conditions in order to determine whether transport or metabolism is rate limiting in the parasite purine uptake experiments. In their recent paper Quashie et al., (2008) reported that the Km for hypoxanthine uptake in intact parasites was 0.34 μM [21]; this is 1000-fold higher apparent affinity than we have measured for the hypoxanthine Ki in the present work. In parasites, hypoxanthine enters the purine salvage pathway through hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRT) which has a Km for hypoxanthine of 0.46 μM [39], similar to that reported by Quashie et al. [21]. Furthermore, guanine also enters the purine salvage pathway via HGXPRT and thus competes with hypoxanthine both at HGXPRT and at the transporter. Uptake competition experiments between unlabeled guanine and [3H]hypoxanthine in intact parasites yielded a guanine Ki of 0.11 μM [21], similar to the reported guanine Km for PfHGXPRT of 0.30 μM [39]. Thus, there is a striking concordance between the affinities reported by Quashie et al., (2008) and the affinities of the primary metabolic enzyme, HGXPRT, suggesting that in the intact parasites, metabolism and not transport is the rate limiting step in purine uptake experiments. Performing the experiments in glucose deprived parasites will reduce the effects of metabolism on the uptake process and may provide data on the properties of the transmembrane transporters but even there substrate uptake for adenosine and inosine plateaus at a distribution ratio that is five times the level expected for an equilibrative transporter suggesting that metabolism is affecting the uptake experiments [5]. It should be noted, however, that PfENT1 is not the only transport pathway for nucleosides and nucleobases in the parasite plasma membrane because PfENT1 knockout parasites can incorporate some exogenous purines, suggesting that at least one alternative purine entry route exists but its molecular basis is unknown [6, 11]. The immucillins may employ this transport pathway.
Unlike the immucillins, for which the parasite enzyme target is established, it is unclear how tubercidins block parasite growth. Tubercidin has been shown to inhibit parasite growth during in vitro culture in RPMI media supplemented with human serum with an IC37 of 0.4 – 0.7 μM [16, 40]. The parasite does not contain adenosine kinase, although the erythrocyte does contain adenosine kinase that tubercidins will competitively inhibit. Erythrocyte adenosine kinase is not vital to parasite survival. Tubercidins can gain access to the parasite cytosol through PfENT1 mediated transport but the subsequent target is unknown. While PfENT1 exhibits a tenfold greater apparent affinity for iodotubercidin over tubercidin, these two compounds exhibit identical antimalarial activities [7]. Further studies are warranted to advance our understanding of the target for these compounds.
Our studies of PfENT1 mediated transport of immucillins and tubercidins have implications for the identification of structural determinants within the purine moiety that affect PfENT1’s substrate specificity. Both 7-deazaadenosine (tubercidin) and 9-deazahypoxanthine are transported by PfENT1 with affinities similar to their natural analogues (adenosine and hypoxanthine, respectively). The nitrogens at positions 7 and 9 are not essential for substrate recognition by PfENT1. By contrast, alteration of the nucleoside ribose moiety in 1 and MT-tubercidin (10) decreases PfENT1’s affinity for these substrates or ablates transport altogether in the case of the immucillins. The effect of these modifications upon transport suggests that PfENT1 substrate recognition involves the purine ribose moiety, despite the fact that PfENT1 is capable of transporting nucleobases that lack a ribose ring. Thus, while the nucleoside ribose ring affects substrate recognition, PfENT1’s ability to transport nucleobases suggests that determinants in the purine ring are also vital to substrate recognition.
PfENT1 has potential as a target for antimalarial drug development because PfENT1 knockouts are lethal at physiologic concentrations of purines. Further characterization of the structure of PfENT1 and determinants of substrate specificity may provide valuable insights for rational drug design.
Supplementary Material
Acknowledgments
We thank I.J. Frame for expert technical assistance and Dr. Alan Finkelstein for helpful discussions. This work was supported in part by the National Institutes of Health [Grant AI064933 (to MHA) and AI49512 (to VS)] and by a contract from the New Zealand Foundation for Science, Research and Technology.
The abbreviations used are
- ADA
adenosine deaminase
- AK
adenosine kinase
- DEPC
diethylpyrocarbonate
- HGXPRT
hypoxanthine guanine xanthine phosphoribosyltransferase
- HPLC
high performance liquid chromatography
- ImmH
ImmucillinH
- PfENT1
Plasmodium falciparum Equilibrative Nucleoside Transporter 1
- PNP
purine nucleoside phosphorylase
- 9DHx
9-deazahypoxanthine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carter NS, Landfear SM, Ullman B. Nucleoside transporters of parasitic protozoa. Trends Parasitol. 2001;17:142–5. doi: 10.1016/s1471-4922(00)01806-7. [DOI] [PubMed] [Google Scholar]
- 2.de Koning HP, Bridges DJ, Burchmore RJ. Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol Rev. 2005;29:987–1020. doi: 10.1016/j.femsre.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Divo AA, Geary TG, Davis NL, Jensen JB. Nutritional requirements of Plasmodium falciparum in culture. I. Exogenously supplied dialyzable components necessary for continuous growth. J Protozool. 1985;32:59–64. doi: 10.1111/j.1550-7408.1985.tb03013.x. [DOI] [PubMed] [Google Scholar]
- 4.Downie MJ, Saliba KJ, Broer S, Howitt SM, Kirk K. Purine nucleobase transport in the intraerythrocytic malaria parasite. Int J Parasitol. 2008;38:203–9. doi: 10.1016/j.ijpara.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Downie MJ, Saliba KJ, Howitt SM, Broer S, Kirk K. Transport of nucleosides across the Plasmodium falciparum parasite plasma membrane has characteristics of PfENT1. Mol Microbiol. 2006;60:738–48. doi: 10.1111/j.1365-2958.2006.05125.x. [DOI] [PubMed] [Google Scholar]
- 6.El Bissati K, Zufferey R, Witola WH, Carter NS, Ullman B, Ben Mamoun C. The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc Natl Acad Sci U S A. 2006;103:9286–91. doi: 10.1073/pnas.0602590103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cassera MB, Hazleton KZ, Riegelhaupt PM, et al. Erythrocytic adenosine monophosphate as an alternative purine source in Plasmodium falciparum. J Biol Chem. 2008;283:32889–99. doi: 10.1074/jbc.M804497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reyes P, Rathod PK, Sanchez DJ, Mrema JE, Rieckmann KH, Heidrich HG. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol. 1982;5:275–90. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 10.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 11.El Bissati K, Downie MJ, Kim SK, et al. Genetic evidence for the essential role of PfNT1 in the transport and utilization of xanthine, guanine, guanosine and adenine by Plasmodium falciparum. Mol Biochem Parasitol. 2008;161:130–9. doi: 10.1016/j.molbiopara.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewandowicz A, Ringia EA, Ting LM, et al. Energetic mapping of transition state analogue interactions with human and Plasmodium falciparum purine nucleoside phosphorylases. J Biol Chem. 2005;280:30320–8. doi: 10.1074/jbc.M505033200. [DOI] [PubMed] [Google Scholar]
- 13.Shi W, Ting LM, Kicska GA, et al. Plasmodium falciparum purine nucleoside phosphorylase: crystal structures, immucillin inhibitors, and dual catalytic function. J Biol Chem. 2004;279:18103–6. doi: 10.1074/jbc.C400068200. [DOI] [PubMed] [Google Scholar]
- 14.Kicska GA, Tyler PC, Evans GB, Furneaux RH, Kim K, Schramm VL. Transition state analogue inhibitors of purine nucleoside phosphorylase from Plasmodium falciparum. J Biol Chem. 2002;277:3219–25. doi: 10.1074/jbc.M105905200. [DOI] [PubMed] [Google Scholar]
- 15.Kicska GA, Tyler PC, Evans GB, Furneaux RH, Schramm VL, Kim K. Purine-less death in Plasmodium falciparum induced by immucillin-H, a transition state analogue of purine nucleoside phosphorylase. J Biol Chem. 2002;277:3226–31. doi: 10.1074/jbc.M105906200. [DOI] [PubMed] [Google Scholar]
- 16.Coomber DW, O’Sullivan WJ, Gero AM. Adenosine analogues as antimetabolites against Plasmodium falciparum malaria. Int J Parasitol. 1994;24:357–65. doi: 10.1016/0020-7519(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 17.Martin RE, Henry RI, Abbey JL, Clements JD, Kirk K. The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 2005;6:R26. doi: 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter NS, Ben Mamoun C, Liu W, et al. Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum. J Biol Chem. 2000;275:10683–91. doi: 10.1074/jbc.275.14.10683. [DOI] [PubMed] [Google Scholar]
- 19.Parker MD, Hyde RJ, Yao SY, et al. Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy. Biochem J. 2000;349:67–75. doi: 10.1042/0264-6021:3490067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rager N, Mamoun CB, Carter NS, Goldberg DE, Ullman B. Localization of the Plasmodium falciparum PfNT1 nucleoside transporter to the parasite plasma membrane. J Biol Chem. 2001;276:41095–9. doi: 10.1074/jbc.M107037200. [DOI] [PubMed] [Google Scholar]
- 21.Quashie NB, Dorin-Semblat D, Bray PG, et al. A comprehensive model of purine uptake by the malaria parasite Plasmodium falciparum: identification of four purine transport activities in intraerythrocytic parasites. Biochem J. 2008;411:287–95. doi: 10.1042/BJ20071460. [DOI] [PubMed] [Google Scholar]
- 22.Murkin AS, Tyler PC, Schramm VL. Transition-state interactions revealed in purine nucleoside phosphorylase by binding isotope effects. J Am Chem Soc. 2008;130:2166–7. doi: 10.1021/ja7104398. [DOI] [PubMed] [Google Scholar]
- 23.Ugarkar BG, DaRe JM, Kopcho JJ, et al. Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J Med Chem. 2000;43:2883–93. doi: 10.1021/jm000024g. [DOI] [PubMed] [Google Scholar]
- 24.Furneaux RH, Tyler PC. Improved syntheses of 3H,5H-Pyrrolo[3,2-d]pyrimidines. J Org Chem. 1999;64:8411–12. doi: 10.1021/jo990903e. [DOI] [PubMed] [Google Scholar]
- 25.Jespersen T, Grunnet M, Angelo K, Klaerke DA, Olesen SP. Dual-function vector for protein expression in both mammalian cells and Xenopus laevis oocytes. Biotechniques. 2002;32:536–40. doi: 10.2144/02323st05. [DOI] [PubMed] [Google Scholar]
- 26.Jansen M, Akabas MH. State-dependent cross-linking of the M2 and M3 segments: Functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J Neurosci. 2006;26:4492–99. doi: 10.1523/JNEUROSCI.0224-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clinch K, Evans GB, Frohlich RF, et al. Third-generation immucillins: Syntheses and bioactivities of acyclic immucillin inhibitors of human purine nucleoside phosphorylase. J Med Chem. 2009;52:1126–43. doi: 10.1021/jm801421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Dumont JN. Oogenesis in Xenopus laevis (Daudin). I. Stages of oocyte development in laboratory maintained animals. J Morphol. 1972;136:153–79. doi: 10.1002/jmor.1051360203. [DOI] [PubMed] [Google Scholar]
- 30.Kelly SM, Butler JP, Macklem PT. Control of cell volume in oocytes and eggs from Xenopus laevis. Comp Biochem Physiol A Physiol. 1995;111:681–91. doi: 10.1016/0300-9629(95)00046-a. [DOI] [PubMed] [Google Scholar]
- 31.Petrescu I, Lascu I, Goia I, et al. Phosphorylation and hydrolysis of 7-deazaadenine nucleotides by rat liver and beef heart mitochondria. Biochemistry. 1982;21:886–93. doi: 10.1021/bi00534a012. [DOI] [PubMed] [Google Scholar]
- 32.Sauve AA, Cahill SM, Zech SG, et al. Ionic states of substrates and transition state analogues at the catalytic sites of N-ribosyltransferases. Biochemistry. 2003;42:5694–705. doi: 10.1021/bi034003a. [DOI] [PubMed] [Google Scholar]
- 33.Werner A, Siems W, Schmidt H, et al. Determination of nucleotides, nucleosides and nucleobases in cells of different complexity by reversed-phase and ion-pair high-performance liquid chromatography. J Chromatogr. 1987;421:257–65. doi: 10.1016/0378-4347(87)80406-1. [DOI] [PubMed] [Google Scholar]
- 34.Desai SA, Bezrukov SM, Zimmerberg J. A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature. 2000;406:1001–5. doi: 10.1038/35023000. [DOI] [PubMed] [Google Scholar]
- 35.Huber SM, Duranton C, Lang F. Patch-clamp analysis of the “new permeability pathways” in malaria-infected erythrocytes. Int Rev Cytol. 2005;246:59–134. doi: 10.1016/S0074-7696(05)46003-9. [DOI] [PubMed] [Google Scholar]
- 36.Ting LM, Shi W, Lewandowicz A, et al. Targeting a novel Plasmodium falciparum purine recycling pathway with specific immucillins. J Biol Chem. 2005;280:9547–54. doi: 10.1074/jbc.M412693200. [DOI] [PubMed] [Google Scholar]
- 37.Downie MJ, Kirk K, Mamoun CB. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryot Cell. 2008;7:1231–7. doi: 10.1128/EC.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirk K, Howitt SM, Broer S, Saliba KJ, Downie MJ. Purine uptake in Plasmodium: transport versus metabolism. Trends Parasitol. 2009;25:246–9. doi: 10.1016/j.pt.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Queen SA, Vander Jagt D, Reyes P. Properties and substrate specificity of a purine phosphoribosyltransferase from the human malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol. 1988;30:123–33. doi: 10.1016/0166-6851(88)90105-3. [DOI] [PubMed] [Google Scholar]
- 40.Scott HV, Rieckmann KH, O’Sullivan WJ. Synergistic antimalarial activity of dapsone/dihydrofolate reductase inhibitors and the interaction of antifol, antipyrimidine and antipurine combinations against Plasmodium falciparum in vitro. Trans R Soc Trop Med Hyg. 1987;81:715–21. doi: 10.1016/0035-9203(87)90004-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.