

Abstract
We report on a patient with trisomy 21, microophthalmia, neonatal diabetes mellitus, hypopituitarism, and a complex structural brain anomaly who was a member of a large bilineal family with eye anomalies. The patient inherited a different mutation in PAX6 from each parent and is the only known living and second reported patient with compound heterozygosity for mutations in PAX6. PAX6 is a transcription factor involved in eye and brain development, and has roles in pancreatic and pituitary development. Clinical evaluation of the propositus and his parents demonstrated the effects of mutations of differing severity in multiple individuals.
Keywords: PAX6, Microophthalmia, Aniridia, Neonatal Diabetes Mellitus
Introduction
PAX6, located on chromosome 11p13, is a highly evolutionarily conserved transcription factor involved in ocular and neural development [Hanson et al., 1999; Simpson and Price, 2002]. Heterozygous deletions or loss-of-function mutations in PAX6 can result in ocular anomalies [Jordan et al., 1992; Robinson et al., 2008]. Truncating mutations in the open reading frame are more commonly associated with aniridia; missense mutations occur with variable eye anomalies including keratopathy, congenital optic nerve defects, cataracts, isolated foveal hypoplasia, iris hypoplasia and (rarely) microophthalmia [Tzoulaki et al., 2005].
PAX6 is expressed in the pancreas, and mutations in PAX6 may result in abnormal glucose metabolism and defective processing of proinsulin [Wen et al., 2009]. In the human embryo, PAX6 is expressed in the pituitary gland, and in mice, appears to play a role in pituitary development and function [Terzic and Saraga-Babic, 1999; Kioussi et al., 1999; Bentley et al., 1999]. Finally, heterozygous mutations in PAX6 may also result in auditory processing deficits related to corpus callosum anomalies [Bamiou et al., 2007].
One individual has previously been described with compound heterozygous nonsense mutations in PAX6 [Glaser et al., 1994]. That patient, who died at 1 week of age, had anophthalmia with fused eyelids, choanal atresia, microcephaly and a complex brain anomaly described as a large midline cavity with an absent corpus callosum, hypoplastic brainstem, near-absent olfactory bulbs, and polymicrogyria. Facial dysmorphisms included a small, malformed nose, a high-arched palate, and micrognathia. No other organ anomalies were noted on full autopsy, and endocrinologic abnormalities were not described.
We describe a 4-year-old male who additionally had trisomy 21 and who is the second reported patient (and the only to survive the neonatal period) with mutations in both PAX6 alleles. We also depict a 4-generation pedigree in which there was clinical and molecular evidence that numerous relatives were PAX6 mutation heterozygotes. This family provides evidence for the wide phenotypic spectrum associated with mutations in PAX6. The father's missense mutation resulted in much milder ophthalmologic anomalies than the mother's nonsense mutation, while the presence of both mutations in the propositus (and likely, his deceased brother) resulted in severe ophthalmologic, neurologic, and endocrinologic manifestations, the last of which has not previously been described in a human with two PAX6 mutations.
Methods
The patient and his parents participated in our comprehensive clinical study on holoprosencephaly and related neurological disorders at the National Human Genome Research Institute, National Institutes of Health. Appropriate consent was obtained for all participants, including for photo publication. Sequence analysis for SHH, ZIC2, SIX3, and TGIF, the 4 most common holoprosencephaly-associated genes, was performed by methods previously described [Roessler et al., 1996; Brown et al., 1998; Wallis et al., 1999; Gripp et al., 2000]. Sequence analysis of PAX6 was performed commercially (GeneDx, Gaithersburg, MD).
Clinical Report
When evaluated by us, the propositus was a 4-year-old Caucasian male with prenatally-diagnosed trisomy 21. He was initially diagnosed as having lobar holoprosencephaly. His complex medical history additionally included bilateral microophthalmia, choanal atresia, severe developmental delay, and renal dysplasia with recurrent urinary tract infections. Evidence for hypopituitarism included central hypothyroidism, secondary adrenal insufficiency, and a history of cryptorchidism and micropenis (now status-post testosterone treatment) making gonadotropin deficiency likely. He had neonatal-onset insulin-dependent diabetes mellitus which was very difficult to control, but abdominal MRI revealed no pancreatic anomalies.
Review of the propositus's brain MRI showed structural abnormalities not consistent with holoprosencephaly because of the lack of hemispheric fusion, but included multiple anomalies including agenesis of the corpus callosum, midline interhemispheric cyst, hypoplastic pons and vermis (absent inferiorly), possible Dandy-Walker malformation, dysplastic tectum, pituitary and hypothalamic hypoplasia, and a globular (though not fused) basal ganglia. The thalamic nuclei were well-separated by a large third ventricle. Microcephaly and asymmetric microophthalmia were also evident (Fig 1).
Figure 1.
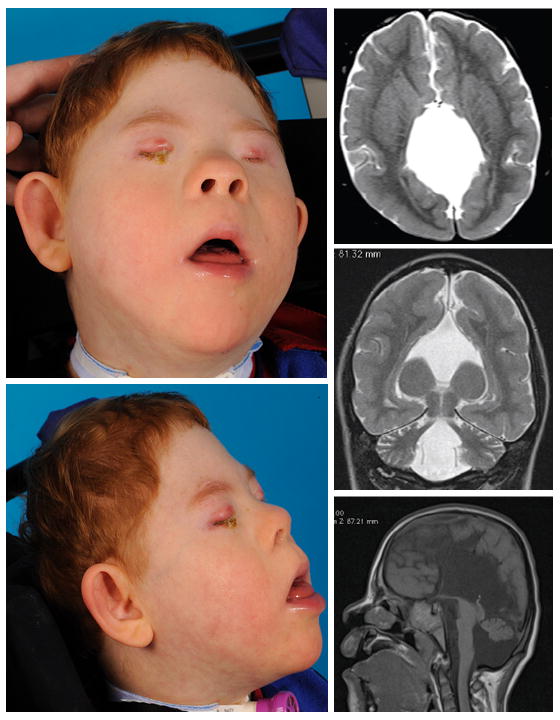
Propositus with compound heterozygosity for mutations in PAX6. Facial features were consistent with trisomy 21, and were also notable for extreme microcephaly, microophthalmia, and a smooth philtrum. Axial, coronal, and sagittal (from top) brain MRI demonstrates complex structural brain anomaly in propositus.
On physical examination, the child displayed physical features consistent with his diagnosis of trisomy 21, as well as bilateral severe microophthalmia, extreme microcephaly (head circumference 50th centile for a 1-month-old with Down syndrome), and a smooth philtrum (Fig 1).
The propositus's mother had a history of aniridia, but reported no other medical problems. She had an extensive family history of autosomal dominant aniridia, though no previous genetic study had been initiated (Fig 2). On physical examination, no extraocular anomalies were appreciated. Ophthalmological examination showed bilateral aniridia, glaucoma, and corneal opacifications, as well as a dense cataract in the right eye (Fig 3). While she had not been previously diagnosed with diabetes, she had an elevated fasting glucose on our evaluation.
Figure 2.
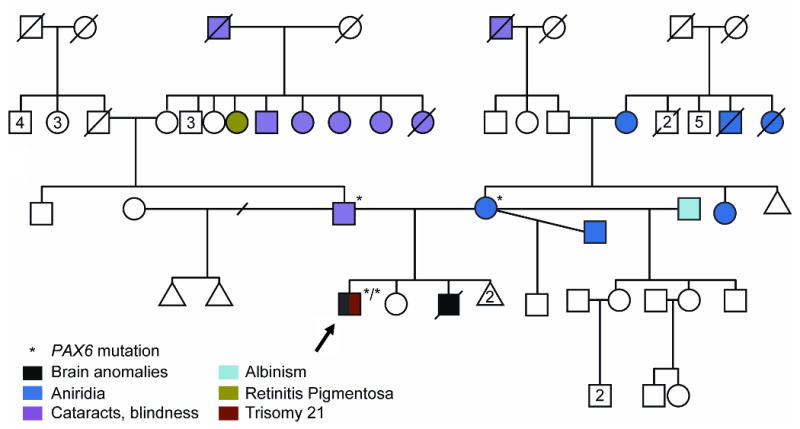
Family pedigree.
Figure 3.
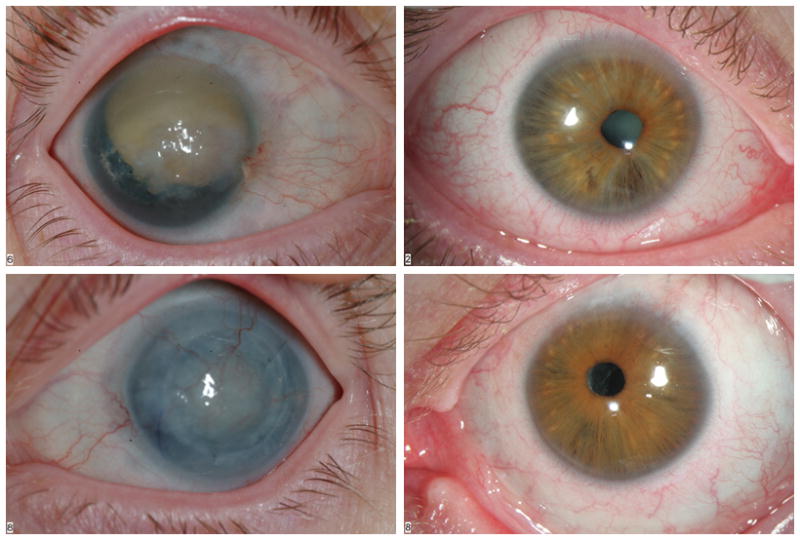
Photos of parents' ocular findings (mother on left, father on right).
The propositus's father reported a history of cataracts in early childhood and eventual blindness, as well as hearing loss. He also had an extensive family history of similar visual problems and hearing loss (Fig 2). On examination, he had a high palate and dental crowding in addition to ocular anomalies. Ophthalmological examination showed bilateral microcornea, a right eye cataract, and left aphakia (absent lens) (Fig 3). He also had subtle iris hyoplasia and corectopia.
The propositus's brother, on whom DNA was not available for testing, was described as having very similar structural brain anomalies to the propositus. He additionally had neonatal diabetes mellitus and anophthalmia, and died in infancy.
Molecular Testing
Sequence analysis for mutations in the four most common holoprosencephaly-associated genes (SHH, ZIC2, SIX3, and TGIF) was negative.
The patient's father had a nonconservative missense mutation (c.112C>T, resulting in p.R38W) in the paired box domain of PAX6. This mutation has previously been reported in a patient with microphthalmia and aniridia [Henderson et al., 2007]. This is a highly evolutionarily conserved residue which mediates sequence-specific DNA binding [Xu et al., 1999; Hanson et al., 1999].
The patient's mother had a nonsense mutation (c.718C>T, resulting in p.R240X) in the homeobox domain of PAX6. This mutation has previously been reported in a patient with aniridia. The premature insertion of a stop codon in the homeobox domain is predicted to result in nonsense-mediated decay and a consequently functionally null allele [Wilson et al., 1995; Tzoulaki et al., 2007].
The propositus inherited both the maternal and paternal mutations in PAX6.
Discussion
This is the second reported and the only known surviving patient with mutations in both PAX6 alleles. Analysis if the findings provides a unique example of the phenotypic effects of compound heterozygosity of mutations of PAX6. The propositus's hypopituitarism, diabetes mellitus, and brain and ophthalmologic anomalies can all be explained by the PAX6 mutations. The mutations also explain the ophthalmologic phenotype in his parents, and additionally may explain impaired glucose tolerance in the propositus's mother.
The previously reported patient, who had a nonsense mutation in each PAX6 allele, survived to the eighth day of life [Glaser et al., 1994]. The propositus's brother, who was described as having near-identical brain and ophthalmologic anomalies as the propositus, died in early infancy, and was not available for genetic testing. However, given the similar phenotypes and the 25% chance that the parents would give birth to a child with both mutations, it is likely that the deceased sibling inherited both mutant alleles. The morbidity associated with inheriting two mutations and the fact that the propositus additionally had trisomy 21 makes his survival and relative good health an especially rare event.
Finally, this case highlights the importance of a multidisciplinary approach which includes the evaluation of multiple family members in the diagnosis of unusual and complex patients.
Acknowledgments
We are extremely grateful to the family presented in this report. This research was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
References
- Bamiou DE, Free SL, Sisodiya SM, Chong WK, Musiek F, Williamson KA, van Heyningen V, Moore AT, Gadian D, Luxon LM. Auditory interhemispheric transfer deficits, hearing difficulties, and brain magnetic resonance imaging abnormalities in children with congenital aniridia due to PAX6 mutations. Arch Pediatr Adolesc Med. 2007;161:463–469. doi: 10.1001/archpedi.161.5.463. [DOI] [PubMed] [Google Scholar]
- Bentley CA, Zidehsarai MP, Grindley JC, Parlow AF, Barth-Hall S, Roberts VJ. Pax6 is implicated in murine pituitary endocrine function. Endocrine. 1999;10:171–177. doi: 10.1385/ENDO:10:2:171. [DOI] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994;7:463–471. doi: 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massagué J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- Hanson I, Churchill A, Love J, Axton R, Moore T, Clarke M, Meire F, van Heyningen V. Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet. 1999;8:165–172. doi: 10.1093/hmg/8.2.165. [DOI] [PubMed] [Google Scholar]
- Henderson RA, Williamson K, Cumming S, Clarke MP, Lynch SA, Hanson IM, FitzPatrick DR, Sisodiya S, van Heyningen V. Inherited PAX6, NF1 and OTX2 mutations in a child with microphthalmia and aniridia. Eur J Hum Genet. 2007;15:898–901. doi: 10.1038/sj.ejhg.5201826. [DOI] [PubMed] [Google Scholar]
- Jordan T, Hanson I, Zaletayev D, Hodgson S, Prosser J, Seawright A, Hastie N, van Heyningen V. The human PAX6 gene is mutated in two patients with aniridia. Nat Genet. 1992;1:328–332. doi: 10.1038/ng0892-328. [DOI] [PubMed] [Google Scholar]
- Kioussi C, O'Connell S, St-Onge L, Treier M, Gleiberman AS, Gruss P, Rosenfeld MG. Pax6 is essential for establishing ventral-dorsal cell boundaries in pituitary gland development. Proc Natl Acad Sci U S A. 1999;96:14378–14382. doi: 10.1073/pnas.96.25.14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DO, Howarth RJ, Williamson KA, van Heyningen V, Beal SJ, Crolla JA. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am J Med Genet Part A. 2008;146A:558–569. doi: 10.1002/ajmg.a.32209. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Simpson TI, Price DJ. Pax6; a pleiotropic player in development. Bioessays. 2002;24:1041–1051. doi: 10.1002/bies.10174. [DOI] [PubMed] [Google Scholar]
- Terzić J, Saraga-Babić M. Expression pattern of PAX3 and PAX6 genes during human embryogenesis. Int J Dev Biol. 1999;43:501–508. [PubMed] [Google Scholar]
- Tzoulaki I, White IM, Hanson IM. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 2005;6:27. doi: 10.1186/1471-2156-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- Wen JH, Chen YY, Song SJ, Ding J, Gao Y, Hu QK, Feng RP, Liu YZ, Ren GC, Zhang CY, Hong TP, Gao X, Li LS. Paired box 6 (PAX6) regulates glucose metabolism via proinsulin processing mediated by prohormone convertase 1/3 (PC1/3) Diabetologia. 2009;52:504–513. doi: 10.1007/s00125-008-1210-x. [DOI] [PubMed] [Google Scholar]
- Wilson DS, Guenther B, Desplan C, Kuriyan J. High resolution crystal structure of a paired (Pax) class cooperative homeodomain dimer on DNA. Cell. 1995;82:709–719. doi: 10.1016/0092-8674(95)90468-9. [DOI] [PubMed] [Google Scholar]
- Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999;13:1263–1275. doi: 10.1101/gad.13.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]