

Abstract
Hereditary folate malabsorption is a rare inborn error of metabolism due to mutations in the proton-coupled folate transporter (PCFT). Clinical presentation of PCFT deficiency may mimic severe combined immune deficiency (SCID). We report a 4-month-old female who presented with failure to thrive, normocytic anemia, Pneumocystis jirovecii pneumonia and systemic cytomegalovirus infection. Immunological evaluation revealed hypogammaglobulinemia, absent antibody responses, and lack of mitogen-induced lymphocyte proliferative responses. However, the absolute number and distribution of lymphocyte subsets, including naïve T cells and recent thymic emigrants, were normal, arguing against primary SCID. Serum and cerebrospinal fluid folate levels were undetectable. A homozygous 1082-1G>A mutation of the PCFT gene was found, resulting in skipping of exon 3. Parenteral folinic acid repletion resulted in normalization of anemia, humoral and cellular immunity, and full clinical recovery. PCFT mutations should be considered in infants with SCID-like phenotype, as the immunodeficiency is reversible with parenteral folinic acid repletion.
Keywords: Hereditary folate malabsorption, PCFT, folic acid, severe combined immunodeficiency, recent thymic emigrants
INTRODUCTION
Folates are essential cofactors in the human metabolism of purines, pyrimidines, and amino acids, and thus are crucial for the development and function of several organs and systems. Folate deficiency leads to defective thymidine synthesis with resulting DNA instability characterized by increased DNA strand breaks, uracil misincorporation and defective repair of double-strand breaks [1]. Folate deficiency in humans has been associated with a variety of clinical disorders, including neural tube defects, cardiovascular diseases and possibly an increased risk of malignancy.
Hereditary folate malabsorption (HFM; OMIM 229050) is a rare, autosomal recessive inborn error of metabolism, characterized by impaired absorption of folates in the gut and through the choroid plexus into the cerebrospinal fluid (CSF). Two different molecular mechanisms account for folate transport in humans. The reduced folate carrier (SLC19A1) is a ubiquitously expressed membrane protein that works as a facilitative transporter that optimally functions at neutral pH and, although it is present in the gut, intestinal folate absorption occurs independent of its presence [2]. The second mechanism is dependent on the proton-coupled folate transporter (PCFT, SLC46A1), which is expressed on the surface epithelial cells of the duodenum and upper jejunum as well as in choroid plexus epithelial cells, and functions as a high-affinity folate transporter with optimal transport activity at low pH [3]. Recently, loss-of-function mutations in the PCFT gene were identified in patients with HFM [4-6].
HFM patients present very early in life with megaloblastic anemia, failure to thrive, diarrhea, infections, and seizures, and can develop severe neurodevelopmental defects unless they are treated promptly and aggressively [7]. Hypogammaglobulinemia and other immunological defects have been reported in a few patients [6, 8, 9], but the immunological phenotype of HFM has not been fully characterized.
In this report we describe a female child who presented with clinical and immunological features suggestive of severe combined immunodeficiency (SCID). Identification of a homozygous mutation in the PCFT gene prompted folate repletion, with full normalization of the clinical and laboratory abnormalities.
MATERIALS AND METHODS
Blood samples
Blood samples were obtained from the patient, her parents and healthy controls upon informed consent in agreement with the Studies of Immunological Deficiency Syndromes protocol approved by the institutional review board at Children’s Hospital Boston.
Sequencing of the PCFT gene
Whole genomic DNA was isolated from peripheral blood using the Puregene DNA purification Kit (Gentra System, Minneapolis, MN), and quantified using a NanoDrop ND-1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE). All exons and flanking splice-sites of the SLC46A1 gene were amplified using primers reported in supplementary Table 1. Reaction products were purified by treating the PCR mixture with Exonuclease I (New Englands, Biolabs, Ipswich, MA) and shrimp alkaline phosphatase (Roche Diagnostics, Indianapolis, IN). The sequence of both DNA strands was determined on an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, CA) using fluorescent dye-terminator chemistry, and the results were analyzed using the Sequencher 4.8 software (Gene Codes Corporation, Ann Arbor, MI).
RT-PCR
RNA was isolated from the patient’s primary fibroblasts using TRIzol® Reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized from 1 μg RNA by using the iScript cDNA Synthesis Kit from Bio-Rad (Hercules, CA). RT-PCR was performed as previously described [4].
Immunophenotyping of peripheral blood mononuclear cells (PBMCs)
Peripheral blood mononuclear cells (PBMCs) were isolated using a Ficoll gradient. Immunophenotyping was performed using the following anti-human monoclonal antibodies (mAb) and their isotype controls: anti-CD3-PerCP, anti-CD4-PerCP, anti-CD8-FITC, anti-CD31-PE, anti-CD45RA-FITC or APC, anti-CD11a-PE, anti-CCR7-PE, anti-IgD-FITC, anti-CD27-PE, anti-CD19-PerCP, anti-TCRab-FITC, anti-TCRgd-PE, MultiTEST kit CD3-FITC/CD8-PE/CD45-PerCP/CD4-APC, and MultiTEST kit CD3-FITC/CD16+CD56-PE/CD45-PerCP/CD19-APC. All of the mAbs were purchased from BD Biosciences (Becton-Dickinson, Mountain View, CA). Flow cytometry was performed using a FACScalibur flow cytometer (Becton-Dickinson, Mountain View, CA) and data were analysed using the FlowJo 8.3.3 software (TreeStar, Ashland, OR).
Lymphocyte proliferation assays
Lymphocyte proliferation responses to phytohemagglutinin (PHA), concanavalin A (ConA) and pokeweed mitogen (PWM) were assayed as previously reported [10]. Analysis of lymphocyte proliferation in response to stimulation with anti-CD3 mAb (with or without exogenous IL-2) was performed by carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution as described [11]. Briefly, PBMCs from the patient and from an age-matched healthy control were labeled at a concentration of 10 × 106/ml with 7.5mM CFSE (Molecular Probes, Carlsbad, CA) in phosphate buffered saline, 0.1% bovine serum albumin for 5min at 37°C. After two washes in RPMI medium supplemented with 20% fetal calf serum, 106 cells were distributed in round bottom tubes in triplicate for each condition: unstained, unstimulated, stimulated with anti-CD3 (100 ng/ml; OKT3 ATCC CRL 8001, Manassas, VA) or with anti-CD3 (100 ng/ml) and IL-2 (30 ng/ml; R & D Systems, Minneapolis, MN). After 4 days of culture at 37°C, cell division was assessed by the dilution of CFSE staining measured as reduction of mean of fluorescence intensity (MFI) by flow cytometry on a FACScalibur instrument (Becton-Dickinson, Mountain View, CA). Data were analyzed by the FlowJo 8.3.3 software (TreeStar, Ashland, OR) upon gating for CD3+ cells.
Analysis of maternal T-cell engraftment
Genotyping analysis for maternal T-cell engraftment was performed with the AmpFℓSTR® Profiler Plus® ID PCR Amplification Kit (Applied Biosystems, Foster City, CA).
Analysis of T-cell repertoire diversity
DNA was isolated from peripheral blood and analyzed by a polymerase chain reaction technique using primers conjugated with fluorescent dyes that hybridize to T cell receptor gamma chain (TCRγ) gene V segment 1-8, 9, 10, 11 (Vg1-8, Vg9, Vg10, Vg11) and joining region (Jg1 and Jg2) (InVivoScribe Technologies, BIOMED, San Diego, CA). The DNA was also amplified with control primers to multiple gene segments (64 to 600 bp in length) to assess the integrity and quality of the isolated DNA. The PCR products were analyzed by capillary gel electrophoresis. The DNA was analyzed for TCR beta clonal rearrangements by Southern Blot technique.
RESULTS
Case report
A 3-month-old female, born to non-consanguineous parents of Puerto Rican descent, developed frequent emesis and failure to thrive. Her weight dropped from the 25th percentile to the 5th percentile. At 4 months of age, she developed cough, coryza, low-grade fever, irritability, and increased emesis without diarrhea, for which she was hospitalized at another institution. Chest X-ray revealed diffuse interstitial pneumonia. Laboratory studies showed severe normocytic anemia (hemoglobin 6.7 g/dL, MCV 76 μm3) with 0.4% reticulocyte count, mild neutropenia (1.4 ×109 cells/L) with hypersegmentation, eosinophilia (1.1 ×109 cells/L) and a normal absolute lymphocyte count (3.4 ×109 cells/L). Serum folate levels were undetectable. She was treated with antibiotics, a packed red blood cell transfusion and a single replacement dose of intravenous folinic acid (2 mg/kg) prior to transfer to Children’s Hospital Boston.
Upon admission, bronchoalveolar lavage revealed Pneumocystis jirovecii. In addition, active cytomegalovirus (CMV) infection was documented by p65 antigenemia, PCR positivity in the bone marrow, and positive staining for CMV in endoscopic biopsies of esophagus and duodenum. Testing for HIV by ELISA was negative. Treatment was started with trimethoprim-sulfamethoxazole and ganciclovir. High-frequency ventilation was initially required due to respiratory distress, but gradual improvement allowed extubation after 12 days. The clinical course was complicated by Klebsiella pneumoniae bacteriemia, requiring treatment with ceftriaxone.
Bone marrow aspirate performed 10 days after the initial dose of folinic acid was normal except for decreased iron staining and rare neutrophil hypersegmentation, without evidence of megaloblastic hematopoiesis. At that time, blood folate levels were still low. Continued treatment with intravenous folate resulted in normalization of plasma levels. However a lumbar puncture disclosed that 5-methyl-tetra-hydrofolate (5-MTH), the major form of folate in the CSF, was undetectable. The patient was then switched to parenteral folinic acid to facilitate CSF penetration. Progressive clinical improvement, with normalization of hematological and immunological parameters was observed. CSF 5-MTH improved to 33 nmol/L on 5 mg IM folinic acid daily. Dosing has been increased to 7.5 mg daily of IM folinic acid with the goal of normalizing CSF 5-MTH. At the age of 18 months, she has had no more infections and has reached all developmental milestones appropriate for her age.
Identification of a homozygous PCFT splice site mutation
Genomic sequencing of the PCFT gene revealed a homozygous 1082-1G>A mutation in the splice acceptor of intron 2. Sequencing of cDNA synthesized from the patient’s fibroblast RNA confirmed skipping of exon 3, resulting in a predicted in-frame deletion of 28 amino acids. Both parents were heterozygous for the same splice junction mutation (Figure 1).
Figure 1.

Chromatograms of DNA sequence of the PCFT gene showing a homozygous missense G>A mutation at the splice junction of intron 2 and exon 3 in the patient and a heterozygous mutation in both parents.
Immunological phenotype
Severe abnormalities of both cellular and humoral immunity were demonstrated at admission (Table 1), with low serum IgG and IgM levels, absent isohemagglutinins, and lack of antibody response to tetanus toxoid (TT), Haemophilus influenzae b and heptavalent pneumococcal vaccines after two doses of immunization. The absolute number and distribution of T, B and NK lymphocytes were normal, however in vitro lymphocyte proliferative responses to PHA, ConA and PWM were abrogated. The patient was started on intravenous immunoglobulin replacement therapy to maintain an IgG trough level over 500 mg/dl.
Table 1.
Immunologic status of the patient before and after folate replacement therapy.
| Laboratory test | Before folate replacement (4 months of age) |
After folate replacement (8 months of age) |
|---|---|---|
| Folate status | ||
| Serum folate (5.9 – 20.2 ng/ml) | < 1 | 79.6 |
| CSF 5-methyltetrahydrofolate (40-240 nmol/L) | < 5 | 33 |
| Hematology | ||
| Hemoglobin (g/dL) | 6.7 | 9.9 |
| Hematocrit (%) | 19.2 | 28.6 |
| Leukocytes (×109/L) | 6.1 | 8.9 |
| Neutrophils (×109/L) | 1.4 | 1.7 |
| Lymphocytes (×109/L) | 3.4 | 6.7 |
| Eosinophils (×109/L) | 1.1 | 0 |
| Platelets (×109/L) | 242 | 390 |
| Serum immunoglobulins | ||
| IgG (mg/dl) | 119 | 576 |
| IgG1 (mg/dl) | 60 | 354 |
| IgG2 (mg/dl) | 26 | 82 |
| IgG3 (mg/dl) | 26 | 65 |
| IgG4 (mg/dl) | < 1 | 3 |
| IgM (mg/dl) | 15 | 71 |
| IgA (mg/dl) | 18 | 8 |
| IgE (units/ml) | 2 | 5 |
| Vaccine titers (protective titers) | ||
| Tetanus (IU/ml)a | 0.08 | 0.58 |
| H. influenzae b PRP (ng/ml)a | < 110 | 640b |
| Pneumococcal heptavalent vaccine serotypesa | 0 / 7 | 7/7 |
| Isohemagglutinin titers A and B | negative | |
| Lymphocyte subsets | ||
| CD3+ cells/μL (%) | 7,187 (83%) | 3,827 (70%) |
| CD4+ cells/μL (%) | 5,596 (62%) | 1,595 (29%) |
| CD8+ cells/μL (%) | 1,893 (21%) | 3,774 (69%) |
| CD19+ cells/μL (%) | 962 (12%) | 654 (12%) |
| CD16+56+ cells/μL (%) | 299 (4%) | 951 (17%) |
| Lymphocyte proliferation ( (cpm) | ||
| PHA | 117 | 87,644 |
| ConA | 155 | 60,843 |
| PWM | 100 | 16,746 |
Protective antibodies titers are as follows: Tetanus toxoid: >0.15 IU/ml; Hib PRP: >1,000 ng/ml; anti-pneumococcus: >1 mcg/ml
After a fourth dose of Hib vaccine at 15 months of age, PRP titers increased to 3760 ng/ml.
Further characterization of lymphocyte subsets disclosed a decreased proportion of both unswitched (IgD+) and switched (IgD-) CD27+ memory B cells (0.5% and 2.4% of CD19+ B cells, respectively). The vast majority (97.7%) of circulating T cells expressed an αβ T-cell receptor (TCR). The proportion of CD4+ and CD8+ T cells with a CD45R0+ memory/activated phenotype was increased (81.7% and 81.3%, respectively). This prompted evaluation of possible maternal T cell engraftment, which was ruled out by genotyping of informative loci in the patient and her mother.
The demonstration of an increased proportion of T cells with a memory/activated phenotype, along with markedly decreased proliferation to mitogens and hypogammaglobulinemia, was suggestive of leaky SCID. To address this possibility, enumeration of true naïve (CCR7+CD45RA+) CD4+ T cells and of CD31+CD45RA+CD4+ recent thymic emigrants, and analysis of the diversity of T-cell repertoire were performed. True naïve CD4+ T cells were present in the patient in adequate numbers (3811 cells/mm3, 68.1% of CD4+ T cells) (Fig. 2A). In addition, 43.5% of the patient’s CD4+ T cells co-expressed CD45RA and CD31, indicating normal thymic output (Fig. 2B). Analysis of T cell receptor repertoire diversity revealed normal distribution of the TCR gamma and beta variable chains (data not shown). Overall, these data argued against significant defects in T-cell development or peripheral expansion of selected clonotypes.
Figure 2.
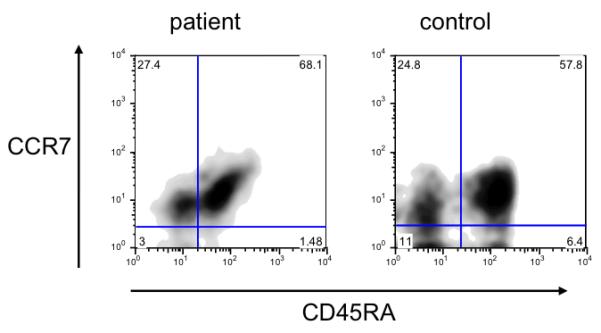
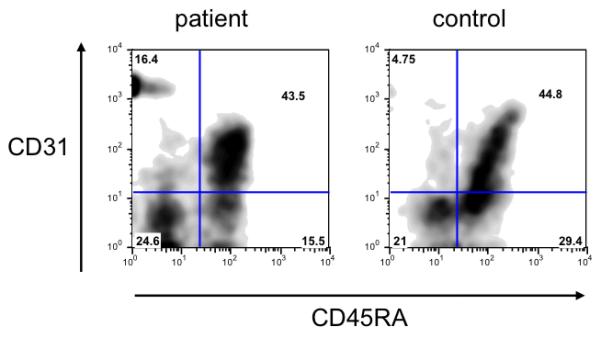
Flow cytometry analysis of CCR7+CD45RA+ true naïve T cells (A) and CD31+CD45RA+ recent thymic emigrants (B) in patient and control CD4+ T cells.
Previous studies had shown that folate starvation in vitro causes S-phase cell cycle arrest and induces apoptosis [12]. To further investigate whether the severe defect of in vitro lymphocyte proliferation could reflect increased apoptosis, we analyzed proliferation to anti-CD3 (with or without IL-2) using a CFSE dilution assay. As shown in Fig. 3, after 96 hours of culture, a large proportion of the patient’s CD3+ lymphocytes had died, and the increased apoptosis was only partially rescued by addition of exogenous IL-2. However, upon gating on live CD3+ lymphocytes, CFSE dilution was observed both in the patient and in the control, indicating that the patient’s cells were able to divide normally upon stimulation with anti-CD3 alone or with IL-2 (Fig. 4).
Figure 3.

Flow cytometry scatter analysis with gating on live cells of patient and control CD3+ cells after 96 hours of culture in non-stimulated media, stimulated with anti-CD3 or with anti-CD3 and IL-2.
Figure 4.

Lymphocyte proliferation analysis by CFSE dilution of patient and control CD3+ cells after 96 hours of culture in unstimulated media, stimulated with anti-CD3 or with anti-CD3 and IL-2.
Intravenous repletion of plasma folate resulted in progressive improvement in hematopoiesis, normalization of IgG and IgM, and normalization of lymphocyte proliferation to mitogens. Intravenous immunoglobulin was discontinued once folate repletion therapy was instituted and lymphocyte proliferation to mitogens had normalized. Following a third immunization with TT, conjugated pneumococcal and Haemophilus influenzae b (Hib) vaccines, protective titers to TT and pneumococcus were achieved, while PRP titers only achieved protective levels after a fourth Hib immunization. Low serum IgA levels persist despite folate repletion therapy (Table 1).
DISCUSSION
Hereditary folate malabsorption (HFM) is an extremely rare disorder with less than 30 cases reported in the literature, and even fewer patients having proven mutations of the PCFT gene. The homozygous 1082-1G>A mutation identified in our patient has been previously reported to cause intracellular trapping of the PCFT protein and disruption of transporter activity [4].
The profound humoral and cellular immunodeficiency detected in our patient, prompted us to rule out SCID and to begin the work up for potential bone marrow transplantation. The evaluation of thymic output by flow cytometry of truly naïve T cells and recent thymic emigrants, along with the demonstration of a polyclonal T-cell repertoire, were crucial to rule out a diagnosis of SCID.
The immune system appears to be particularly sensitive to folate deficiency. In vitro studies have shown that folate deprivation impairs T lymphocyte proliferation, and induces cell cycle arrest in the S-phase and apoptosis [12]. These defects appear to be more severe in CD8+ than in CD4+ lymphocytes. Furthermore, the lymphocyte proliferative defect is reversible, as shown by the fact that lymphocytes cultured without folate for one week resumed proliferation when folate was added to the medium [13]. In keeping with these observations, the immunologic recovery of our patient after parenteral folate repletion was dramatic, providing strong in vivo evidence of the importance of folates for lymphocyte function in humans.
Among the patients with HFM reported in the literature, 10 were found to carry mutations in the PCFT gene, including our case. Of these, six suffered from severe or recurrent infections and hypogammaglobulinemia [6, 14]. Their clinical manifestations of immunodeficiency are depicted in Table 2. The severity of immune defects among these patients was variable, and apart from immunoglobulin serum levels, no detailed studies of immune function were consistently performed. In the patient reported by Corbeel et al., lymphocyte subsets and lymphocyte proliferation studies to mitogens were normal [15]. In another case, a reduced lymphocyte proliferation response to tetanus was observed, although only after one dose of tetanus vaccine, making interpretation of this data difficult [8]. Prior to the identification of the PCFT, Urbach et al. reported an infant with congenital malabsorption of folic acid and recurrent infections, who was found to have normal IgG, high IgM, undetectable IgA, as well as decreased erythrocyte rosette formation and poor responses to PHA, ConA and PWM [16].
Table 2.
Clinical manifestations of immunodeficiency in 10 HFM patients with proven PCFT mutations.
| Clinical Manifestations | |
|---|---|
| Pneumocystis jirovecii pneumonia | 40% |
| Protracted diarrhea | 30% |
| Bacteremia | 20% |
| Recurrent respiratory infections | 10% |
| UTI Pseudomonas aeruginosa | 10% |
| Severe RSV infection | 10% |
| Systemic CMV infection | 10% |
Although few studies have analyzed in detail cellular immunity in HFM, these patients seem to be highly susceptible to P. jirovecii pneumonia (PJP). Our patient presented with PJP, and she showed deterioration of respiratory function shortly after initiation of folate replacement therapy. It has been previously reported that folate supplementation in patients with HFM and PJP may result in the ability to mount inflammatory responses, and thus may cause initial worsening of the clinical picture [8]. Furthermore, in patients with AIDS, adjunctive folinic acid to trimethoprimsulfamethoxazole for PJP has been associated with an increased risk of therapeutic failure and death [17]. It has been proposed that although P. jirovecii synthesizes its folates de novo, it may be able to use exogenous folates to thrive [18].
Several previously reported patients with HFM had siblings who died around 3-4 months of age with protracted diarrhea or overwhelming infection, including cytomegalovirus and PJP [9, 15, 16, 19, 20]. This observation suggests that presentation of HFM with SCID-like features may be more common than usually thought. The onset of severe infections and other clinical manifestations of HFM around 3 months of age may reflect progressive depletion of folate stores that are acquired transplacentally during pregnancy.
The diagnosis of HFM in our patient was particularly difficult due to the absence of megaloblastic anemia at presentation. Combined deficiencies of either folate or B12 deficiency with a concurrent iron deficiency are known to mask the macrocytosis generally seen with megaloblastic anemia. In fact, replacement of one deficiency can result in emergence of the typical phenotype of the untreated deficiency [21]. Our patient had both iron and folate deficiencies at presentation. Folinic acid replacement 10 days prior to bone marrow studies likely resulted in normalization of bone marrow morphology, masking the signs of megaloblastic anemia. Parenteral folate replacement was unable to normalize the plasma to CSF ratio (normally 3:1) as has been seen in several cases in the literature [8, 9, 19, 22, 23]. In contrast, high parenteral doses of folinic acid were required to improve CSF folate levels, as previously reported in other cases.
Treatment of HFM requires long-term replacement of plasma and CSF folate levels. Replacement with parenteral folate has resulted in normal hematologic and immunologic responses, but persistence of neurologic symptoms if already present. Methionine replacement has been tried with some success and one case report suggests adequate dosing of oral folinic acid can result in sufficient CSF levels and improved neurologic outcomes [9, 15]. Although optimal treatment for HFM is not yet clear, IM folinic acid has been used most often, with excellent hematologic, immunologic, and neurologic outcomes if treatment is started in a timely manner [8, 19, 22, 23].
This study shows that HFM due to PCFT mutations can present with life-threatening infections due to profound immunological defects that may mimic SCID. We propose that HFM should be considered in the differential diagnosis of SCID, as the immune defects are fully reversible with adequate folate repletion.
Supplementary Material
Acknowledgments
This research was partly funded by NHLBI K24 grant HL004184.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duthie SJ, Hawdon A. DNA instability (strand breakage, uracil misincorporation, and defective repair) is increased by folic acid depletion in human lymphocytes in vitro. FASEB J. 1998;12:1491–1497. [PubMed] [Google Scholar]
- 2.Wang Y, Rajgopal A, Goldman ID, Zhao R. Preservation of folate transport activity with a low-pH optimum in rat IEC-6 intestinal epithelial cell lines that lack reduced folate carrier function. Am. J. Physiol. Cell Physiol. 2005;288:C65–71. doi: 10.1152/ajpcell.00307.2004. [DOI] [PubMed] [Google Scholar]
- 3.Wollack JB, Makori B, Ahlawat S, Koneru R, Picinich SC, Smith A, et al. Characterization of folate uptake by choroid plexus epithelial cells in a rat primary culture model. J. Neurochem. 2008;104:1494–1503. doi: 10.1111/j.1471-4159.2007.05095.x. [DOI] [PubMed] [Google Scholar]
- 4.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 5.Lasry I, Berman B, Straussberg R, Sofer Y, Bessler H, Sharkia M, et al. A novel loss-of-function mutation in the proton-coupled folate transporter from a patient with hereditary folate malabsorption reveals that Arg 113 is crucial for function. Blood. 2008;112:2055–2061. doi: 10.1182/blood-2008-04-150276. [DOI] [PubMed] [Google Scholar]
- 6.Zhao R, Min SH, Qiu A, Sakaris A, Goldberg GL, Sandoval C, et al. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood. 2007;110:1147–1152. doi: 10.1182/blood-2007-02-077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitehead VM. Acquired and inherited disorders of cobalamin and folate in children. Br. J. Haematol. 2006;134:125–136. doi: 10.1111/j.1365-2141.2006.06133.x. [DOI] [PubMed] [Google Scholar]
- 8.Malatack JJ, Moran MM, Moughan B. Isolated congenital malabsorption of folic acid in a male infant: insights into treatment and mechanism of defect. Pediatrics. 1999;104:1133–1137. doi: 10.1542/peds.104.5.1133. [DOI] [PubMed] [Google Scholar]
- 9.Geller J, Kronn D, Jayabose S, Sandoval C. Hereditary folate malabsorption: family report and review of the literature. Medicine (Baltimore) 2002;81:51–68. doi: 10.1097/00005792-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Stone KD, Feldman HA, Huisman C, Howlett C, Jabara HH, Bonilla FA. Analysis of in vitro lymphocyte proliferation as a screening tool for cellular immunodeficiency. Clin Immunol. 2009;131:41–49. doi: 10.1016/j.clim.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007;2:2049–2056. doi: 10.1038/nprot.2007.296. [DOI] [PubMed] [Google Scholar]
- 12.Courtemanche C, Elson-Schwab I, Mashiyama ST, Kerry N, Ames BN. Folate deficiency inhibits the proliferation of primary human CD8+ T lymphocytes in vitro. J. Immunol. 2004;173:3186–3192. doi: 10.4049/jimmunol.173.5.3186. [DOI] [PubMed] [Google Scholar]
- 13.Courtemanche C, Huang AC, Elson-Schwab I, Kerry N, Ng BY, Ames BN. Folate deficiency and ionizing radiation cause DNA breaks in primary human lymphocytes: a comparison. FASEB J. 2004;18:209–211. doi: 10.1096/fj.03-0382fje. [DOI] [PubMed] [Google Scholar]
- 14.Sofer Y, Harel L, Sharkia M, Amir J, Schoenfeld T, Straussberg R. Neurological manifestations of folate transport defect: case report and review of the literature. J. Child. Neurol. 2007;22:783–786. doi: 10.1177/0883073807304004. [DOI] [PubMed] [Google Scholar]
- 15.Corbeel L, Van den Berghe G, Jaeken J, Van Tornout J, Eeckels R. Congenital folate malabsorption. Eur J Pediatr. 1985;143:284–290. doi: 10.1007/BF00442302. [DOI] [PubMed] [Google Scholar]
- 16.Urbach J, Abrahamov A, Grossowicz N. Congenital isolated folic acid malabsorption. Arch Dis Child. 1987;62:78–80. doi: 10.1136/adc.62.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safrin S, Lee BL, Sande MA. Adjunctive folinic acid with trimethoprimsulfamethoxazole for Pneumocystis carinii pneumonia in AIDS patients is associated with an increased risk of therapeutic failure and death. J. Infect. Dis. 1994;170:912–917. doi: 10.1093/infdis/170.4.912. [DOI] [PubMed] [Google Scholar]
- 18.Then RL. Antimicrobial dihydrofolate reductase inhibitors--achievements and future options: review. J. Chemother. 2004;16:3–12. doi: 10.1179/joc.2004.16.1.3. [DOI] [PubMed] [Google Scholar]
- 19.Jebnoun S, Kacem S, Mokrani CH, Chabchoub A, Khrouf N, Zittoun J. A family study of congenital malabsorption of folate. J. Inherit. Metab. Dis. 2001;24:749–750. doi: 10.1023/a:1012905823879. [DOI] [PubMed] [Google Scholar]
- 20.Poncz M, Colman N, Herbert V, Schwartz E, Cohen AR. Therapy of congenital folate malabsorption. J. Pediatr. 1981;98:76–79. doi: 10.1016/s0022-3476(81)80541-0. [DOI] [PubMed] [Google Scholar]
- 21.Spivak JL. Masked megaloblastic anemia. Arch. Intern. Med. 1982;142:2111–2114. [PubMed] [Google Scholar]
- 22.Min SH, Oh SY, Karp GI, Poncz M, Zhao R, Goldman ID. The clinical course and genetic defect in the PCFT gene in a 27-year-old woman with hereditary folate malabsorption. J Pediatr. 2008;153:435–437. doi: 10.1016/j.jpeds.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanzkowsky P. Congenital malabsorption of folate. Am. J. Med. 1970;48:580–3. doi: 10.1016/0002-9343(70)90007-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.