

Abstract
Insulin coordinately up-regulates lipogenic gene transcription via induction of sterol regulatory element binding protein-1c (SREBP-1c). Conversely, polyunsaturated fatty acids (PUFA) decrease lipogenic gene transcription via suppression of SREBP-1c. We therefore examined the ability of n-3 PUFA to mitigate induction of SREBP-1c and its downstream lipogenic targets by insulin in primary rat hepatocyte cultures. Insulin induced expression of SREBP-1c mRNA 5–6 fold as well as rat SREBP-1c promoter activity. These effects were prevented by the n-3 fatty acids eicosapentaenoic acid (20:5 n-3; EPA) and docosahexaenoic acid (22:6 n-3, DHA), but not by the monounsaturated fatty acid oleic acid (18:1 n-6, OLA). N-3 fatty acids also effectively prevented insulin induction of the downstream lipogenic enzyme targets fatty acid synthase (FAS) and acetyl carboxyl coenzyme acetyltransferase-1 (ACC-1), and reduced de novo lipogenesis. The SREBP-1c promoter contains an insulin response unit consisting of tandem LXRα response elements (LXREs) as well as sites for NF-Y, Sp1, and SREBP-1c itself. The LXREs were identified as a primary site mediating suppression of SREBP-1c transcription by n-3 PUFA. DHA effectively prevented LXRα-dependent activation of both the wild type SREBP-1c promoter and the synthetic LXRE-driven promoter, and significantly blunted LXRα-dependent activation of a Gal4-LXRα chimeric protein thus demonstrating that n-3 PUFA effectively mitigate induction of SREBP-1c by insulin via reduced trans-activation of LXRα.
Keywords: Sterol response element binding protein-1c, insulin, polyunsaturated fatty acids, docosahexaenoic acid, eicosapentaenoic acid, liver x receptor alpha
INTRODUCTION
A major metabolic action of insulin in the liver is to increase de novo lipogenesis (DNL), which in turn increases the pool of fatty acids available for incorporation into triglyceride (TG). In the liver, DNL is governed by the expression and activity of the lipogenic enzymes, fatty acid synthase (FAS) and acetyl carboxyl coenzyme acetyltransferase-1 (ACC-1) [1]. Expression of these enzymes is controlled by the major lipogenic regulator, SREBP-1c [2–4]. Therefore, perturbation of SREBP-1c expression has a direct effect on DNL and subsequent hepatic triglyceride production [5, 6].
SREBP-1c is a member of the sterol response element binding protein (SREBP) family of transcription factors that are basic helix-loop-helix leucine zipper transcription factors [7]. There are three members of the SREBP family of transcription factors, SREBP-1c, 1a, and SREBP-2. SREBP-1c and 1a are encoded by the same gene and differ only in the composition of their first exons [8]. SREBP-1c is the major isoform in the liver and adipose tissue and its expression is positively and negatively regulated at the transcriptional and posttranslational levels by insulin and glucagon, respectively [9–12].
In hyperinsulinemic states, including obesity and type II diabetes mellitus, dysregulation of SREBP-1c results in hepatic overproduction of fatty acid and triglyceride with resulting dyslipidemia and atherogenic lipid profile [13, 14]. Previous studies from our laboratory have demonstrated that SREBP-1c expression is increased in livers of the JCR:LA-cp rat, a model of obesity and hyperinsulinemia, and that this up-regulation is associated with increased production of very-low-density-lipoprotein (VLDL) and TG [15]. In a murine model of obesity, the leptin receptor deficient ob/ob mouse, hepatic steatosis or “fatty liver” is ameliorated by the absence of SREBP-1c [16]. N-3 PUFA have been shown to reduce lipogenic enzyme expression via down-regulation of SREBP-1c in rodent hyperinsulinemic models [17–19]. These data form the basis for use of n-3 PUFA supplements in treating hypertriglyceridemia associated with human states of hyperinsulinemia, such as type 2 diabetes mellitus [20, 21].
The molecular mechanisms by which n-3 PUFA mitigate insulin induction of SREBP-1c are not fully understood. Although n-3 (and other) PUFA appear to regulate SREBP-1c via both transcriptional and posttranscriptional mechanisms [22–25], in the present studies, we focused on defining the molecular mechanisms by which n-3 PUFA mitigate transcriptional up-regulation of SREBP-1c by insulin [18, 22, 26–29].
Some but not all previous studies suggested that suppression of SREBP-1c transcription by n-3 PUFA was LXRα dependent, however, the mechanism of PUFA repression of insulin induction of SREBP-1c promoter activity has not been investigated. In this regard, we have previously shown that activation of SREBP-1c transcription by insulin is mediated by a multi-component insulin response element including binding sites for not only LXRa but also NF-Y, Sp1, and SREBP-1c itself [30]. With regards to PUFA repression of SREBP-1c expression, in general, prior studies point to both LXRα [25, 31] and SRE [28, 32] response elements as mediating the suppressive effect of PUFA on the SREBP-1c promoter. When parallels are made between transcriptional control of SREBP-1c and FAS, whose promoter contains an insulin responsive region that is similar to that of SREBP-1c insofar as it contains SREBP-1c, NF-Y, and Sp1 binding sites, and PUFA repression of FAS transcription appears to involve both SREBP-1c and NF-Y binding [33, 34]. Thus, the possibility arises that PUFA may repress insulin-induced SREBP-1c expression through LXRα, SREBP-1c, or NF-Y dependent mechanisms. Therefore we conducted a systematic investigation of the molecular mechanisms by which PUFA repress insulin-induced SREBP-1c transcription.
The present work was designed to determine the molecular mechanisms through which n-3 PUFAs decrease insulin-induced expression of SREBP-1c and DNL in the liver. We examined whether the n-3 PUFAs, EPA and DHA, are equally effective at decreasing insulin-induced expression of SREBP-1c and its downstream lipogenic targets, and if this inhibition involves transcriptional regulation. We identified the LXRα response elements within the SREBP-1c promoter as the major mediators of PUFA suppression of insulin-induced SREBP-1c transcription and showed that the effect of n-3 PUFA is mediated through decreased trans-activating potential of LXRα
MATERIALS AND METHODS
Fatty acid preparation
Oleic acid (OLA), EPA, and DHA were purchased (Nu-Chek Prep) and stored at −20°C as either 25 or 50 mg/ml ethanol stock solutions. Fatty acid/BSA complexes were made by conjugating the fatty acid sodium salt to delipidated bovine serum albumin (BSA, Sigma-Aldrich) to make a working concentration of 5 mM fatty acid prior to use. The final molar FA/BSA ratio was 4.3:1 for each fatty acid.
Primary hepatocyte isolation and culture
Rat primary hepatocytes were isolated according to previously reported methods [15, 35]. Isolated hepatocytes were plated on rat tail collagen (BD Biosciences) coated tissue culture plates (6-well plates for luciferase assays and 6-cm plates for RNA isolation and DNL assays) at a density of 1 × 106 cells per well of the 6-well plate and 3 × 106 cells per 6-cm plate. Hepatocytes were seeded in RPMI-1640 media (Lonza) containing 10% FBS (Sigma), glucose (20 mM), insulin (100 nM), penicillin, streptomycin, and fungizone for 3–4 hours to allow for plate attachment. Following seeding, adherent cells were washed with PBS and subjected to experimental treatments.
For real-time PCR and DNL assays, cells were incubated overnight in serum-free RPMI-1640 (Lonza) prior to incubation for 24 hours with delipidated BSA (0.15%; Sigma), OLA (100 μM), EPA (100 μM), or DHA (100 μM) in the presence or absence of insulin (100 nM; Gibco). At the end of 24 hours, cells were either harvested for RNA isolation or subjected to lipid synthesis studies.
Real-time PCR
Hepatocytes were isolated and treated as described above. At the end of the 24 hour treatment, cultures were harvested, total RNA isolated with the RNeasy kit (Qiagen), and cDNA synthesized (SuperScript First Strand Synthesis, Invitrogen) from 1 μg of total RNA. Primers for mRNA detection were designed with Universal Probe Library (Roche). Amplification of target cDNA was detect by Sybr Green and analyzed by the ΔΔCt method. Cyclophilin D was used as the reference gene.
2-14C acetate incorporation into hepatic lipids
Hepatocytes were cultured and treated as described above. At the end of 24 hours treatment, cells were washed and pulsed with 2-14C acetate (4 μCi) for three hours. Following three hours of incubation with 2-14C acetate, media was removed, cells washed twice with cold PBS, and then harvested in cold PBS. Individual hepatic lipid fractions were isolated by thin layer chromatography (TLC) and incorporation of 2-14C acetate was measured as previously described [15].
Luciferase promoter reporter assays
Rat primary hepatocytes were transfected overnight in serum free RPMI-1640 media using Lipofectin (Invitrogen) following seeding. Cells were transfected with a luciferase reporter vector (pGL3 basic, Promega) containing the full-length, 1.5 kb rat SREBP-1c promoter (1.5 kb SREBP-1c Luc) [9], the full-length rat SREBP-1c promoter with mutated binding sites for either NF-Y, LXRα, or SREBP-1c [30], or three LXRα response elements (pLXREx3) [36] and a Renilla control vector (pRL-null; Promega) for normalization. Following transfection, cells transfected with 1.5 kb SREBP-1c - Luc were treated with delipidated BSA (0.15%), OLA (100 μM), EPA (100 μM) or DHA (100 μM) in the presence or absence of insulin (100 nM) in serum free RPMI-1640 media (Lonza) for 24 hours. Cells transfected with the mutated full-length SREBP-1c promoter were treated with either BSA (0.15%) or DHA (100 μM) in the presence of insulin (100 nM) for 24 hours. Cells transfected with pLXREx3 - Luc were treated with either BSA (0.15%) or DHA (100 μM) in the presence or absence of the LXRα agonist, T0901317 (5 μM; Sigma). At the end of 24 hours, cells were lysed and both firefly luminescence and renilla luminescence were measured (Dual-Glo Luciferase Reporter Assay, Promega). Firefly luminescence was normalized to renilla luminescence and values expressed as the ratio of treatment to BSA + vehicle control value.
Gal4-LXRα luciferase assays
Rat primary hepatocytes were transfected overnight in serum free medium with either a plasmid containing a Gal4 DNA binding domain- LXRα ligand binding domain chimeric protein (generous gift from Dr. Terry Unterman), a luciferase reporter plasmid (pGL3 basic; Promega) containing five Gal4 binding sites, and a Renilla control vector (pRL-null; Promega) for normalization. Following transfection, cells were treated with BSA (0.15%) or DHA (100 μM) in the presence of T0901317 (0, 1, 5, or 10 μM) in serum free RPMI-1640 for 24 hours. At the end of 24 hours, cells were lysed and both firefly luciferase and renilla luminescence were measured (Dual-Glo Luciferase Reporter Assay, Promega). Luciferase luminescence was normalized to renilla luminescence and values expressed as the ratio of treatment to BSA + vehicle value.
RESULTS
Effect of PUFA on expression of SREBP-1c and its downstream lipogenic target genes
In order to assess the impact of n-3 PUFA exposure on induction of lipogenic enzymes by insulin in vitro, the effect of EPA and DHA on induction of SREBP-1c expression and SREBP-1c target genes, FAS and ACC-1, was assessed by real-time PCR in rat primary hepatocyte cultures. To determine that the suppression of lipogenic enzymes is an effect of n-3 PUFA, the monounsaturated fatty acid, OLA, was included as a control. Insulin treatment of rat primary hepatocytes resulted in a 5.5 fold increase in SREBP-1c mRNA compared to control (Figure 1A). While co-treatment with OLA (100 μM) had no significant effect on expression co-treatment with EPA (100 μM) or DHA (100 μM) significantly reduced basal and insulin-induced SREBP-1c expression.
Figure 1.
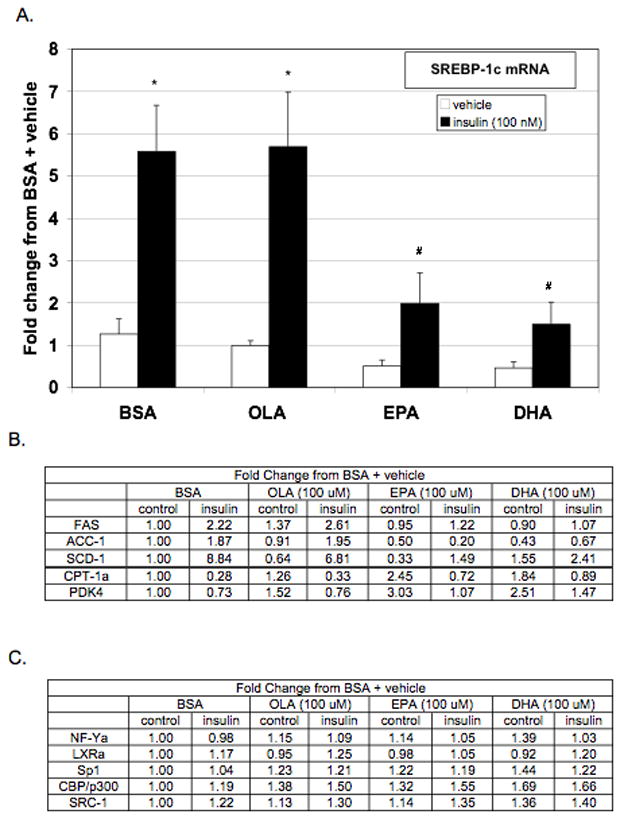
Real-time PCR analysis of hepatic lipogenic gene expression. Rat primary hepatocytes were incubated with or without insulin (100 nM) in the presence or absence of BSA (0.15%), OLA (100 μM), EPA (100 μM), or DHA (100 μM). After 24 hours of treatment, cells were lysed and total RNA isolated. (A.) Expression of SREBP-1c, (B.) SREBP-1c target genes, (C.) and known transactivators of SREBP-1c were assessed by real-time PCR. Values expressed as the fold change from vehicle + BSA control and are the mean of at least four hepatocyte preparations (n ≥ 4). * P ≤ 0.05 vs. vehicle + BSA, # P ≤ 0.05 vs. insulin + BSA.
Analysis of SREBP-1c target genes, FAS, ACC-1, and SCD-1 revealed a similar effect on expression by EPA and DHA (Figure 1B). Co-treatment with either EPA or DHA decreased insulin induction of SREBP-1c target genes in primary hepatocytes. To determine if the reductions in SREBP-1c, FAS, ACC, and SCD-1 were due to a nonspecific reduction in cellular transcription, expression of the carnitine palmitoyltransferase-1 alpha (CPT-1α) and pyruvate dehydrogenase kinase 4 (PDK4) were assessed. CPT-1a transfers the fatty acyl group from CoA to carnitine for translocation across the mitochondrial membrane. It is a rate-controlling step in the beta-oxidation of long chain fatty acid [37]. PDK4 regulates the activity of the pyruvate dehydrogenase complex (PDC), which catalyzes the formation of acetyl-CoA from pyruvate [38]. Insulin decreases the expression of these genes and n-3 PUFA increases their expression [39–41]. Indeed, insulin decreased and n-3 PUFAs increased the expression of both CPT-1α and PDK4 indicating that the n-3 PUFA repression of lipogenic genes is not due to nonspecific transcriptional repression.
To determine if n-3 PUFAs blunted SREBP-1c expression by reducing the expression of known transactivators, mRNA levels of NF-Y, LXRα, Sp1, CBP/p300, and SRC-1 were measured by real-time PCR (Figure 1C). Insulin treatment did not increase the expression of these transactivators and n-3 PUFAs did not decrease their expression. Therefore, altered expression of coactivators for SREBP-1c does not appear to mediate n-3 PUFA repression although PUFA could alter protein levels of these transactivators and/or their trans-activating capacity.
PUFA mediated repression of insulin-induced DNL
To determine whether the observed decrease in expression of lipogenic genes by PUFA was physiologically relevant, the effect of PUFAs on insulin-induced hepatic DNL was assessed in rat primary hepatocytes by measuring 2-14C acetate incorporation into individual hepatic lipid fractions. Incorporation of labeled acetate into the TG fraction of cellular lipids was an indicator of DNL (Figure 2A) and the incorporation into all lipid fractions, TG, phospholipids (PL), and free fatty acid (FFA) was measured as an indicator of total cellular lipid content (Figure 2B). Insulin treatment resulted in a significant increase of 2-14C acetate into the TG portion of the hepatic lipid fraction and an increased incorporation into total cellular lipids. Treatment with the monounsaturated fatty acid, OLA, had little effect on insulin-induced incorporation of labeled acetate into TG or total cellular lipids. However, EPA and DHA significantly decreased insulin-induced 2-14C acetate incorporation into the hepatic TG fractions as well as into total cellular lipids. Therefore, these data demonstrate that EPA and DHA can significantly reduce hepatic DNL associated with hyperinsulinemic states.
Figure 2.
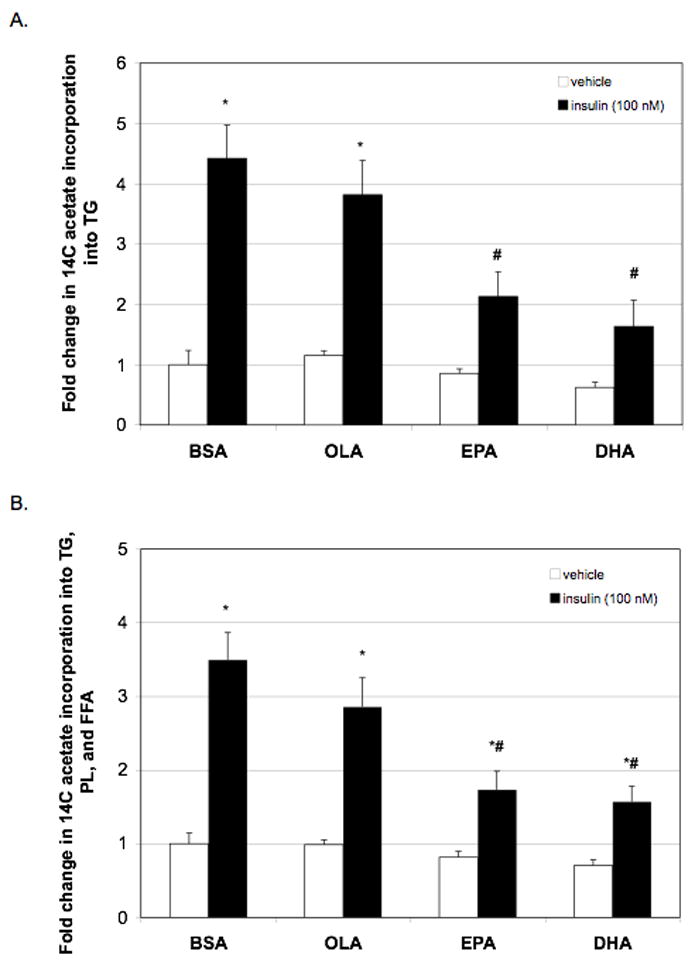
Effect of PUFA on lipid metabolism as determined by 2-14C acetate into TG and total cellular lipids. Rat primary hepatocytes were incubated with or without insulin (100 nM) in the presence or absence of BSA (0.15%), OLA (100 μM), EPA (100 μM), or DHA (100 μM). After 24 hours of treatment, cells were pulsed with 2-14C acetate (4 μCi) and 1 mM unlabelled acetate for 3 hours to allow for incorporation into cellular lipid. (A.) Hepatocyte lipids were subjected to methanol:chloroform extraction followed by separation by TLC. Individual lipid fraction bands were collected and counted. Incorporation into TG is shown as an indicator of DNL. (B.) Incorporation of 2-14C acetate into TG, PL, and FFA were added together as an indicator of effect of PUFA on incorporation into total cellular lipids. Data are expressed the fold change (disintegrations per minute normalized to protein) from BSA + vehicle and are the mean ± SEM of 4 hepatocyte preparations (n ≥ 4). * P ≤ 0.05 vs. vehicle + BSA, # P ≤ 0.05 vs. insulin + BSA
Effect of PUFA on transcriptional activity of the rat SREBP-1c promoter
To assess whether PUFA repression of insulin-induced SREBP-1c expression is due to decreased transcription, rat SREBP-1c promoter activity was assessed using luciferase reporter assays. The full-length rat SREBP-1c promoter (1.5 kb SREBP-1c – Luc) was transfected into primary rat hepatocytes treated with insulin and various fatty acids (Figure 3). Insulin stimulated promoter activity by approximately 3.5 fold. Co-treatment with OLA had no significant effect on insulin-induced promoter activity. However, co-treatment with either EPA or DHA significantly reduced insulin-induced SREBP-1c promoter activity. Therefore, the observed effect of PUFA on SREBP-1c mRNA can be attributed to decreased gene transcription.
Figure 3.
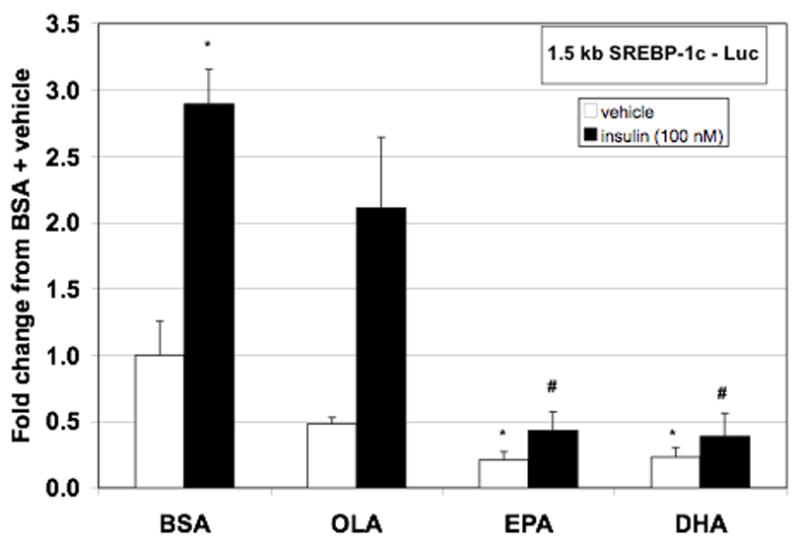
EPA and DHA effectively attenuate insulin induction of 1.5 kb SREBP-1c - Luc. Hepatocytes were transfected with both full-length rat SREBP-1c promoter luciferase construct and renilla control. Following transfection, cells were incubated with or without insulin (100 nM) in the presence or absence of BSA (0.15%), OLA (100 μM), EPA (100 μM), or DHA (100 μM). After 24 hours of treatment, cells were lysed, luciferase activity measured and normalized to renilla luminescence. Data are expressed as the fold change in normalized luciferase from vehicle + BSA control and are the mean of at least five hepatocyte preparations (n ≥ 5). * P ≤ 0.05 vs. vehicle + BSA, # P ≤ 0.05 vs. insulin + BSA
Identification of the PUFA responsive site in the SREBP-1c promoter
The insulin response unit of the rat SREBP-1c promoter consists of binding sites for Sp1, LXRα NF-Y, and SREBP-1. In order to identify the site or sites that mediate n-3 PUFA repression of SREBP-1c promoter activity, the response elements were disrupted by site-directed mutagenesis and DHA mediated repression of SREBP-1c promoter activity in the presence of insulin treatment was assessed (Figure 4). Treatment with DHA strongly inhibited induction of the wild-type full-length SREBP-1c promoter construct. Mutation of the SRE or NF-Y binding sites did not alter DHA repression of SREBP-1c promoter activity. However, mutation of both LXREs in the SREBP-1c promoter resulted in a significant loss of DHA mediated repression of SREBP-1c promoter activity. These data implicate alterations in LXRα signaling as the mechanism through which n-3 PUFA decreases insulin-induced promoter activity.
Figure 4.
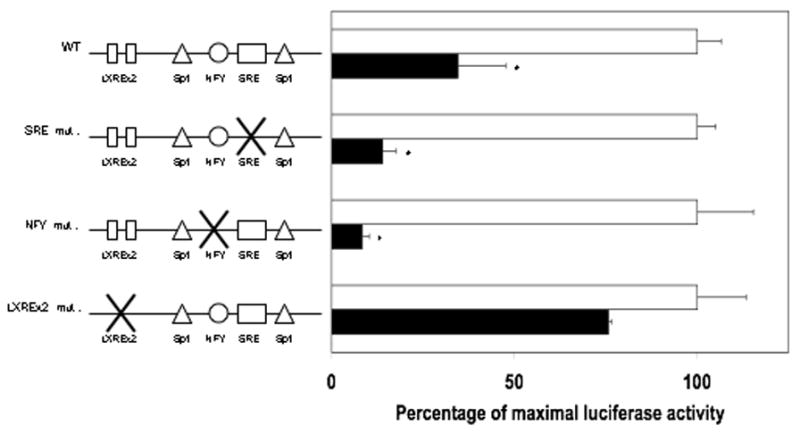
DHA repression of 1.5 kb SREBP-1c – Luc activity is attenuated by LXRE mutation. Hepatocytes were transfected with full-length wild-type (WT), SRE mutant, NFY mutant, or LXREx2 mutant rat SREBP-1c promoter luciferase constructs and null renilla control. Following transfection, cells were incubated with insulin (100 nM) in the presence or absence of BSA (0.15%) or DHA (100 μM). After 24 hours of treatment, cells were lysed, luciferase activity measured and normalized to renilla luminescence. Maximal insulin-induced activity in the presence of BSA for each construct is expressed as 100% activity. DHA mediated repression is expressed as the percentage of the corresponding maximal insulin induction. Data are the mean of at least four hepatocyte preparations (n ≥ 4). * P ≤ 0.05 vs. insulin + BSA for each construct
Repression of LXRα agonist-activated promoter activity
The LXRE’s within the rat SREBP-1c promoter appear to mediate DHA repression of insulin-induced SREBP-1c transcription. Therefore, we assessed the ability of DHA to prevent activation of the SREBP-1c promoter by the LXRα agonist, T0901317 (Figure 5A). T0901317 (5 μM) increased SREBP-1c promoter activity approximately 11-fold compared to vehicle + BSA, but this increased activity was blunted by co-treatment with DHA. In order to determine if DHA will decrease the activity of the LXRα/RXR transcriptional complex, we utilized a luciferase reporter construct that was driven by three LXRα response elements (Figure 5B). As with the full length SREBP-1c promoter, T0901317 increased the activity of the pLXREx3 - Luc reporter approximately 12 fold. This activity was decreased by co-treatment with DHA. These data indicate that DHA repression of the T0901317-driven full-length SREBP-1c promoter activity can be attributed to DHA repression of LXRα/RXR heterodimer transcriptional activity.
Figure 5.
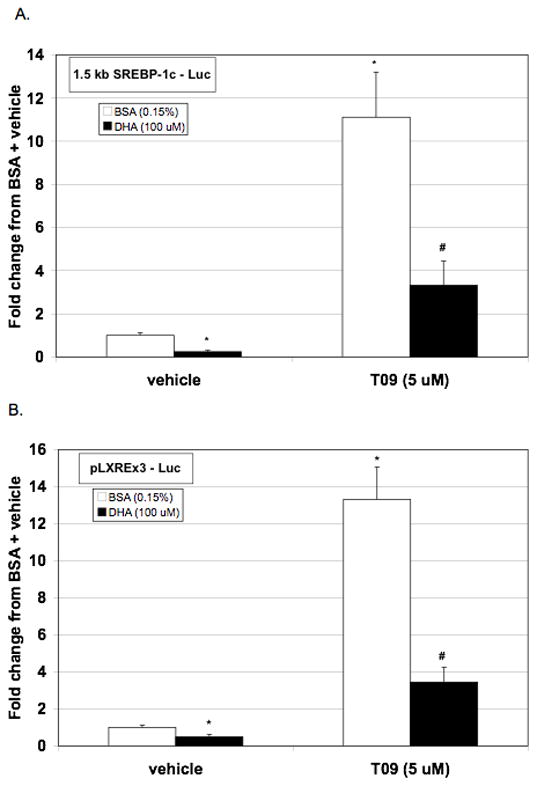
DHA attenuates LXR-agonist induced activation of 1.5 kb SREBP-1c – Luc and of pLXREx3 - Luc. Hepatocytes were transfected with either (A.) 1.5 kb SREBP-1c – Luc or (B.) pLXREx3 - Luc and null renilla control. Following transfection, cells were incubated with or without T0901317 (5 μM) in the presence or absence of BSA (0.15%) or DHA (100 μM). After 24 hours of treatment, cells were lysed, luciferase activity measured and normalized to renilla luminescence. Data are expressed as the fold change in normalized luciferase from BSA + vehicle control and are the mean of at least four hepatocyte preparations (n ≥ 4). * P ≤ 0.05 vs. vehicle + BSA, # P ≤ 0.05 vs. T0901317 + BSA
Antagonism of LXRα trans-activating properties
In order to determine the effect of PUFA on ligand-induced trans-activating potential of LXRα independent of the formation of the LXRα/RXR transcriptional complex, we utilized a Gal4-LXRα construct consisting of the DNA binding domain of Gal4 fused to the ligand binding domain/activation domain of LXRα (Figure 6). Treatment with T0901317 resulted in a concentration-related increase in Gal4-LXRα activity with maximal activity at the highest concentration of T0901317 (10 μM). Addition of DHA significantly reduced Gal4-LXRα activity at each concentration of T0901317. Maximal T0901317-induced Gal4-LXRα activity in the presence of DHA was approximately half of that observed without DHA. Therefore, DHA appears to repress LXRα agonist induced trans-activation.
Figure 6.
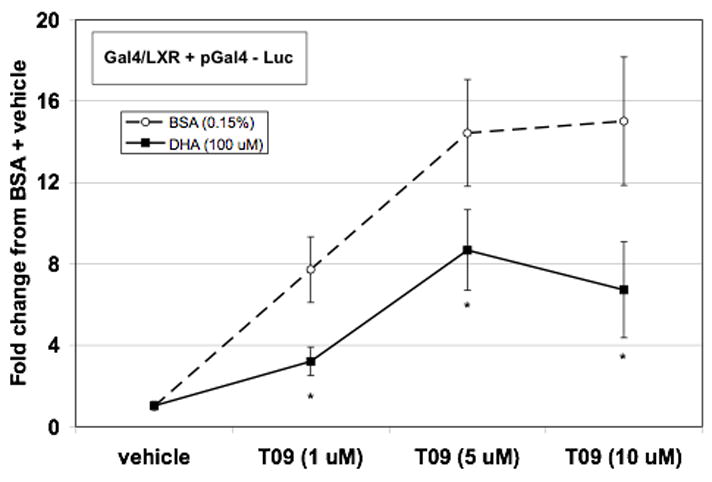
DHA effectively reduces transactivating capacity of LXRα. Hepatocytes were transfected with plasmids expressing Gal4-LXRα, Gal4 promoter luciferase construct, and null renilla control. Following transfection, cells were incubated with T0901317 (0,1, 5, or 10 μM) in the presence or absence of BSA (0.15%) or DHA (100 μM). After 24 hours of treatment, cells were lysed, luciferase activity measured and normalized to renilla luminescence. Data are expressed as the fold change in normalized luciferase from BSA + vehicle control and are the mean of at least five hepatocyte preparations (n ≥ 5). * P ≤ 0.05 vs. T0901317 + BSA
DISSCUSSION
Administration of fish oils rich in PUFAs either alone or as an adjunct therapy with statin administration is an effective therapy for dyslipidemia associated with type 2 diabetes [21, 42]. EPA and DHA are the major PUFAs contained in fish oils. Therefore, in the present work we determined the molecular mechanisms through which the dietary n-3 PUFAs, EPA and DHA, decrease insulin-induced expression of lipogenic genes as well as DNL in the liver using rat primary hepatocytes as a model. We demonstrated that insulin-induced expression of FAS, ACC-1, and SCD-1 as well as the master regulator of hepatic lipogenic genes, SREBP-1c, are significantly decreased following treatment with either EPA or DHA in vitro. This decrease in mRNA of lipogenic genes is coupled with a reduction in DNL as indicated by reduced incorporation of 2-14C acetate into the TG fraction of hepatocyte lipids. These data are in agreement with in vivo n-3 PUFA treatment studies previously reported by our lab demonstrating that menhaden oil feeding significantly decreases hepatic DNL as indicated by 3H water incorporation into hepatic and plasma TG fractions of the corpulent, hyperinsulinemic JCR:LA-cp rat as well as decreases in SREBP-1c and FAS expression [17]. Taken together, these data demonstrate PUFA mediated reductions in DNL during hyperinsulinemic states is a mechanism by which PUFA may decrease dyslipidemia and hepatic steatosis in pathological conditions such as type 2 diabetes and metabolic syndrome [43].
SREBP-1c has been identified as mediating the coordinate down-regulation of lipogenic enzyme transcription by PUFA [18, 29]. N-3 PUFA exert their effects on SREBP-1c via both transcriptional and posttranscriptional mechanisms. Regarding mRNA stability, n-6 and n-3 PUFA accelerate decay of the SREBP-1c transcript in rat hepatocytes [24]. Although initial studies primarily identified posttranscriptional mechanisms as mediating down-regulation of SREBP-1c by PUFA [29], subsequent work has demonstrated a transcriptional component to the effect of PUFA as well [9, 25, 31]. In light of these findings and given the central role of SREBP-1c in induction of lipogenesis in hyperinsulinemic states, we examined the mechanisms by which PUFA exert their negative influence on SREBP-1c transcription. We sought to identify the proteins involved in the n-3 PUFA (EPA and DHA) inhibition of SREBP-1c in rat primary hepatocyte cultures. We employed a strategy of site-directed mutagenesis of critical elements of the insulin response unit of the SREBP-1c promoter along with use of synthetic promoter constructs to identify the LXRα response elements of the promoter as the major site of the inhibitory effect of PUFA. Our evidence indicates that this effect is mediated through reduced trans-activating capacity of LXRα.
Insulin-induced transcription of SREBP-1c is achieved through the combinatorial actions of LXRα, NF-Y, Sp1, and SREBP-1c itself [30]. Prior studies have identified the LXRα response elements of the SREBP-1c promoter as potential sites for PUFA repression of unstimulated promoter activity [25, 31]. Studies of PUFA repression of transcription in other promoter contexts also identified the NF-Y and SRE binding sites as potential mediators of inhibitory effects of PUFA [28, 33]. In this context, we examined the potential role of the LXRα, NF-Y, and SREBP responsive elements in repression of insulin-induced SREBP-1c transcription by the n-3 PUFA EPA and DHA. In our studies, loss of DHA-mediated repression was specifically observed in a SREBP-1c promoter construct containing mutated LXRα response elements but not in those containing mutant NF-Y and SRE sites. Thus, alterations in LXRα signaling are implicated in DHA repression of insulin-induced SREBP-1c transcription.
Based on these findings, we next examined the effect of DHA to inhibit activation of the SREBP-1c promoter by the selective LXRα agonist T091317. We also examined the effect of DHA to inhibit activation of a synthetic promoter construct consisting of three LXRα response elements in a luciferase reporter vector (pLXREx3). The n-3 PUFA DHA significantly blunted T0901317 induction of both the full-length rat SREBP-1c promoter and the pLXREx3 construct, providing further evidence that PUFA exert their effect via attenuation of LXRα These data corroborate previous findings of antagonism by PUFA of induction of SREBP-1c expression and promoter activity in response to overexpression or activation of LXRα in immortalized cell lines [25, 31]
Our finding that DHA reduces the trans-activating capacity of a chimeric protein containing LXRα ligand binding and trans-activation domains linked to a Gal4 DNA binding domain indicate that n-3 PUFA likely reduces SREBP-1c transcription via reduced trans-activation of LXRα. The underlying molecular mechanisms mediating this effect of PUFA remain incompletely defined. LXRα is a ligand-activated nuclear receptor and requires binding of endogenous sterols and/or fatty acids to become transcriptionally active. Using a cell-free fluorescence polarization assay, Ou et al. determined that the n-6 PUFA arachidonic acid (AA) competitively antagonizes 24(S),25-epoxycholesterol mediated activation of LXRα as well as showing a reduction in T0901317-induced Gal4-LXRα activity in HEK293 cells [31]. Competitive antagonism of a synthetic LXRE-driven reporter construct by EPA in HEK293 cells has also been reported [25]. Thus it appears likely that PUFA reduce transcription of SREBP-1c at least in part by reducing the trans-activating capacity of LXRα. On the other hand, Pawar et al. found, in rat primary hepatocytes, that higher concentrations of n-3 fatty acid (EPA) than those used in the current study (0.5 vs. 0.1 mM) were required to antagonize T0901317 driven Gal4-LXRα activity [32]. These findings, as well as the inability of EPA to suppress mRNA of known LXRα-regulated genes (ABCG5 and ABCG8) in FTO-2B cells, led these investigators to conclude that LXRα is not a target for fatty acid antagonism in rat liver [32]. In contrast, both the present data and that of Ou et al [31] support the hypothesis that PUFA repress SREBP-1c expression via attenuation of LXRα dependent activation of transcription. Differing outcomes of these studies may be attributed to differences in experimental design including the species of PUFA utilized, tissue culture conditions, and methods used to prepare PUFA.
Attenuation of LXRα activity has been attributed to competitive inhibition of ligand binding to LXRα by PUFA [25, 31]. In the present studies the effect of DHA was not overcome by the maximal concentration of T0901317 as would be expected if competitive antagonism of ligand binding were responsible for the effect. Therefore, our data corroborate previous findings with AA and EPA in that DHA significantly decreases the trans-activating property of LXRα. However, our data suggest that interference with LXRα agonist binding may not be a primary mechanism for the effect of DHA on LXRα. An alternative possibility is that DHA interferes with LXRα signaling by altering the composition of the LXRα/RXR transcriptional complex, including co-repressors and co-activators of LXRα. In obese, hyperinsulinemic JCR:LA-cp rats, menhaden oil feeding significantly increased the expression of Nr0B2 (alternatively known as small heterodimer partner, SHP) when compared to control diet [17]. SHP has been documented to interact with LXRα and decrease its transcriptional activity [44, 45]. Therefore, PUFA induction of SHP and or other repressors, or reduced association of known co-activators of LXRα including SRC-1 and CBP/p300 may lead to decreased LXRα transcriptional activity [36].
Previous observations of differential regulation of LXRα target genes by PUFA are particularly intriguing. LXRα regulates a wide range of genes related to lipogenesis (SREBP-1c, FAS), cholesterol transport (ABCG5, ABCG8) and bile acid metabolism (CYP7A1). Fish oil feeding in the whole animal or PUFA treatment of rat hepatoma cells effectively decreases hepatic expression of lipogenic genes whereas the expression of more traditional LXRα target genes such as ABCG5, ABCG8, or CYP7A1 are unchanged [17, 32]. This suggests that the effect of PUFA is influenced by the specific promoter context.
In conclusion, we have demonstrated that mitigation of the insulin induction of SREBP-1c by n-3 polyunsaturated fatty acids (EPA and DHA) is, at least in part, due to transcriptional down regulation. We present evidence in support of the hypothesis that down regulation of SREBP-1c transcription by PUFA results from attenuated trans-activation of the ligand-activated nuclear receptor LXRα. Further study is needed to determine the molecular mechanisms of this effect. In this regard, understanding the mechanisms underlying attenuation of insulin-induced SREBP-1c transcription by PUFA and other factors has important implications for development of effective therapeutic modalities for the dyslipidemia that accompanies hyperinsulinemic states including obesity and type II diabetes mellitus.
Supplementary Material
Acknowledgments
These studies were supported by NIDDK RO1-DK75504-01 (MBE, RR, CY), by an individual NIDDK postdoctoral fellowship award to GH (NIDDK 1F32DK083210-01), by USPHS GR HL 07641-14, and in part by Merit Review Grants from the Department of Veteran’s Affairs (MBE, RR). We thank Poonam Kumar and Emily Kern for technical assistance and Dr. William L. Taylor of the UTHSC Molecular Resource Center for his invaluable assistance. The pCMV-Gal4-LXRα expression vector was a kind gift of Dr. Terry Unterman (Dept. of Physiology and Biophysics, University of Illinois, Chicago). RR is a Senior Research Career Scientist of the Department of Veterans Affairs. XD is an MREP awardee of the Department of Veteran’s affairs Research Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Girard J, Perdereau D, Foufelle F, Prip-Buus C, Ferre P. Regulation of lipogenic enzyme gene expression by nutrients and hormones. FASEB J. 1994;8:36–42. doi: 10.1096/fasebj.8.1.7905448. [DOI] [PubMed] [Google Scholar]
- 2.Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 3.Shimano H. Sterol regulatory element-binding protein-1 as a dominant transcription factor for gene regulation of lipogenic enzymes in the liver. Trends Cardiovasc Med. 2000;10:275–278. doi: 10.1016/s1050-1738(00)00079-7. [DOI] [PubMed] [Google Scholar]
- 4.Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, Gotoda T, Ishibashi S, Yamada N. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem. 1999;274:35832–35839. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 5.Foretz M, Guichard C, Ferre P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A. 1999;96:12737–12742. doi: 10.1073/pnas.96.22.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Liepvre X, Berthelier-Lubrano C, Spiegelman B, Kim JB, Ferre P, Foufelle F. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 8.Hua X, Wu J, Goldstein JL, Brown MS, Hobbs HH. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics. 1995;25:667–673. doi: 10.1016/0888-7543(95)80009-b. [DOI] [PubMed] [Google Scholar]
- 9.Deng X, Cagen LM, Wilcox HG, Park EA, Raghow R, Elam MB. Regulation of the rat SREBP-1c promoter in primary rat hepatocytes. Biochem Biophys Res Commun. 2002;290:256–262. doi: 10.1006/bbrc.2001.6148. [DOI] [PubMed] [Google Scholar]
- 10.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96:13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99:838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Park EA, Raghow R, Elam MB. Posttranslational processing of SREBP-1 in rat hepatocytes is regulated by insulin and cAMP. Biochem Biophys Res Commun. 2005;332:174–180. doi: 10.1016/j.bbrc.2005.04.112. [DOI] [PubMed] [Google Scholar]
- 13.Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obesity (Silver Spring) 2006;14(Suppl 1):41S–49S. doi: 10.1038/oby.2006.281. [DOI] [PubMed] [Google Scholar]
- 14.Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46:733–749. doi: 10.1007/s00125-003-1111-y. [DOI] [PubMed] [Google Scholar]
- 15.Elam MB, Wilcox HG, Cagen LM, Deng X, Raghow R, Kumar P, Heimberg M, Russell JC. Increased hepatic VLDL secretion, lipogenesis, and SREBP-1 expression in the corpulent JCR:LA-cp rat. J Lipid Res. 2001;42:2039–2048. [PubMed] [Google Scholar]
- 16.Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 17.Deng X, Elam MB, Wilcox HG, Cagen LM, Park EA, Raghow R, Patel D, Kumar P, Sheybani A, Russell JC. Dietary olive oil and menhaden oil mitigate induction of lipogenesis in hyperinsulinemic corpulent JCR:LA-cp rats: microarray analysis of lipid-related gene expression. Endocrinology. 2004;145:5847–5861. doi: 10.1210/en.2004-0371. [DOI] [PubMed] [Google Scholar]
- 18.Kim HJ, Takahashi M, Ezaki O. Fish oil feeding decreases mature sterol regulatory element-binding protein 1 (SREBP-1) by down-regulation of SREBP-1c mRNA in mouse liver. A possible mechanism for down-regulation of lipogenic enzyme mRNAs. J Biol Chem. 1999;274:25892–25898. doi: 10.1074/jbc.274.36.25892. [DOI] [PubMed] [Google Scholar]
- 19.Yahagi N, Shimano H, Hasty AH, Amemiya-Kudo M, Okazaki H, Tamura Y, Iizuka Y, Shionoiri F, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J Biol Chem. 1999;274:35840–35844. doi: 10.1074/jbc.274.50.35840. [DOI] [PubMed] [Google Scholar]
- 20.Davidson MH. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am J Cardiol. 2006;98:27i–33i. doi: 10.1016/j.amjcard.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Goldberg RB, Sabharwal AK. Fish oil in the treatment of dyslipidemia. Curr Opin Endocrinol Diabetes Obes. 2008;15:167–174. doi: 10.1097/MED.0b013e3282f76728. [DOI] [PubMed] [Google Scholar]
- 22.Nakatani T, Kim HJ, Kaburagi Y, Yasuda K, Ezaki O. A low fish oil inhibits SREBP-1 proteolytic cascade, while a high-fish-oil feeding decreases SREBP-1 mRNA in mice liver: relationship to anti-obesity. J Lipid Res. 2003;44:369–379. doi: 10.1194/jlr.M200289-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Cho H, O’Malley S, Park JH, Clarke SD. Dietary polyunsaturated fats regulate rat liver sterol regulatory element binding proteins-1 and -2 in three distinct stages and by different mechanisms. J Nutr. 2002;132:3333–3339. doi: 10.1093/jn/132.11.3333. [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Teran-Garcia M, Park JH, Nakamura MT, Clarke SD. Polyunsaturated fatty acids suppress hepatic sterol regulatory element-binding protein-1 expression by accelerating transcript decay. J Biol Chem. 2001;276:9800–9807. doi: 10.1074/jbc.M008973200. [DOI] [PubMed] [Google Scholar]
- 25.Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T, Nakakuki M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Takahashi A, Sone H, Osuga Ji J, Gotoda T, Ishibashi S, Yamada N. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem. 2002;277:1705–1711. doi: 10.1074/jbc.M105711200. [DOI] [PubMed] [Google Scholar]
- 26.Botolin D, Wang Y, Christian B, Jump DB. Docosahexaneoic acid (22:6,n-3) regulates rat hepatocyte SREBP-1 nuclear abundance by Erk- and 26S proteasome-dependent pathways. J Lipid Res. 2006;47:181–192. doi: 10.1194/jlr.M500365-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem. 2001;276:4365–4372. doi: 10.1074/jbc.M007273200. [DOI] [PubMed] [Google Scholar]
- 28.Worgall TS, Sturley SL, Seo T, Osborne TF, Deckelbaum RJ. Polyunsaturated fatty acids decrease expression of promoters with sterol regulatory elements by decreasing levels of mature sterol regulatory element-binding protein. J Biol Chem. 1998;273:25537–25540. doi: 10.1074/jbc.273.40.25537. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Nakamura MT, Cho HP, Clarke SD. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J Biol Chem. 1999;274:23577–23583. doi: 10.1074/jbc.274.33.23577. [DOI] [PubMed] [Google Scholar]
- 30.Cagen LM, Deng X, Wilcox HG, Park EA, Raghow R, Elam MB. Insulin activates the rat sterol-regulatory-element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1 and NF-Y cis-acting elements. Biochem J. 2005;385:207–216. doi: 10.1042/BJ20040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, Brown MS. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A. 2001;98:6027–6032. doi: 10.1073/pnas.111138698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawar A, Botolin D, Mangelsdorf DJ, Jump DB. The role of liver X receptor-alpha in the fatty acid regulation of hepatic gene expression. J Biol Chem. 2003;278:40736–40743. doi: 10.1074/jbc.M307973200. [DOI] [PubMed] [Google Scholar]
- 33.Teran-Garcia M, Adamson AW, Yu G, Rufo C, Suchankova G, Dreesen TD, Tekle M, Clarke SD, Gettys TW. Polyunsaturated fatty acid suppression of fatty acid synthase (FASN): evidence for dietary modulation of NF-Y binding to the Fasn promoter by SREBP-1c. Biochem J. 2007;402:591–600. doi: 10.1042/BJ20061722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teran-Garcia M, Rufo C, Nakamura MT, Osborne TF, Clarke SD. NF-Y involvement in the polyunsaturated fat inhibition of fatty acid synthase gene transcription. Biochem Biophys Res Commun. 2002;290:1295–1299. doi: 10.1006/bbrc.2002.6341. [DOI] [PubMed] [Google Scholar]
- 35.Thorngate FE, Raghow R, Wilcox HG, Werner CS, Heimberg M, Elam MB. Insulin promotes the biosynthesis and secretion of apolipoprotein B-48 by altering apolipoprotein B mRNA editing. Proc Natl Acad Sci U S A. 1994;91:5392–5396. doi: 10.1073/pnas.91.12.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng X, Yellaturu C, Cagen L, Wilcox HG, Park EA, Raghow R, Elam MB. Expression of the rat sterol regulatory element-binding protein-1c gene in response to insulin is mediated by increased transactivating capacity of specificity protein 1 (Sp1) J Biol Chem. 2007;282:17517–17529. doi: 10.1074/jbc.M702228200. [DOI] [PubMed] [Google Scholar]
- 37.McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- 38.Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. 2002;42:249–259. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- 39.Holness MJ, Bulmer K, Smith ND, Sugden MC. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem J. 2003;369:687–695. doi: 10.1042/BJ20021509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon HS, Harris RA. Mechanisms responsible for regulation of pyruvate dehydrogenase kinase 4 gene expression. Adv Enzyme Regul. 2004;44:109–121. doi: 10.1016/j.advenzreg.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 41.Park EA, Mynatt RL, Cook GA, Kashfi K. Insulin regulates enzyme activity, malonyl-CoA sensitivity and mRNA abundance of hepatic carnitine palmitoyltransferase-I. Biochem J. 1995;310(Pt 3):853–858. doi: 10.1042/bj3100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maki KC, McKenney JM, Reeves MS, Lubin BC, Dicklin MR. Effects of adding prescription omega-3 acid ethyl esters to simvastatin (20 mg/day) on lipids and lipoprotein particles in men and women with mixed dyslipidemia. Am J Cardiol. 2008;102:429–433. doi: 10.1016/j.amjcard.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 43.Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, Ishibashi S, Osuga J, Yamada N, Shimano H. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology. 2003;38:1529–1539. doi: 10.1016/j.hep.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 44.Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J. The small heterodimer partner interacts with the liver X receptor alpha and represses its transcriptional activity. Mol Endocrinol. 2002;16:2065–2076. doi: 10.1210/me.2001-0194. [DOI] [PubMed] [Google Scholar]
- 45.Shang Q, Pan L, Saumoy M, Chiang JY, Tint GS, Salen G, Xu G. The stimulatory effect of LXRalpha is blocked by SHP despite the presence of a LXRalpha binding site in the rabbit CYP7A1 promoter. J Lipid Res. 2006;47:997–1004. doi: 10.1194/jlr.M500449-JLR200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.