

Abstract
In previous studies we reported that key antioxidant and DNA repair genes are regulated differently in normal bronchial epithelial cells (NBEC) of lung cancer cases compared to non-lung cancer controls. In an effort to develop a biomarker for lung cancer risk, we evaluated transcript expression of 14 antioxidant, DNA repair and transcription factor genes in NBEC (HUGO names CAT, CEBPG, E2F1, ERCC4, ERCC5, GPX1, GPX3, GSTM3, GSTP1, GSTT1, GSTZ1, MGST1, SOD1, and XRCC1). A test comprising these 14 genes accurately identified the lung cancer cases in two case-control studies. The receiver operating characteristic (ROC) area under the curve (AUC) was 0.82 (95% CI, 0.68 – 0.91) for the first case-control set (25 lung cancer cases and 24 controls), and 0.87 (95% CI, 0.73 – 0.96) for the second set (18 cases and 22 controls). For each gene comprised by the test, the key difference between cases and controls was altered distribution of transcript expression among cancer cases compared to controls, with more lung cancer cases expressing at both extremes among all genes (K-S test, D=0.0795; P=0.041). A novel statistical approach was used to identify for each gene the lower and upper boundaries of transcript expression that optimally classifies cases and controls. Based on the data presented here, there is increased prevalence of lung cancer diagnosis among individuals that express a threshold number of key antioxidant, DNA repair and transcription factor genes at either very high or very low level in normal airway epithelium.
Keywords: Lung Cancer risk, biomarker, DNA repair, bronchial epithelial cells, airway epithelial cells
Introduction
Lung cancer is the leading cause of cancer mortality in both men and women in the United States with cigarette smoking being the primary known risk factor (1–4). Lung cancer is associated with a low survival rate in part because it typically is at an advanced stage when first detected and treated (5, 6). Studies to improve post-diagnosis outcome of lung cancer through early detection by means of low-dose spiral coaxial tomography (CT) screening and surgical intervention are promising (7–9). However, because as many as 90 million active or ex-smokers in the United States alone are candidates for screening the potential cost is very high and may be prohibitive (5). Additionally, CT screening studies completed thus far are associated with a high incidence of false positive findings which may lead to unnecessary follow-up diagnostic testing, including biopsies and surgical procedures, with associated risk and emotional and financial cost to the patient (10). Based on demographic criteria it is possible to identify a group of individuals for whom the 10 year risk for lung cancer is more than 20% (11, 12). Even in a group as selected as this, subjecting all individuals to close monitoring would be costly, associated with many false positive results, and lead to increased testing, some of which would take place in false positive individuals (13). A molecular genetic biomarker that identifies the subset of individuals at greatest risk for lung cancer within a demographically defined high risk group will enable even more focused selection for closer monitoring and further reduction in risk of false positive findings. Further, if CT screening is validated, limiting screening to the individuals with the highest demographic and biological risk will lead to marked reduction in costs of implementation (5, 14). Similarly, an accurate lung cancer risk biomarker will enable design of more effective chemoprevention trials.
There is reason to believe that genetic variability in key metabolic pathways is a primary determinant of inter-individual variation in lung cancer risk (15, 16). Prior work from this laboratory has focused on identifying differences in such pathways in primary normal bronchial epithelial cells (NBEC) of lung cancer cases compared to controls (17–20). These studies were guided by the hypothesis that increased risk for lung cancer is in part due to suboptimal regulation and/or function of a threshold number of key genes that protect NBEC from DNA damage. If true, genetic risk may manifest through differences in transcript expression profiles of these genes in NBEC between cases and controls. From these earlier investigations we identified a set of anti-oxidant and DNA repair genes that were differentially expressed in NBEC of cancer individuals compared to controls (21). More recently we determined that in a group of non-lung cancer control individuals there was significant inter-gene correlation of transcript expression values among 14 key DNA repair, antioxidant and transcription factor genes in NBEC samples (22). Conversely, these genes were not correlated in NBEC samples among individuals diagnosed with lung cancer. Moreover, the distribution of transcript expression among these 14 genes was different among those diagnosed with lung cancer compared to controls, with a higher frequency of more extreme values among the lung cancer cases. We hypothesized that the observed difference in expression between cases and controls might serve as the basis for a lung cancer risk test. Toward the goal of developing a test for lung cancer risk, we analyzed data from 49 individuals included in a previously reported study (22), as well as data from an independent set of 40 individuals.
Methods
Patients and Clinical Specimens
Patients were recruited at the University of Toledo Medical Center (UTMC) according to a protocol approved by the UTMC institutional review board. Inclusion criteria were willingness and ability to give informed consent, scheduled for diagnostic bronchoscopy, and age between 18 and 90. Exclusion criteria were HIV, Hepatitis B or TB infection or medical instability. Pregnant women and prisoners were excluded. NBEC samples were collected from a total of 90 patients. For each participating subject, BEC were obtained by 3–5 cytology brush biopsies of normal bronchium according to previously described methods (22). For lung cancer patients, sampling of normal bronchial epithelium was performed in the lung not involved with cancer. Of the 90 enrolled subjects included in this study (49 in the first case-control set, 41 in the second), gene expression data were obtained for 89 subjects (RNA from the BEC sample for one subject in the second case-control set failed reverse transcription due to faulty equipment). Patient information is presented in Supplementary Tables 1 and 2. In the first case-control set (Supplementary Table 1), normal BEC samples from 49 individuals, including 25 lung cancer and 24 non-lung cancer individuals, were evaluated. In the second case-control set (Supplementary Table 2), 40 normal BEC samples (18 lung cancer and 22 non-lung cancer subjects) were evaluated. We reviewed the charts of all patients. For each lung cancer patient, lung cancer diagnosis and subtype identification were determined in the Department of Pathology at UTMC by histopathological examination of tumor biopsy samples obtained at the time of bronchoscopy or at a separate biopsy procedure. For each non-lung cancer control, absence of lung cancer was determined by CT scan, bronchoscopy and (if biopsies conducted) pathology reports. Subjects in the first case-control set were recruited between 1997 and 2004 while those in the second case-control set were recruited between 1999 and 2008. There were no patient adverse events resulting from collecting the normal BEC samples.
Transcript Expression Measurement
Total RNA was extracted from normal BEC using TriReagent and reverse transcribed using M-MLV reverse transcriptase and oligo-dT primers as previously described (23–25). Standardized Reverse Transcriptase (StaRT)-PCR was used for transcript expression measurement in these studies. The fourteen genes measured in both sample sets were (with HUGO names provided in parentheses and used hereafter) catalase (CAT), CCAAT/enhancer binding protein gamma (CEBPG), E2F1 transcription factor (E2F1), excision repair cross-complementing rodent repair deficiency, complementation group 4 (ERCC4), excision repair cross-complementing rodent repair deficiency, complementation group 5 (ERCC5), glutathione peroxidase 1 (GPX1), glutathione peroxidase 3 (GPX3), glutathione S-transferase mu 3 (GSTM3), glutathione S-transferase pi 1 (GSTP1), glutathione S-transferase theta 1 (GSTT1), glutathione S-transferase zeta 1 (GSTZ1), microsomal glutathione S-transferase 1 (MGST1), superoxide dismutase 1 (SOD1), and X-ray repair complementing defective repair in Chinese hamster cells 1 (XRCC1). According to StaRT-PCR protocol (23–25), a known number of copies of an internal standard for each gene within a standardized mixture of internal standards (SMIS) was included in each PCR reaction. For the first case-control set (N=49) analysis was conducted at UTMC with StaRT-PCR reagents that were either obtained commercially (Gene Express, Inc, Wilmington, NC) or prepared according to previously described methods (23–25). For the second case-control set (N=40) analysis was conducted at Gene Express, Inc. using newly generated StaRT-PCR reagents prepared under Good Laboratory Practice conditions including carefully established standard operating procedures.
Statistical Analysis
When two groups are distinguished by a difference in central tendency of a variable, a single cut-point for the variable should be identified to classify each sample (Figure 1A). However, in our prior studies we observed that cases and controls did not differ significantly by central tendency, but instead differed by kurtosis, and to some degree, variation. Importantly, alteration in kurtosis or increased variation, but not change in central tendency, results in two inflection points on an ROC curve of the frequency distribution (Figure 1B). These two inflection points symbolize two cutpoints, a lower and upper boundary, which best classify cases and control. Here we rationalize that multiple mechanisms may result in either extremely low, or high expression of a given “Risk” gene, and that either low or high is indicative of suboptimal functionality of the gene in that pathway. Thus, when two groups are distinguished by a difference in distribution of a variable, two-cut points may offer the best criteria for sample classification (Figure 1B). In this study, the two best cut-points were derived using receiver operating characteristic (ROC) curve-based analysis using a modification of the recently described Youden Index (J) method (26). The Youden Index is equal to:
and was obtained from ROC analysis of transcript expression for each gene compared to the “truth” state of cancer or non-cancer. Using the Youden Index each cut-point was determined as the log transcript expression that yielded the maximum or minimum index representing the lower or upper boundary of transcript expression range associated with lower likelihood for lung cancer diagnosis (Figure 1B; ROC Plot). A cross-validation step (all possible combinations of leave-5-out) was applied to reduce spuriously derived cut-points (See Supplementary PERL files Cutpointmapper.pl and Combinatorialcreation.pl). The frequency mean of cut-points derived from cross-validation was taken as the final cut-point to be used in subsequent steps.
Figure 1. Schematic of two cut-point analysis to identify an informative gene with altered kurtosis or range in transcript expression between two populations. (One column width).
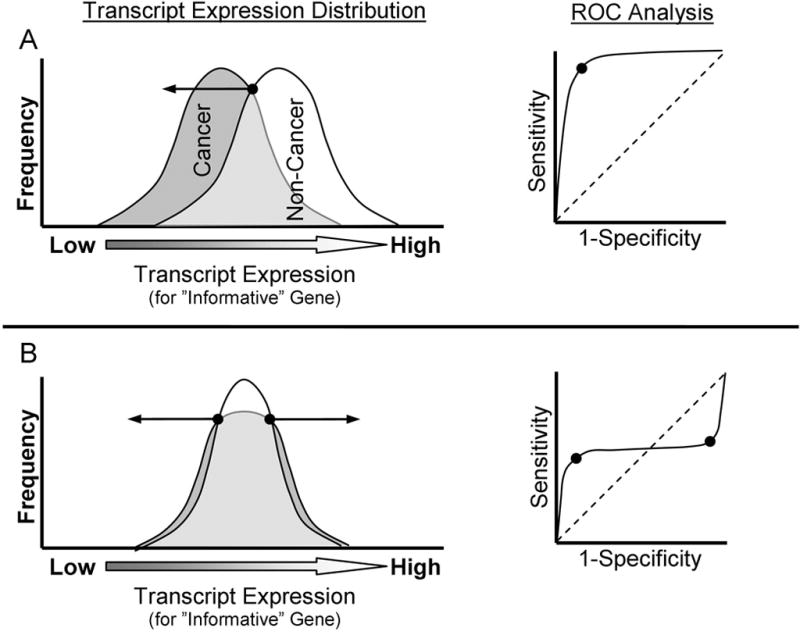
Shown in A and B are two depictions of case-control transcript expression frequency distribution plots for a trait of interest (e.g. Cancer [shaded] and Non-cancer [white]). Arrows stemming from the points on the frequency distribution plots indicate the range of values associated with higher prevalence of cancer diagnosis, and are derived from Reciever Operator Characterstics (ROC) identification of inflection point(s). A) The most common approach to identify informative genes is identification of difference in mean transcript expression between cases and controls (t-score criterion). B) However, for a set of genes with high prior likelihood of involvement in lung carcinogenesis, statistically significant difference in central tendency of transcript expression was not observed in normal airway tissue between lung cancer cases and controls (Mullins, 2005). Instead, lower prevalence of cancer cases were observed in the central region of the transcript expression distribution, with increased dispersion of cancer cases to extreme transcript expression levels. Using typical ROC analysis, ROC area under the curve was ~0.50 for each of the genes investigated, which may signify lack of informativeness in some discovery algorithms. However, using the approach described in methods section, ROC analysis identified two inflection points for each of these genes’ transcript expression profiles corresponding to the lower and upper transcript expression boundaries optimally separating cases from controls.
In each subject the score for each gene was specified as “1” if the log transcript expression value fell above or below the low-prevalence range based on the two cut-points or specified as “0” if the log transcript expression value fell within the low-prevalence range. The sum of scores for the component genes yielded each subject’s composite Risk Test Value (RTV), which could range from 0 (no genes outside the low prevalence range) to 14 (all genes expressed in extreme ranges). ROC analysis was then used to assess the performance of the composite marker RTV in classifying cases and controls (27). The Wald test was used to determine significance of odds ratio confidence intervals. The RTV for the 14-gene composite biomarker also was assessed for significant covariation with age or smoking history using Pearson’s correlation and assessed for association with gender or race (Caucasian vs. other) using Student’s t-test.
A multivariable risk model analysis for the proportion of patients that have cancer was conducted using logistic regression. This approach enabled simultaneous adjustment for significant covariates. The covariates considered in the model included age, smoking history, gender, race and the RTV. In univariate logistic screening to determine which variables to include in the competition for the final model, selection was based on the P-value of the covariate being less than 0.10. All variables significant by univariate analysis and pairwise interaction terms comprised the initial risk model. The final model was obtained by manual backward elimination in which the term with the highest P-value was removed from the model at each step and the model was rerun. The final model contained terms with P-values of 0.05 or less. Logistic regression was used to assess the odds ratio for the RTV in a subset of individuals of age 50 or over and smoking history of 20 pack years or greater and in this subset adjusted for age, smoking history and gender.
Kolmorogov-Smirnov test was used to test for significant difference between case and control groups (each comprising the combined data sets from first and second study) with respect to the composite distribution of transcript expression values (Figure 2).
Figure 2. Lung Cancer cases have a lower prevalence of median transcript expression than non-cancer controls. (One column width).
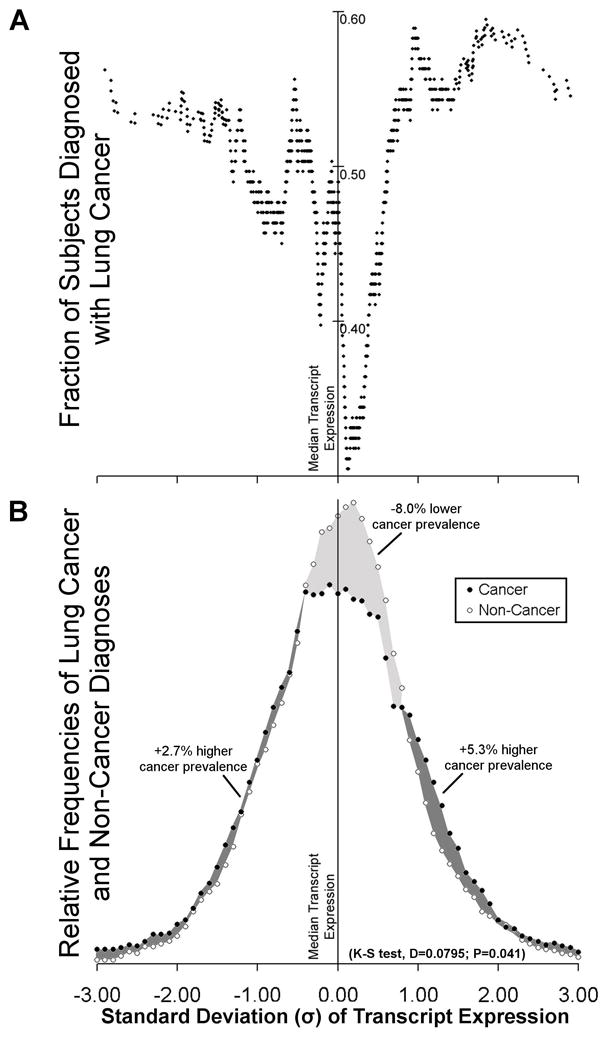
Median transcript expression measurement was determined for each gene among all 89 individuals from both case-control sets. Each transcript expression measurement was then converted to units of Standard Deviation (σ) from the median transcript expression value (Supplementary Table 3; z-score transformation). Both Panels A and B share the same x-axis. A) For each gene, the average fraction of individuals diagnosed with lung cancer relative to non-cancer controls across all transcript expression windows was normalized to 0.5 before binning. Using the composite value from all fourteen genes the moving average of subjects diagnosed with lung cancer was plotted in windowed increments of nearest transcript expression measurements (see Supplementary Table 3 for data analysis). B) Frequency histogram of lung cancer and non-cancer diagnoses of the data plotted in panel A. Area under the frequency distribution curves for lung-cancer and non-cancer populations was normalized to 100% for each category. Darker shading indicates areas where transcript expression exhibits greater prevalence of lung cancer cases compared to controls. Lighter shading indicates areas where transcript expression regions exhibit lower prevelance of lung cancer cases compared to controls. Percentages of change in cancer prevalence shown are calculated from the net difference in area under the curve between lung cancer and non-cancer cases in each of the three shaded areas. K-S test for significant difference in composite transcript expression distribution for lung cancer cases and controls is shown.
Results
Greater dispersion in transcript expression is associated with lung cancer diagnosis
Composite analysis of the fractional distribution of lung cancer cases and controls relative to the median transcript expression for the 14 genes (CAT, CEBPG, E2F1, ERCC4, ERCC5, GPX1, GPX3, GSTM3, GSTP1, GSTT1, GSTZ1, MGST1, SOD1, and XRCC1) enabled understanding of the average effect of each gene. This demonstrated higher fraction of cancer cases at extreme transcript expression ranges. Conversely, there was a narrower unimodal distribution of controls centered over the median expression value (Figure 2A). On average each gene exhibited 8.0% lower prevalence of lung-cancer diagnoses relative to non-cancer controls in the median transcript expression regions, and a cumulative 8.0% increase spread approximately evenly in each transcript expression extreme, both low and high (Figure 2B). This altered transcript expression frequency distribution for cancer cases (Kurtosis = +0.767), compared to controls (Kurtosis = +0.439), was significant (D=0.0795; P=0.041) by Kolmorogov-Smirnov test (Figure 2B and Supplementary Table 3). Because the genes studied exhibited difference in transcript expression dispersion (kurtosis) and to some extent range but little difference in central tendency (Figure 2), two cut-points were found to best differentiate between cases and controls in the two groups (Figure 1B). For each gene in each case-control set, two cut-point values were identified by ROC analysis using the Cutpointmapper.pl algorithm (Figure 3, Supplementary Table 4 and Statistical Methods). For each individual gene, these cut-points distinguished cancer from non-cancer control groups with an accuracy of between 0.53 and 0.75, where 0.50 is no better than guessing (normalized to a fractional scale of 0 to 1). The non-cancer transcript expression control range identified by analysis of the second case-control set closely matched the corresponding range identified through analysis of the first case-control set for all genes (Figure 3).
Figure 3. Two transcript expression cut-points best separate cancer from non-cancer. (one column width).
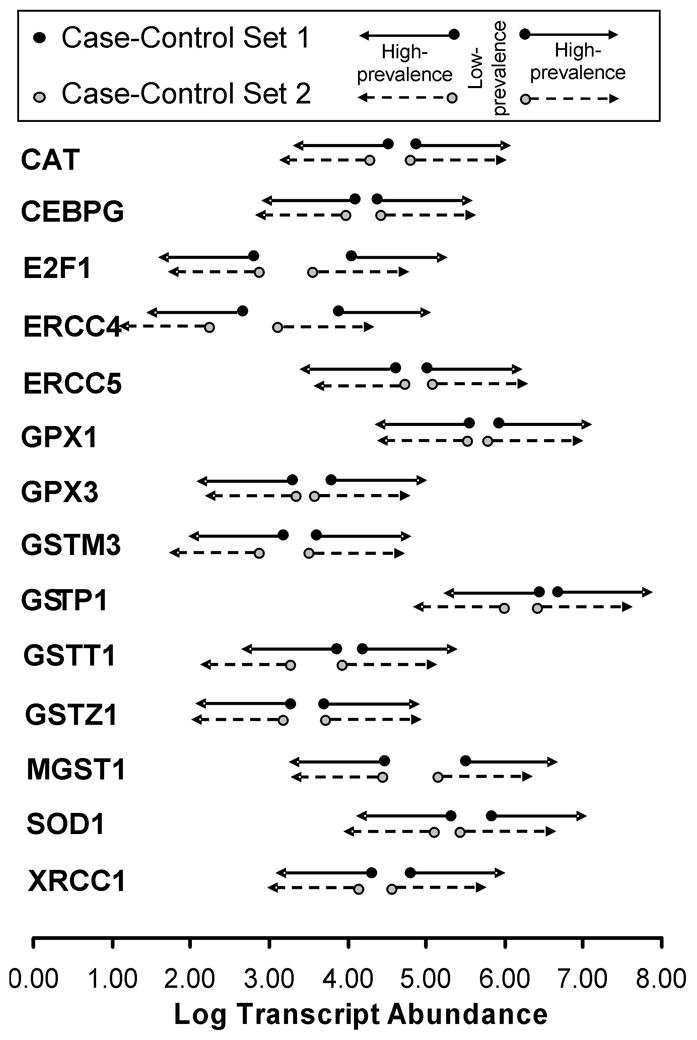
Using the modified Youden Index (J) method, for each of the 14 antioxidant, DNA repair and transcription factor genes, two cut-points were identified that best separated cancer from non-cancer (Supplementary Table 4). Cut-point levels are displayed in units of Log10 transformed target gene transcript abundance molecules per 106 ACTB transcript molecules. Arrows stemming from the points indicate the range of values with higher likelihood of cancer diagnosis compared to the ranges between the two cut-points, which are indicative of the range of values associated with lower likelihood of cancer diagnosis. Genes are listed in HUGO gene nomenclature format.
An accurate biomarker for lung cancer diagnosis comprises multiple genes
Figure 2A and B demonstrate average effect of a single gene on classification of cancer versus non-cancer. The combination of single gene effects from each of the 14 genes leads to the high accuracy of the LCRT. For each subject, each of the 14 genes was assigned a zero (0) or one (1) depending on whether the transcript expression for that gene was inside a range indicative of non-cancer (0) or outside that range (1). The sum of the values assigned to each gene for each subject was then used as a Risk Test Value (RTV; see Methods) and ROC analysis was used to compare the RTV to known cancer status. The ROC area under the curve (AUC) for the first case-control set (N=49) was 0.82 (95% CI, 0.68 – 0.91) (Figure 4). The best RTV cut-off (>8 genes) for separating cancer case group from non-cancer control group had an accuracy of 80% and an odds ratio of 12.8 (95% CI, 3.2 – 50.9; P<0.001). When applied to the second case-control set (N=40), the ROC AUC was 0.87 (95% CI, 0.73 – 0.96) and the same cut-off had an accuracy of 80% and an odds ratio of 15.8 (95% CI, 3.3 – 74.3; P<0.001). For the combined case-control sets (N=89), the ROC AUC was 0.84 (95% CI, 0.75–0.91), the accuracy of the cut-off was 0.80 and the odds ratio was 13.9 (95% CI, 5.0 – 38.8; P<0.001). For the subset of individuals of age 50 or over with 20 pack-year smoking history or greater (N=49) using logistic regression the odds ratio for the same cut-off was 8.17 (95%CI, 2.13 – 31.4; P=0.002). In this subset adjusted for age, smoking history and gender the odds ratio for this cut-off was 8.31 (95%C, 1.8 – 37.7; P=0.006).
Figure 4. ROC analysis of the 14-gene composite lung cancer marker. (two column width).
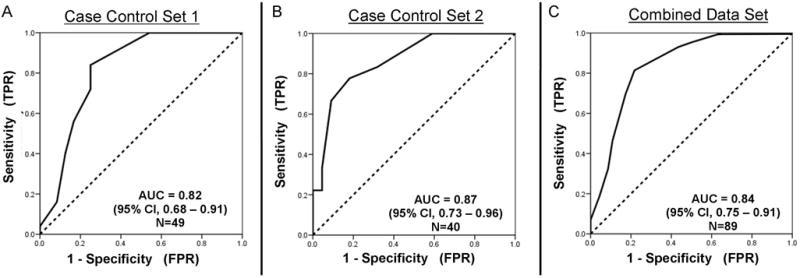
ROC analysis was used to assess the ability of RTV to correctly classify each subject into the cancer or non-cancer group in the first case-control set (panel A), second set (panel B) or combined sets (panel C). AUC = Area Under the receiver Curve.
Risk Test Value is independent of important demographic factors associated with lung cancer prevalence
There was no association between RTV and age (P=0.09) or race (P=0.99) in either individual set or the combined set but males had a higher RTV (~1 index point) than females in the combined set (P<0.05). Smoking history was not significantly associated with RTV for either case-control set but was (P<0.05) for the combined sets (N=89). However, the R2 value for smoking history in the combined set was 0.11, indicating that it explained only 11% of the variance in RTV.
Multivariate Analysis
In univariate analysis, smoking history (P<0.001), age (P=0.003), gender (P=0.002) and the 14-gene RTV (P<0.001) predicted cancer status for the combined case-control set (N=89). Although race did not predict cancer status (P=0.20), the numbers were too small to reach firm conclusion. Many of the subjects were long-time smokers and so there was a significant correlation (P=0.009) between age and smoking history. Following backward elimination, the strongest multivariate model was the age x RTV interaction term (P<0.001) (Figure 5). The age x RTV term was more predictive than the Smoking History x RTV term only because there were fewer cancers at lower ages yet the incidence of cancer spanned the whole range of smoking history.
Figure 5. Lung cancer discrimination using Risk Test Value and age. (two column width).
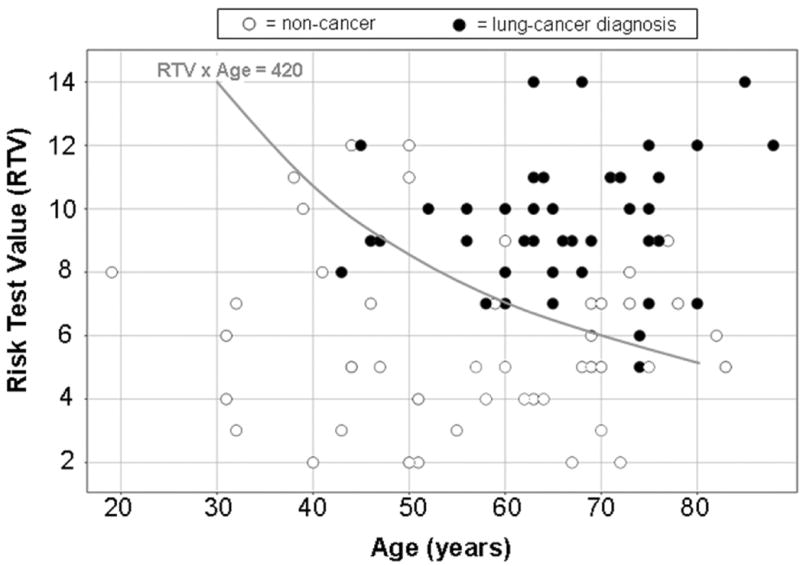
Plotted is the multigene Risk Test Value (RTV) as a function of age (years) for the combined set of 89 bronchial epithelial cell samples. RTV x age = 420 gave the best discrimination between lung cancer case samples and controls.
Discussion
The data presented in this study validate our previously reported observation that there is increased dispersion around the transcript expression median (kurtosis) for a set of 14 antioxidant, DNA repair and transcription factor genes in the NBEC of lung cancer cases compared to non-lung cancer controls (Figure 2) (22). These data suggest that susceptibility to lung cancer may be characterized as expression at relatively extreme levels (either high or low for each gene) for a combination of key genes in cellular pathways responsible for DNA repair, antioxidant protection and transcription regulation in normal airway epithelium (15, 16). Further, these results support previous studies indicating that inter-individual variation in risk for lung cancer may be in part dependent on DNA repair gene and antioxidant gene function (28–32). Inter-individual variation in antioxidant and DNA repair protection may be particularly important as a determinant in the previously reported relationship between chronic inflammation and lung cancer risk (33–35).
The observed higher transcript expression dispersion appears to be a corollary of prior observations of differences in normal airway epithelium inter-gene transcript expression correlation between cases and controls (22). The two-cutpoint approach described here may aid in identification of clinically useful diagnostics for other diseases from genes exhibiting altered transcript expression dispersion between cases and controls (Figures 2 and 3). For example, a very similar transcript expression distribution phenomenon for a set of bone morphogenic, inflammatory and transcription factor genes was observed in the peripheral blood of newly diagnosed polyarticular juvenile rheumatoid arthritis patients compared to healthy donor controls (36). This suggests that transcript expression dispersion may be an important parameter to assess as an indicator of risk for a variety of diseases. The ability to observe this phenomenon may be in part dependent on the transcript abundance analytical tool used. In studies directly comparing the analytical performance of RT-PCR methods compared to microarray platforms, RT-PCR methods had 2–3 orders of magnitude greater linear dynamic range and 1–2 orders of magnitude lower detection threshold (23). The extended measurement range of RT-PCR may be necessary to observe the inter-group difference in transcript abundance dispersion described here.
The set of genes that separates lung cancer cases from non-lung cancer controls in this study have different characteristics from a set of genes recently reported to have similar classification capabilities discovered through high density microarray analysis by Spira et al (37). One difference is that 12 of the 14 genes reported here are key anti-oxidant or DNA repair genes while the remaining two are transcription factors expressed in normal airway epithelium. In contrast, the set of genes described in the Spira report comprises primarily signal transduction and small molecule transport genes (Supplementary Figure 1). A second difference is that, as described above, each of the genes comprised by the multigene test reported here has increased dispersion among the lung cancer cases rather than altered mean level. In contrast, each of genes reported by Spira et al have altered central tendency of expression. These differences likely result from the different investigative and analytical approaches taken. The set of genes reported here were discovered initially through quantitative PCR analysis of key genes known through previous studies to have a key role in protection of the airway epithelium from DNA damage secondary to oxidants and carcinogens in cigarette smoke (22). In contrast, in the Spira study an initially unsupervised high density microarray analysis was used (38). Because these different sets of genes have very different functions (Supplementary Figure 1), it is reasonable to hypothesize that each set may play an independent role in determining lung cancer risk. If so, the most accurate test for lung cancer risk may be derived by combining genes from each set.
The observed increased dispersion in gene expression in normal bronchial epithelial cells of lung cancer cases reported here could result from inheritance at the germ cell level, acquisition of genetic alterations in somatic cells in the airway epithelium, previously described as a field effect (33–35, 39–42)or through a combination of both. Based on the data reported here, it is likely that germ cell inheritance plays a role. For example, field effect is typically observed in all smokers, not just those with lung cancer (39). In contrast, the increased variation in antioxidant and DNA repair gene expression reported here separated the lung cancer case group from the non-lung cancer control group and was largely unrelated to cigarette smoking. This indicates that there is inter-individual variation in the field effect of cigarette smoking and, although there are multiple possible explanations, it is reasonable to hypothesize that the basis for this variation is germ cell inheritance. This hypothesis is supported by accumulating evidence for germ cell inheritance of particular cis-element SNPs that cause inter-individual variation in regulation of the genes comprised by this test and that are associated with increased lung cancer risk. Specifically, particular polymorphisms in the regulatory region of ERCC5 are associated with increased dispersion of transcript expression around its median expression value and altered prevalence of lung cancer diagnosis (unpublished data). Recently a polymorphism in the regulatory region of XRCC1 was found to be significantly associated with altered XRCC1 expression and increased lung cancer risk (31). Both ERCC5 and XRCC1 are among the 14 genes comprised by the multigene test reported here. We hypothesize that germ cell inheritance of particular alleles may be associated with increased range and dispersion of transcript expression in the other genes comprised by this test as well.
Previous studies of lung cancer risk support the conclusion that many genes contribute to determining risk for lung cancer, and that the regulation/function of each gene is affected by numerous inter-individual genetic differences (43). In this study, assessing for alteration in transcript expression dispersion facilitated identification of molecular genetic tests for risk of lung cancer by more effectively taking into account the likely subtle but cumulative genetic etiology of complex disease risk. Application of analytical and statistical methods employed here may be a robust way to directly assess function of the many pathways involved in maintaining a particular normal phenotype. The findings described here are consistent with the hypothesis that in high risk individuals A) a threshold number of key protective genes function at a sub-optimal level in NBEC and B) chronic inhalation of cigarette smoke causes sub-optimally protected NBEC to experience higher levels of DNA damage, and subsequently higher risk of malignant transformation. One explanation for increased range/dispersion of expression for each gene in the lung cancer group is that in some individuals sub-optimal function of protein product(s) induces feed-back signals that upregulate transcription while in other genes a sub-optimal regulatory apparatus causes inappropriate down regulation of transcript expression.
The multigene lung cancer risk test value (RTV) based on the two-cutoff per gene approach reported here quantifies determinants of lung cancer diagnosis independent of the well-documented demographic factors, smoking and age. Thus, even among individuals in the sub-set over the age of 50 and with greater than 20 pack years smoking history, the odds ratio that an individual with a positive multigene test would have lung cancer was >8. It is important to note that the best RTV cutoff value for classifying lung cancer cases and controls significantly decreased with increasing age (Figure 5) and smoking history. For example, based on the data in Figure 5, a heavy smoker (>20 pack years) with an RTV of 6 may not have sufficient risk at age 50 to warrant increased surveillance (Age x RTV < 420), yet would be predicted to have sufficient risk upon reaching the age of 75 (Age x RTV > 420) (Figure 5). Since many of the subjects were long-time smokers and there was a significant correlation between age and smoking history it is clear that the age term included some of the predictive effects of smoking history as well as age specific phenomena in the case-control sets assessed here. Larger prospective studies will need to be done in order to better distinguish the effects of RTV at various age intervals with and without smoking history effects for predicting lung cancer risk. The modest correlation between gender and RTV is also intriguing. One possible interpretation is that this might contribute to the observed differences in male to female ratio for lung cancer among smokers compared to non-smokers (44). The gender effect observed here will be explored more fully in larger case-control and prospective studies in addition to a more thorough analysis of the effect of race on RTV and lung cancer diagnosis.
Case control studies are a powerful method to identify risk factors that contribute to disease with rare incidence (28–30, 45). However, as with any case-control study the data reported here may be subject to misinterpretation due to unknown factors that were not controlled. For example, one interpretation is that increased variation in antioxidant and DNA repair gene expression in NBEC is the result, rather than a cause of the cancer. However, this interpretation is not supported by the available evidence. For example, because the normal airway epithelial samples were obtained from the lung opposite the one harboring the cancer, local signaling is less likely to explain observed differences in transcript expression profiles in NBEC. In addition, none of the patients had yet received radiation, chemotherapy or other intervention, which might be associated with altered transcript expression levels. Moreover, evidence acquired thus far indicates that the antioxidant and target genes measured in this study are not inducible by cigarette smoke or other oxidant exposure (21, 22). Rather, the evidence indicates that these genes are subject to inter-individual variation in constitutive expression. In order to directly test the hypothesis that the expression patterns observed here are a cause of, rather than an effect of lung cancer, a larger prospective nested case control trial will need to be done. Prospective validation of the multigene test described here will enable identification of the subset of individuals at highest risk for lung cancer so that they may be more closely monitored for early detection or selected for entry into promising early detection and/or chemoprevention studies. This test was measured in NBEC obtained through bronchoscopy. This procedure is at least as safe and well-tolerated as colonoscopy which is now commonly used for individuals over the age of 50 to screen for early-stage colon carcinoma (46).
Supplementary Material
Acknowledgments
The authors would like to thank Albert Levine and Christine C. Johnson, for their constructive comments and review.
Footnotes
Conflict of Interest Statement: James C. Willey has significant equity interest in Gene Express, Inc., which produces and markets StaRT-PCRTM reagents used in this study.
Authors' contributions
TB, ELC, DM and JCW all contributed to the design, interpretation and writing of the manuscript as a whole. YY, DH, and JCW each contributed to the acquisition of primary bronchial epithelial cell samples used in this study. ELC and DM contributed to the measurement of gene-expression measurements for case-control set 1. EP, DJO, BA and JCA contributed to the measurement of gene-expression measurements for case-control set 2. PLR and SK provided biostatistical analysis and review. TB, PLR and JW contributed significantly to the rationale, development and implementation of a two cut-point statistical approach for discriminating cases and controls. JCW was responsible for overall conduct and direction of the study and final approval of the manuscript. All authors read and approved the final manuscript.
References
- 1.Alberg AJ, Samet JM. Epidemiology of lung cancer. Chest. 2003;123:21S–49S. doi: 10.1378/chest.123.1_suppl.21s. [DOI] [PubMed] [Google Scholar]
- 2.Gloeckler Ries LA, Reichman ME, Lewis DR, Hankey BF, Edwards BK. Cancer survival and incidence from the Surveillance, Epidemiology, and End Results (SEER) program. Oncologist. 2003;8:541–52. doi: 10.1634/theoncologist.8-6-541. [DOI] [PubMed] [Google Scholar]
- 3.Doll R, Hill AB. A study of the aetiology of carcinoma of the lung. Br Med J. 1952;2:1271–86. doi: 10.1136/bmj.2.4797.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shields PG. Molecular epidemiology of lung cancer. Ann Oncol. 1999;10 (Suppl 5):S7–11. doi: 10.1093/annonc/10.suppl_5.s7. [DOI] [PubMed] [Google Scholar]
- 5.Ganti AK, Mulshine JL. Lung cancer screening. Oncologist. 2006;11:481–7. doi: 10.1634/theoncologist.11-5-481. [DOI] [PubMed] [Google Scholar]
- 6.Fry WA, Phillips JL, Menck HR. Ten-year survey of lung cancer treatment and survival in hospitals in the United States: a national cancer data base report. Cancer. 1999;86:1867–76. doi: 10.1002/(sici)1097-0142(19991101)86:9<1867::aid-cncr31>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 7.Sone S, Nakayama T, Honda T, et al. Long-term follow-up study of a population-based 1996–1998 mass screening programme for lung cancer using mobile low-dose spiral computed tomography. Lung Cancer. 2007;58:329–41. doi: 10.1016/j.lungcan.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 8.Unger M. A pause, progress, and reassessment in lung cancer screening. N Engl J Med. 2006;355:1822–4. doi: 10.1056/NEJMe068207. [DOI] [PubMed] [Google Scholar]
- 9.Henschke CI, Yankelevitz DF, Libby DM, Pasmantier MW, Smith JP, Miettinen OS. Survival of patients with stage I lung cancer detected on CT screening. N Engl J Med. 2006;355:1763–71. doi: 10.1056/NEJMoa060476. [DOI] [PubMed] [Google Scholar]
- 10.Bach PB, Jett JR, Pastorino U, Tockman MS, Swensen SJ, Begg CB. Computed tomography screening and lung cancer outcomes. Jama. 2007;297:953–61. doi: 10.1001/jama.297.9.953. [DOI] [PubMed] [Google Scholar]
- 11.Cronin KA, Gail MH, Zou Z, Bach PB, Virtamo J, Albanes D. Validation of a model of lung cancer risk prediction among smokers. J Natl Cancer Inst. 2006;98:637–40. doi: 10.1093/jnci/djj163. [DOI] [PubMed] [Google Scholar]
- 12.Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst. 2007;99:715–26. doi: 10.1093/jnci/djk153. [DOI] [PubMed] [Google Scholar]
- 13.Wilson DO, Weissfeld JL, Fuhrman CR, et al. The Pittsburgh Lung Screening Study (PLuSS): outcomes within 3 years of a first computed tomography scan. Am J Respir Crit Care Med. 2008;178:956–61. doi: 10.1164/rccm.200802-336OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chirikos TN, Hazelton T, Tockman M, Clark R. Cost-effectiveness of screening for lung cancer. Jama. 2003;289:2358. doi: 10.1001/jama.289.18.2358-a. author reply -9. [DOI] [PubMed] [Google Scholar]
- 15.Reid ME, Santella R, Ambrosone CB. Molecular epidemiology to better predict lung cancer risk. Clin Lung Cancer. 2008;9:149–53. doi: 10.3816/CLC.2008.n.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz AG, Prysak GM, Bock CH, Cote ML. The molecular epidemiology of lung cancer. Carcinogenesis. 2007;28:507–18. doi: 10.1093/carcin/bgl253. [DOI] [PubMed] [Google Scholar]
- 17.Crawford EL, Weaver DA, DeMuth JP, et al. Measurement of cytochrome P450 2A6 and 2E1 gene expression in primary human bronchial epithelial cells. Carcinogenesis. 1998;19:1867–71. doi: 10.1093/carcin/19.10.1867. [DOI] [PubMed] [Google Scholar]
- 18.Willey JC, Coy E, Brolly C, et al. Xenobiotic metabolism enzyme gene expression in human bronchial epithelial and alveolar macrophage cells. Am J Respir Cell Mol Biol. 1996;14:262–71. doi: 10.1165/ajrcmb.14.3.8845177. [DOI] [PubMed] [Google Scholar]
- 19.Willey JC, Coy EL, Frampton MW, et al. Quantitative RT-PCR measurement of cytochromes p450 1A1, 1B1, and 2B7, microsomal epoxide hydrolase, and NADPH oxidoreductase expression in lung cells of smokers and nonsmokers. Am J Respir Cell Mol Biol. 1997;17:114–24. doi: 10.1165/ajrcmb.17.1.2783. [DOI] [PubMed] [Google Scholar]
- 20.Willey JC, Crawford EL, Olson D, et al. Expression measurement of genes related to cancer susceptibility in human bronchial epithelial cells. In: Mohr U, editor. Relationships between acute and chronic effects of air pollution. Washington D.C: ILSI Press; 2000. pp. 79–96. [Google Scholar]
- 21.Crawford EL, Khuder SA, Durham SJ, et al. Normal bronchial epithelial cell expression of glutathione transferase P1, glutathione transferase M3, and glutathione peroxidase is low in subjects with bronchogenic carcinoma. Cancer Res. 2000;60:1609–18. [PubMed] [Google Scholar]
- 22.Mullins DN, Crawford EL, Khuder SA, Hernandez DA, Yoon Y, Willey JC. CEBPG transcription factor correlates with antioxidant and DNA repair genes in normal bronchial epithelial cells but not in individuals with bronchogenic carcinoma. BMC Cancer. 2005;5:141. doi: 10.1186/1471-2407-5-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canales RD, Luo Y, Willey JC, et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol. 2006;24:1115–22. doi: 10.1038/nbt1236. [DOI] [PubMed] [Google Scholar]
- 24.Willey JC, Crawford EL, Knight CA, et al. Use of Standardized Mixtures of Internal Standards in Quantitative RT-PCR to Ensure Quality Control and Develop a Standardized Gene Expression Database. In: Bustin SA, editor. A–Z of Quantitative PCR. 1. La Jolla, CA: International University Line; 2004. pp. 545–76. [Google Scholar]
- 25.Willey JC, Crawford EL, Knight CR, et al. Standardized RT-PCR and the standardized expression measurement center. Methods Mol Biol. 2004;258:13–41. doi: 10.1385/1-59259-751-3:13. [DOI] [PubMed] [Google Scholar]
- 26.Perkins NJ, Schisterman EF. The inconsistency of “optimal” cutpoints obtained using two criteria based on the receiver operating characteristic curve. Am J Epidemiol. 2006;163:670–5. doi: 10.1093/aje/kwj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SAS. 9.1 ed. Cary, NC: SAS Institute Inc; 2003. [Google Scholar]
- 28.Kiyohara C, Yoshimasu K. Genetic polymorphisms in the nucleotide excision repair pathway and lung cancer risk: a meta-analysis. Int J Med Sci. 2007;4:59–71. doi: 10.7150/ijms.4.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zienolddiny S, Campa D, Lind H, et al. A comprehensive analysis of phase I and phase II metabolism gene polymorphisms and risk of non-small cell lung cancer in smokers. Carcinogenesis. 2008;29:1164–9. doi: 10.1093/carcin/bgn020. [DOI] [PubMed] [Google Scholar]
- 30.Zienolddiny S, Campa D, Lind H, et al. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis. 2006;27:560–7. doi: 10.1093/carcin/bgi232. [DOI] [PubMed] [Google Scholar]
- 31.Hao B, Miao X, Li Y, et al. A novel T-77C polymorphism in DNA repair gene XRCC1 contributes to diminished promoter activity and increased risk of non-small cell lung cancer. Oncogene. 2006;25:3613–20. doi: 10.1038/sj.onc.1209355. [DOI] [PubMed] [Google Scholar]
- 32.Spitz MR, Wei Q, Dong Q, Amos CI, Wu X. Genetic susceptibility to lung cancer: the role of DNA damage and repair. Cancer Epidemiol Biomarkers Prev. 2003;12:689–98. [PubMed] [Google Scholar]
- 33.Walser T, Cui X, Yanagawa J, et al. Smoking and lung cancer: the role of inflammation. Proc Am Thorac Soc. 2008;5:811–5. doi: 10.1513/pats.200809-100TH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynolds PR, Cosio MG, Hoidal JR. Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cells. Am J Respir Cell Mol Biol. 2006;35:314–9. doi: 10.1165/rcmb.2005-0428OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith CJ, Perfetti TA, King JA. Perspectives on pulmonary inflammation and lung cancer risk in cigarette smokers. Inhal Toxicol. 2006;18:667–77. doi: 10.1080/08958370600742821. [DOI] [PubMed] [Google Scholar]
- 36.Dozmorov I, Knowlton N, Tang Y, et al. Hypervariable genes-- experimental error or hidden dynamics. Nucleic Acids Res. 2004;32:e147. doi: 10.1093/nar/gnh146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spira A, Beane JE, Shah V, et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007;13:361–6. doi: 10.1038/nm1556. [DOI] [PubMed] [Google Scholar]
- 38.Spira A, Beane J, Shah V, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–8. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wistuba II. Genetics of preneoplasia: lessons from lung cancer. Curr Mol Med. 2007;7:3–14. doi: 10.2174/156652407779940468. [DOI] [PubMed] [Google Scholar]
- 40.Guo M, House MG, Hooker C, et al. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin Cancer Res. 2004;10:5131–6. doi: 10.1158/1078-0432.CCR-03-0763. [DOI] [PubMed] [Google Scholar]
- 41.Franklin WA, Gazdar AF, Haney J, et al. Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest. 1997;100:2133–7. doi: 10.1172/JCI119748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 43.Christiani DC. Genetic susceptibility to lung cancer. J Clin Oncol. 2006;24:1651–2. doi: 10.1200/JCO.2005.05.4114. [DOI] [PubMed] [Google Scholar]
- 44.Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer. 2005;117:294–9. doi: 10.1002/ijc.21183. [DOI] [PubMed] [Google Scholar]
- 45.Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–22. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380. N Engl J Med. 2000;343:162–8. doi: 10.1056/NEJM200007203430301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.