

Abstract
(S)-3-((R)-9-bromo-4-methoxy-6-methyl-5,6,7,8-tetrahydro-[1,3]dioxolo[4,5-g]isoquino-lin-5-yl)-6,7-dimethoxyisobenzofuran-1(3H)-one (EM011) is a tubulin-binding agent with significant anticancer activity. Here we show that EM011 modulates microtubule dynamics at concentrations that do not alter the total polymer mass of tubulin. In particular, EM011 decreases the transition frequencies between growth and shortening phases and increases the duration microtubules spend in an idle ‘pause’ state. Using B16LS9 murine melanoma cells, we show that EM011 briefly arrests cell-cycle progression at the G2/M phase by formation of multiple aster spindles. An aberrant mitotic exit without cytokinesis then occurs, leading to the accumulation of abnormal multinucleated cells prior to apoptosis. Our pharmacokinetic studies conformed to a linear dose-response relationship upto 150 mg/kg. However, non-linearity was observed at 300 mg/kg. In a syngeneic murine model of subcutaneous melanoma, better antitumor responses were seen at 150 mg/kg compared to 300 mg/kg of EM011. Unlike currently available chemotherapeutics, EM011 is non-toxic to normal tissues and most importantly, does not cause any immunosuppression and neurotoxicity. Our data thus warrant a clinical evaluation of EM011 for melanoma therapy.
Keywords: microtubule dynamics, tubulin-binding agent, neurotoxicity, pharmacokinetics, apoptosis, melanoma
INTRODUCTION
Microtubule-targeting drugs have been powerful chemotherapeutic agents for treatment of a variety of human cancers1,2. Their activities cause either polymerization and bundling of microtubules (taxanes) or depolymerization and decrease in polymer mass (vincas). Thus, these agents not only halt proliferation of mitotically active cells but also jam up microtubule tracks responsible for axonal transport, causing peripheral neuropathies3-5. Furthermore, due to their lack of specificity for cancer cells, tubulin-binding agents also affect other rapidly dividing cells within healthy tissues resulting in gastrointestinal toxicities (diarrhea, nausea), myelosuppression (leucopenia), and alopecia4. Also, their high lipophilicity demands use of solvents like cremophor, which are associated with adverse side effects. All these shortcomings have driven intense efforts to explore novel tubulin-binding agents that have reduced toxicity, and superior pharmacological profiles.
We recently reported on a semi-synthetic brominated analog6 of the naturally-occurring, tubulin-binding, opiate noscapine. This analog, EM011 ((S)-3-((R)-9-bromo-4-methoxy-6-methyl-5,6,7,8-tetrahydro-[1,3]dioxolo[4,5-g]isoquinolin-5-yl)-6,7-dimethoxyisobenzofuran-1(3H)-one) is ~10-40 fold more active than noscapine, binds tubulin with a higher affinity than noscapine, and does not alter the monomer/polymer ratio of tubulin at concentrations as high as 100 μM7. EM011 inhibits cellular proliferation and causes G2/M arrest followed by apoptotic cell death of various human cancer cells both in vitro and in vivo6-10. Accordingly, we wished to further test the anti-cancer effects of EM011 on a particularly aggressive cancer, malignant melanoma. Malignant melanoma diagnosed at advanced stages has a poor prognosis (median survival of <1 year) for which the armamentarium of drugs is restricted to the five most commonly used ones (cisplatin, dacarbazine, carmustine, temozolomide, and interleukin-2), all of which cause serious toxicities11-14.
In this study, we have analyzed the mechanism by which EM011 most likely blocks mitotic progression and established, using a murine model of subcutaneously implanted melanoma, EM011 as an effective antitumor agent that not only lacks toxicity towards mitotically active normal tissues but also causes no detectable neurotoxicity. Thus, our data provide compelling evidence for a clinical evaluation of EM011 for non-toxic melanoma therapy.
MATERIALS AND METHODS
Reagents and cell culture
EM011 was synthesized from noscapine (Sigma, St. Louis, MO) as described earlier6. Taxol (paclitaxel) was from Sigma. Cell culture reagents were obtained from Mediatech, Cellgro. Murine B16LS9 melanoma cells were grown as described previously15.
Analysis of microtubule dynamics
Analysis of microtubule behavior in interphase cells was performed in a porcine renal cell line stably expressing GFP-α-tubulin, namely LLCPK-1α. Briefly, cells treated with 25 μM EM011 or vehicle (DMSO) for 60 min were imaged every 3 seconds for 2 min using 100X Apochromatic (NA = 1.4) oil lens on the PerkinElmer Ultraview spinning-disk confocal system (mounted on a Zeiss-Axiovert 200M microscope) fitted with a Hamamatsu ORCA-ER digital camera. Microtubule dynamics and other parameters shown in Suppl. Table 1 were calculated as described earlier15. All P values were calculated using Student’s t test.
In vitro cell proliferation, immunofluorescence microscopy, and cell-cycle studies
The cell proliferation assay was performed using sulforhodamine B as described16. Immunofluorescence confocal microscopy and cell-cycle studies were also performed as previously described9.
In vivo analysis of melanoma progression
Pathogen-free 8-10 week-old female C57BL/6J mice were obtained from Jackson Animal Laboratories (Bar Harbor, ME) and housed in the Emory University Animal Care Facility. To determine the ability of orally-delivered EM011 to affect melanoma progression, mice (n=8 mice/group) were administered 150 or 300 mg/kg EM011 in acidified deionized water (pH 4.0) by daily gavage. Control untreated mice received acidified water only. Tumor volumes were determined on alternate days by measuring tumors in three perpendicular diameters as described previously9. In control vehicle-treated groups of each experiment, the rapid growth of subcutaneous tumors required mice euthanasia when tumor volumes exceeded the criteria set by Institutional Animal Care and Use Committee (IACUC) guidelines. This served as an ‘end point’ for control animals. The treated animals with remaining tumors were further followed for long-term survival and were euthanized on day 90. Thus, for survival studies, the end point of drug treatment was 90 days.
Histopathological and immunohistochemical analyses
At the end point for control and treatment groups, liver, kidney, spleen, duodenum, brain, heart, lung, and tumors were formalin-fixed, paraffin-embedded and 5 μm sections were stained with hematoxylin and eosin (H&E). Microscopic evaluation was performed by two pathologists in a blinded-manner. TUNEL staining of tumor-tissue sections was performed as previously described8-10.
Evaluation of immune cells
At the end point of each experimental group as described above, blood was collected from the retro-orbital sinus of mice and peripheral blood mononuclear cells (PBMCs) were isolated using Histopaque-1077 polysucrose density gradient centrifugation. PBMCs were then stained for CD3+, CD4+, CD8+, B220+, and NK1.1+ cells, and data were acquired and analyzed using flow-cytometry.
Pharmacokinetic evaluation and data analyses
Three oral doses (75, 150 and 300 mg/kg) and an intravenous dose (15 mg/kg) of EM011 were used for the pharmacokinetic study. For each dose, 27 mice (male or female) were randomly divided into nine groups (of 3 animals each) corresponding to the time points of blood collection. Samples were collected at 0, 0.25, 0.5, 1, 2, 4, 6, 8, and 24h after oral administration and 0, 0.083, 0.25, 0.5, 0.75, 1, 2, 4, and 6h after intravenous drug administration. Blood (~750 μl) was collected from retro-orbital vein and centrifuged for plasma separation. A simple, rapid and sensitive HPLC method was developed for EM011 estimation in plasma and was applied to the pharmacokinetic study. The protein precipitation method using acetonitrile was used for drug extraction from plasma17. Plasma-concentration data were analyzed with standard non-compartmental methods using the WIN-NONLIN software version 4.1.
Evaluation of neurotoxicity
Dorsal root ganglion cultures and evaluation of neuropathy
For neurotoxicity evaluation, C57BL/6J mice and Sprague Dawley rats were obtained from Jackson and Charles River laboratories, respectively. All animal protocols were IACUC-approved. Dorsal root ganglia (DRGs) from E15 rats were cultured as described previously18-19. After 5 days in culture, the medium was changed to 25 μM EM011 or 30 nM taxol (paclitaxel) or DMSO vehicle solution. Owing to variability in physical characteristics of individual cultures, each DRG imaged before drug exposure served as its own control. Axonal lengths and areas were analyzed as percentage change from day 0 of drug treatment. Normalized data were examined for statistical significance by ANOVA, with post-test correction for multiple comparisons.
Morphometric analyses of dorsal roots
C57BL/6J mice (n=6 per group) were orally administered 300 mg/kg EM011 or acidified water (pH=4) daily for four weeks. Taxol (7.5 mg/ml in cremophor/ethanol) was injected into the jugular vein of taxol-treated animals (positive controls) at a dose of 60 mg/kg every other day for three times. We chose this dose regimen based upon our previous work that showed that 60 mg/kg taxol-treated animals effectively developed peripheral neuropathy within two weeks after the last injection20. L4 dorsal roots were isolated and processed as described20. Cross-sections (720 nm) of dorsal nerve roots were toluidine-blue stained, imaged using an Olympus BX60 microscope and analyzed using Image Pro Version 5.1. Axon interiors were manually marked as solid objects and areas and mean diameters of all myelinated axons were measured by tracing the inner border of myelin sheath. Degenerating axons were identified by lack of axoplasm and presence of myelin ovoids. Means for total number of axons, diameter and area of axons were compared by ANOVA with post-hoc comparison.
Electrophysiological and behavioral measurements
Nerve conduction studies were performed on animals treated with EM011 for four weeks. Taxol was given every other day for three times and measurements were performed two weeks after the last injection using standard equipment (Nicolet, Madison, WI, USA) under chloral-hydrate anesthesia. For hind-limb recording, the recording electrodes were inserted into interosseous muscles of left foot; stimuli were administered transcutaneously at ankle and hip (sciatic notch) close to tibial and sciatic nerve, respectively. A ground electrode was inserted subcutaneously into the tail. For tail-nerve recording, recording electrodes were placed at the base of the tail, keeping anode and cathode ~5 mm apart. Stimuli were administered 4-5 cm distal. A ground electrode was placed in between the stimulus and recording electrodes. The sensory nerve action potential (SNAP) was averaged over 20 stimuli, and the amplitude was recorded. Electrophysiological data before and after treatment were compared for each animal; mean changes were analyzed using a paired t test. To evaluate the sensory-motor function, animals were tested for their ability to maintain balance on a Rotarod apparatus (Columbus Instruments, Columbus, OH, USA). Animals were acclimated to the Rotarod for three consecutive days before the test date. The initial speed was set at 1.6 rpm with an acceleration rate of 4 rpm/min. The test was repeated three times during each testing session with at least 5 min of rest between each test. The best performance of each session was recorded. Percent changes compared to control values at day 0 were calculated and analyzed using ANOVA with post-test comparisons.
RESULTS AND DISCUSSSION
Suppression of microtubule dynamics by low concentrations of tubulin-binding drugs that do not cause net polymer mass changes is responsible for induction of a mitotic block and subsequent apoptosis of the affected cells15,21-24. It is well-appreciated that microtubule dynamics must be tightly regulated for error-free progression through mitosis, and that both accelerated and suppressed dynamics correlate with impaired mitotic spindle function and inhibition of cell proliferation24. EM011, a more potent noscapine analog, binds tubulin without altering its total polymer mass7. Therefore, we first asked if mitotic arrest caused by EM011 in various cancer cells is due to its effects on microtubule dynamics. To determine if dynamic parameters of interphase microtubules are affected by EM011, we followed life-history plots of plus-ends of EM011-treated microtubules in LLCPK-1α cells that stably expressed GFP-alpha-tubulin, thereby ensuring that all cells expressed the chimeric protein at the same level. The advantage of using these cells for microtubule dynamics study is their extreme flatness along the edges, a feature that greatly facilitates the tracking of individual microtubules over time25. Figure 1 shows a gallery of video frames, 3s apart, of plus-ends of several interphase microtubules in vehicle- and EM011-treated cells. As expected, microtubules in control cells alternated between phases of growth and shortening with an intervening ‘pause’ phase. In contrast, EM011 attenuated microtubule dynamics in cells as indicated by unaltered position of their plus-ends (Figure 1, bottom-row, arrows).
Figure 1.
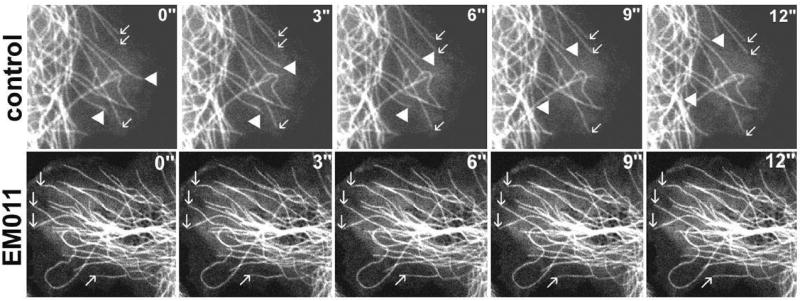
EM011 treatment suppresses microtubule dynamics in living cells. Time-lapse sequences, 3s apart, showing the plus-ends of several microtubules in LLCPK-1α cells in the absence (top row) and presence (bottom row) of 25 μM EM011. Arrows indicate three microtubules that are growing, and solid arrowheads indicate two microtubules that are undergoing shortening events. In the EM011-treated cell (bottom row), arrows indicate four microtubule ends for which the position did not change over a period of 12s.
A quantitative analysis of microtubule growth and shortening, frequencies of catastrophe and rescue, and average duration of pause is shown in Suppl. Table 1. Upon EM011 treatment, a ~82% increase in pause duration was observed. The rate of microtubule growth decreased by ~17%, whereas, shortening rate decreased by ~11%. Dynamicity, which represents the summed gain and loss (exchange) of tubulin subunits at microtubule ends, was significantly reduced by ~56% in drug-treated cells compared to controls. This suggested that EM011 decreases the number of dynamic events in the life-history of a microtubule without affecting its long-term existence. In summary, our results strongly indicate that EM011-induced mitotic arrest results from attenuation of microtubule dynamicity by considerably increasing percentage of time microtubules spend in an idle, paused state.
We have previously shown the in vivo effectiveness of EM011 in xenograft models of human lymphomas and breast cancers in nude mice8-10. However, these cancer types are susceptible to other anti-mitotic treatment regimes that are currently available. Melanomas, on the other hand, are known to be relatively refractory to chemotherapy26. For example, a tumor thickness approaching 4 mm presents a high risk of metastasis, and a diagnosis of metastatic melanoma presents an abysmal median survival of 6-9 months27. We were thus curious to examine if the spectrum of EM011’s anticancer activity spanned the more-aggressive and less-treatable melanomas. Towards this goal, we first evaluated the antiproliferative activity of EM011 by measuring the half-maximal inhibition of cellular proliferation in human and murine melanoma cells. Our results showed that the IC50 of EM011 for four human melanoma cell lines (LOX IMVI, M14, SK-MEL-6 and UACC-257) was in the range of 4-12 μM. However, for murine melanoma B16LS9 cells the IC50 was 23.2 μM, using the standard sulforhodamine B assay. We next tested the effect of EM011 on spindle morphology and cell-cycle progression in the relatively more resistant murine melanoma B16LS9 cells over time using immunofluorescence confocal microscopy. At time 0 of treatment, cells showed intact radial microtubule arrays (Figure 2A). At 12h of EM011-treatment, typical ball-shaped mitotic figures with multiple asters were observed, whereas vehicle-treated cells showed normal bipolar spindles with properly congressed chromosomes. At 24h post-treatment, multinucleated cells were evident. This is, perhaps, due to mutational lesions in checkpoint mechanisms of cancer cells that fail to sustain the mitotic block for long periods of time. After brief periods, such mitotically-arrested cells either succumb to apoptosis directly or undergo aberrant exit from mitosis into a G1-like multinucleate state without cytokinesis. In contrast, vehicle-treated cells showed normal cell-cycle progression with a normal anaphase, characterized by proper separation of sister chromatids towards the two poles. At 48h post-treatment, numerous abnormally large multinucleated cells were evident, along with a few apoptotic cells. The triggering of apoptosis is possibly due to overaccumulation of genotoxic DNA owing to defects in tetraploidy checkpoints of cancer cells that allow them to undergo multiple rounds of DNA synthesis without actual cell division. Apoptotic figures with fragmented chromatin were visible at 72h of treatment (Figure 2A). Our results thus indicate that EM011-induced modulation of microtubule dynamics severely interferes with chromosome congression, resulting in the formation of multipolar spindles and aberrant cell divisions culminating in apoptosis.
Figure 2.

A. EM011 induces spindle abnormalities in melanoma cells. Panels show immunofluorescence confocal micrographs of B16LS9 cells treated with DMSO vehicle (top row) and 25 μM EM011 (bottom row) for 0, 12, 24, 48 and 72h. As expected, vehicle-treated cells showed normal radial arrays of microtubules (time 0). Their cell-cycles progressed normally over time with bipolar spindle formation and complete chromosome congression at the metaphase plate (12h). During anaphase, separation of sister chromatids was evident (24 and 48h) followed by normal telophase and cytokinesis into two daughter cells. In contrast, EM011-treated cells showed perturbed spindle and nuclear morphologies. At 12h of drug treatment, numerous mitotic figures with multiple asters were evident. Cells with multiple asters either succumbed to apoptosis directly (as evident by the appearance of fragmented nuclei) or exited mitoses without cytokinesis as multinucleate interphase cells and continued to traverse the cell-cycle and accumulated more and more DNA that finally triggered apoptosis due to genotoxicity. B. EM011 inhibits cell-cycle progression at mitosis followed by appearance of a characteristic hypodiploid (sub-G1) DNA peak, indicative of apoptosis. Panels (left and middle) depict cell-cycle distribution in a three-dimensional disposition as determined by flow-cytometry of B16LS9 cells treated with 10 and 25 μM EM011 for 0, 24, 48 and 72h. Right panel is a graphical representation of the quantitation of mitotic index (percent G2/M cells) and apoptotic index (percent sub-G1 cells) at the two dose regimes (10 and 25 μM) over the time course of treatment. Values and error bars shown in the graph represent means and standard deviations, respectively, of three independent experiments performed in triplicate. C. Left panel shows representative Alexa-fluor 488-annexin V stained B16LS9 cells upon 25 μM EM011-treatment showing green rims (solid arrowheads) due to phosphatidylserine staining on the outer plasma membrane in early apoptotic cells as opposed to control vehicle-treated cells, where clear green rings are absent. However, some non-specific staining is evident. Right panel shows quantitation of annexin-positive cells upon EM011 treatment for the noted hours as revealed by flow-cytometric analysis. (P<0.05).
We next examined cell-cycle progression of B16LS9 cells at two EM011 doses (10 μM and 25 μM). We included a 10 μM dose to examine if doses lower than the half-maximal dose for inhibition of cellular proliferation could result in a mitotic block. Figure 2B (left and middle) depict cell-cycle profiles over time. At 10 μM EM011, ~53% of cells were arrested in the G2/M phase at 48 h (Figure 2B, right). In contrast, at 25 μM, the percentage of G2/M cells at 24 h increased to ~62% (Figure 2B, right). After 72 h of EM011 exposure, we observed a massive sub-G1 cell population (~72%), indicative of apoptosis, at the 25 μM dose compared to ~35% at the 10 μM dose (Figure 2B, right).
Biochemically, the inner plasma membrane lipid, phosphatidylserine (PS), flips out during early apoptotic stages and once externalized, PS can be specifically detected by annexin-V, a protein with strong-affinity for PS. Thus, to characterize the presence of apoptotic cells, we next visualized EM011-treated melanoma cells stained with Alexa-fluor-488-labeled annexin-V (Figure 2C, left). A green rim was clearly visible on the outer cell periphery indicating early apoptotic cells observable at 48 h post-treatment (Figure 2C, solid arrowheads, left panel). We also quantitated EM011-treated cells flow-cytometrically and observed an increase in annexin-positive cells over time (~65% at 72 h, Figure 2C, right).
Having identified the in vitro efficacy of EM011 in inhibiting cellular proliferation and inducing apoptosis in melanoma cells, we wished to evaluate the in vivo efficacy of EM011 in inhibiting tumor growth. To this end, we examined the ability of EM011 to cause growth inhibition and regression of pre-established subcutaneous melanoma tumors in a syngeneic C57BL/6J immunocompetent murine background. Interestingly, our results showed that orally administered EM011 at 150 mg/kg inhibited growth of melanoma tumors more effectively than 300 mg/kg (Figure 3A). Control group mice were euthanized around day 32 post-inoculation, in compliance with IACUC guidelines.
Figure 3.
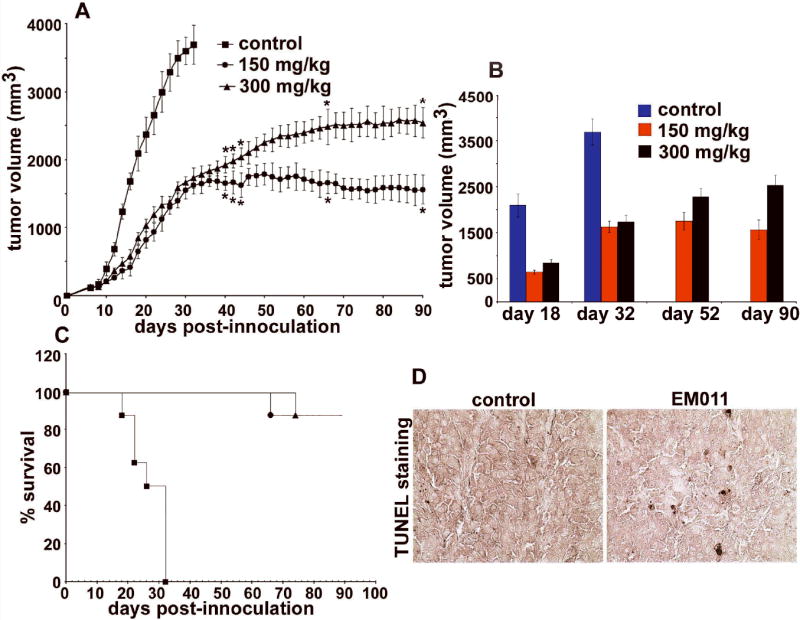
Orally fed EM011 inhibits tumor progression in vivo. Palpable tumors (~100 mm3) were established 6-8 days after injecting 106 B16LS9 melanoma cells (in 0.2 ml of PBS) subcutaneously in mice with a syngeneic background. A shows progression profile of tumor growth in control vehicle-treated mice compared with 150 and 300 mg/kg EM011 treatment. B depicts tumor volume measurements on days 18, 32, 52 and 90 post-inoculation. On day 32, control vehicle-treated mice were euthanized because of overgrown tumors, in compliance with the experimental IACUC protocol. Interestingly, treatment at 150 mg/kg showed better antitumor outcomes compared to 300 mg/kg. C shows Kaplan-Meier analysis of the therapeutic effect of two dose regimes (150 and 300 mg/kg) of EM011. (*, P< 0.05, A-C). D. Immunohistochemical TUNEL-staining of paraffin-embedded tumor sections from mice treated with vehicle (left panel) or EM011 (right panel) for 32 days (end point of control vehicle-treated animals). Right panel shows TUNEL-positive cells (seen as apoptotic brown nuclei) compared to left panel (vehicle-treated control).
Although the initial reduction in tumor volume was not significantly different for the two drug doses (150 and 300 mg/kg) until day 36 post-inoculation, better antitumor outcomes were visible for 150 mg/kg after day 44 post-inoculation. At the end-point for control mice (day 32), EM011-treated mice showed a significant antitumor advantage (1625 ± 124 mm3 upon 150 mg/kg versus 1732 ± 150 mm3 upon 300 mg/kg, average tumor volume ± SE, P<0.05; Figure 3B). On day 90, the reduction in tumor volume was greater for animals treated with 150 mg/kg than those treated at 300 mg/kg (~57% reduction at 150 mg/kg versus ~31% at 300 mg/kg) compared to tumor volume at day 32 for vehicle-treated mice (Figure 3B). To evaluate general health and systemic homeostasis, we monitored progression of body weights of these animals during the course of EM011 treatment. Both doses did not show any apparent weight loss compared to vehicle-treated animals indicating no overall toxicity (data not shown). Kaplan-Meier analysis revealed a significant increase in survival time, with 87.5% (150 mg/kg and 300 mg/kg treated) animals surviving until day 90 (P ≤ 0.05) (Figure 3C). The median survival time of treated mice was increased by ~3 fold (from 32 days to 90 days) (Figure 3C). Surviving mice were followed for 90 days without any signs of new tumors and were sacrificed. We next asked if EM011 inhibited subcutaneous syngeneic tumors by induction of apoptosis. To this end, we microscopically examined TUNEL-stained tumor sections excised from both vehicle-treated control and EM011-treated (150 mg/kg) groups at 32 days (end point of control animals). We observed many more TUNEL-positive cells (Figure 3D) in the tumor sections of mice from the drug-treated group, indicating that tumor inhibition was indeed a result of EM011-induced apoptosis.
To gain further insights and to seek possible explanation for better antitumor outcomes at 300 mg/kg compared to 150 mg/kg, we performed a pharmacokinetic study in mice at different drug doses. We first developed a rapid, sensitive and reproducible HPLC-UV method for EM011 measurement over a linear concentration range that yielded a lower limit of EM011 quantification of 390 ng/ml. This method was then applied to an oral (75, 150 and 300 mg/kg) and an intravenous (15 mg/kg) pharmacokinetic study in mice. EM011 was extracted from mice plasma by acetonitrile using the protein-precipitation method shown by us previously17. All four doses (oral and iv) were well-tolerated and mice did not show any signs of discomfort. Pharmacokinetic parameters for both male and female mice (mean ± SD) at varying doses are summarized in Suppl. Table 2. Our data show that AUC increases linearly from a dose level of 75 mg/kg to 150 mg/kg; however non-linear pharmacokinetics is observed at 300 mg/kg indicating that some aspect of the pharmacokinetic behavior of the drug is saturable. Peak plasma concentrations were attained at about 1 hour, indicating rapid absorption, compatible with lipophilicity and small molecular size of EM011. Since EM011 has a short elimination half life across the dose levels studied, it is likely that the drug and its metabolites clear off rapidly thus avoiding any carry-over side-effects. Furthermore, our results show that the bioavailability at 75 and 150 mg/kg is almost similar (~67%), which is much higher than that of the parent compound noscapine17. Bioavailability calculations depend on oral dosing data and can result in fluctuatingly high/low values if the pharmacokinetic behavior is non-linear (as in our case at 300 mg/kg). This may account for the very high bioavailability at 300 mg/kg. Therefore, in the face of non-linear pharmacokinetics observable at 300 mg/kg, we are limited in drawing a definitive explanation of the mechanism underlying better anti-tumor efficacy at the lower dose (150 mg/kg). It is noteworthy that several other parameters govern antitumor efficacy which can be AUC- and bioavailability- independent, such as drug uptake by the tumor.
Since anticancer drugs are cytotoxic for normal as well as neoplastic cells, improvements in parameters such as clinical benefit, time to progression, overall survival, and quality of life have been considered of utmost value. Thus, we next determined if EM011 caused any toxicity to normal tissues including those with frequently proliferating cells (spleen and gut). To this end, we examined tissue sections of liver, kidney, spleen, duodenum, brain, heart, and lung of tumor-bearing mice by H&E staining (Figure 4A, data shown for 300 mg/kg). EM011 treatment did not cause any detectable pathological abnormalities or any metastatic lesions in these organs at both 150 and 300 mg/kg dose levels.
Figure 4.
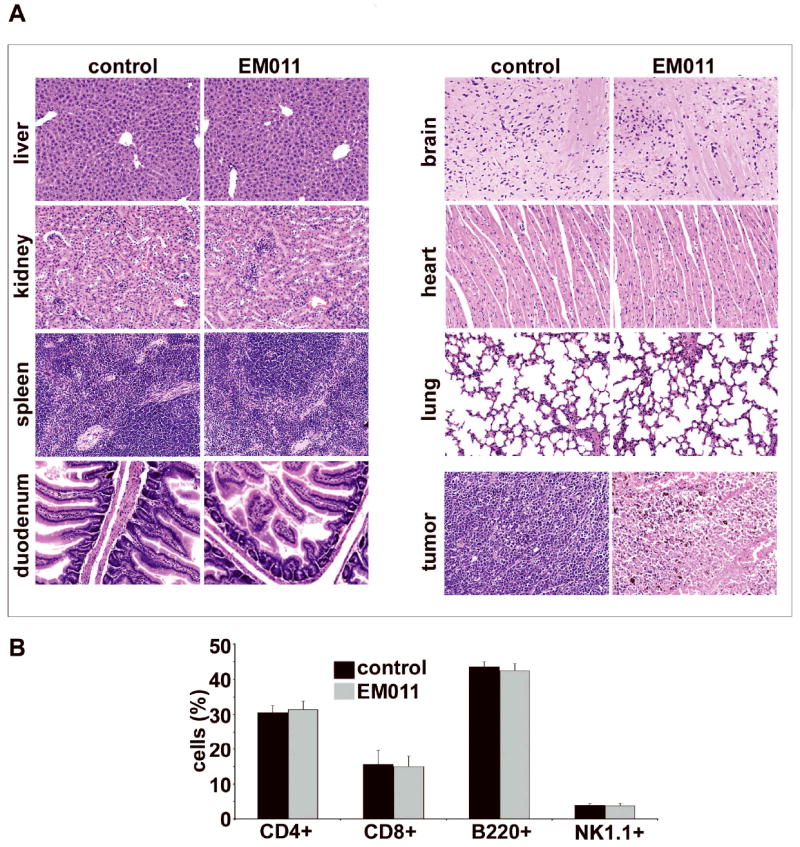
300 mg/kg EM011 fed daily does not cause any detectable pathologic abnormalities in normal tissues. A. Panels show H&E staining of paraffin-embedded tissue sections of the liver, kidney, spleen, duodenum, brain, heart, and lung, from vehicle-treated and EM011-treated mice (magnification, 200X). These tissues were indistinguishable between the EM011-treated and the control groups. The EM011-treated tumor microsections reveal large areas of tumor cell death, consistent with the therapeutic effects of EM011. However, microsections from vehicle-treated tumor tissue show sheets of tumor cells with high-grade pleomorphic nuclei with minimal cell death. EM011 does not cause any obvious hematologic toxicity. B. Bar-graph depicting no alterations in the CD4+, CD8+, B220+, and NK1.1+ cell counts from vehicle- and EM011- treated groups of mice as determined by flow cytometry-based immunophenotyping assays.
Several tubulin-binding drugs are known to cause immunosuppression and weakening of host immune surveillance system28. Therefore, we next evaluated the effect of EM011 on relative counts of immune cells compared to vehicle-treated controls. Even 300 mg/kg EM011 did not perturb CD4+, CD8+, B220+, and NK1.1+ cell counts compared to vehicle-treated controls (Figure 4B). This represents a unique edge of EM011 over currently-available chemotherapeutic drugs that are immunosuppressive.
Peripheral neuropathy is a major dose-limiting complication of commonly used tubulin-binding drugs. It clinically manifests as numbness, pain, loss of balance, and can be severe enough to necessitate cessation of treatment4,29. Therefore, our next concern was to evaluate if EM011 caused neurotoxicity. We included taxol as a positive control since we have previously shown that its intravenous administration at 60 mg/kg in mice caused peripheral neuropathy within two weeks20. To evaluate any potential toxic effects on peripheral nerves, we examined DRG cultures in presence or absence of EM011 (Figure 5A). Cultures exposed to 25 μM EM011 for 11 days did not show loss of axonal length and DRG area; while vehicle-treated controls continued to grow (Figures 5A; left, middle and right panels). However, exposure of DRG cultures to taxol (30 nM) caused significant and progressive loss of axonal length and DRG area, changes that are typically seen with exposure to antimicrotubule drugs such as vincristine or taxol18,29-31.
Figure 5.

Daily oral EM011 treatment does not cause any detectable peripheral neuropathy. A. Left panel shows cultured dorsal root ganglion cells treated with vehicle or EM011 or taxol. DRG cultures were allowed to mature for 5 days to enable development of a lush halo of neurites around the explants. After 5 days, cultures were incubated with vehicle or 25 μM EM011 or 30 nM taxol for up to 11 days post-treatment. The diameter of circular halo of neurites were measured on the initial day of treatment (day 0), and then on day 11. Total axonal length and the DRG halo area were the quantitative parameters used to measure neurotoxicity. Axonal survival was quantified by the longest remaining axon and the area of the remaining DRG halo. The axonal length was measured from the center of the halo to the visible distal ends of the axon in the periphery of the halo. Halo areas were calculated by tracing the outside circumference of the remaining culture halo. Percent change (of day 0) axonal length (middle) and halo area (right) upon vehicle or 25 μM EM011 or 30 nM taxol treatment for 11 days are shown. (Values are mean ± SD, P<0.05). B. Left panel shows pathological assessment of toluidine-stained 5 μm dorsal root sections imaged at 100X magnification. Middle and right panels are bar-graphical representations of the quantitation of mean diameter, area, and number of axons measured using the Image Pro software (Media Cybernetics, Silver Spring, MD). C. EM011 treatment did not cause any neurotoxicity quantifiable by electrophysiological and behavioral evaluations. Measurement of sensory nerve action potential (SNAP) in C57BL/6J mice that were treated with vehicle or 25 μM EM011 or 60 mg/kg taxol. Left panel is a representative recording of SNAP in the tail from EM011-treated mice. Middle panel shows percent change of day 0 SNAP for mice treated with vehicle or EM011 or taxol. (Values are mean ± SD, P<0.05) Right panel shows percent change in Rotarod performance (compared to day 0) of mice treated with vehicle or EM011 or taxol.
We then examined dorsal sensory nerves of control, EM011- and taxol- treated mice for any axonal degeneration (see Methods section for treatment details). EM011 treatment did not result in either tubulovesicular accumulations, as may be seen with impaired axonal transport, or axonal degeneration in the sensory fibers (Figure 5B). Toluidine-staining showed normal myelinated fibers upon EM011 treatment for a four-week period (Figure 5B, left). Analysis of dorsal roots showed that mean axonal diameter, area, and number of axons were comparable among the EM011- and vehicle- controls (Figure 5B; middle and right panels). Taxol, however, resulted in axonal degeneration with significantly reduced mean area, diameter of axons and number of axonal fibers (Figure 5B; middle and right panels). The absence of such pathology upon EM011 treatment suggests that it is non-toxic to peripheral nerves.
We next examined if any signs of functional impairment appear upon a four-week 300 mg/kg EM011 treatment using electrophysiological measures. Figure 5C (left panel) shows a representative recording of tail sensory nerve action potential (SNAP) from an EM011-treated mice. We observed no significant differences in SNAP of EM011- and vehicle-treated mice (Figure 5C, middle panel). In contrast, tail SNAP in taxol-treated animals was significantly reduced (Figure 5C, middle panel). We next asked if sensory-motor function was compromised by EM011 treatment by the Rotarod assay. We observed that EM011 treatment did not affect the ability of mice to maintain balance and stay on the rotating rod, thus exhibiting no loss of function post-treatment (Figure 5C, right). However, the ability of taxol-treated animals to stay on the rotarod was decreased by ~20% (Figure 5C, right). Thus, our results from the electrophysiological and rotarod assessments support absence of any detectable neurotoxicity upon EM011 treatment.
Taken together, our data show that EM011 is effective against melanoma cells as well as syngeneic subcutaneous tumors. In addition, EM011 is non-toxic to normal tissues and does not deplete immune cells at doses effective for tumor inhibition. Another unique advantage of EM011 therapy is absence of neurotoxicity that severely impairs efficacies of currently available chemotherapeutic regimens. Therefore, we believe that EM011 is a safe and effective anticancer drug with a potential for clinical evaluation.
Supplementary Material
Acknowledgments
We thank Adam Marcus, Director, Microscopy Core Facility at Winship Cancer Institute, Emory University for helping with the microtubule dynamics study. We thankfully acknowledge Ming Sheng Wang for providing DRG cultures. We acknowledge the technical assistance of Binfei Zhou and Jyoti Idnani (Lupin labs). We are grateful to Meenakshi Gupta for evaluating animal tissue sections in a blinded-manner. This work was supported by grants to RA from the Department of Defense (PC073104) and the National Cancer Institute at the National Institutes of Health (1K99CA131489) and to HJ from the National Institutes of Health (R01 CA095317).
Footnotes
Novelty and Impact: The manuscript presents evaluation of in vitro efficacy and molecular mechanism of action of a rationally-designed microtubule-modulating anticancer agent in murine melanoma. Unlike currently available drugs, this agent is orally-bioavailable and non-toxic, and in particular, displays absence of neurotoxicity.
References
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8:49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Hilkens PH, ven den Bent MJ. Chemotherapy-induced peripheral neuropathy. J Peripher Nerv Syst. 1997;2:350–361. [PubMed] [Google Scholar]
- 4.Rowinsky EK, Chaudhry V, Cornblath DR, Donehower RC. Neurotoxicity of Taxol. J Natl Cancer Inst Monogr. 1993;15:107–115. [PubMed] [Google Scholar]
- 5.Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9–17. doi: 10.1007/pl00007853. [DOI] [PubMed] [Google Scholar]
- 6.Aneja R, Vangapandu SN, Lopus M, Viswesarappa VG, Dhiman N, Verma A, Chandra R, Panda D, Joshi HC. Synthesis of microtubule-interfering halogenated noscapine analogs that perturb mitosis in cancer cells followed by cell death. Biochem Pharmacol. 2006;72:415–426. doi: 10.1016/j.bcp.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Zhou J, Gupta K, Aggarwal S, Aneja R, Chandra R, Panda D, Joshi HC. Brominated derivatives of noscapine are potent microtubule-interfering agents that perturb mitosis and inhibit cell proliferation. Mol Pharmacol. 2003;63:799–807. doi: 10.1124/mol.63.4.799. [DOI] [PubMed] [Google Scholar]
- 8.Aneja R, Zhou J, Vangapandu SN, Zhou B, Chandra R, Joshi HC. Drug-resistant T-lymphoid tumors undergo apoptosis selectively in response to an antimicrotubule agent, EM011. Blood. 2006;107:2486–2492. doi: 10.1182/blood-2005-08-3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aneja R, Zhou J, Zhou B, Chandra R, Joshi HC. Treatment of hormone-refractory breast cancer: apoptosis and regression of human tumors implanted in mice. Mol Cancer Ther. 2006;5:2366–2377. doi: 10.1158/1535-7163.MCT-06-0205. [DOI] [PubMed] [Google Scholar]
- 10.Aneja R, Liu M, Yates C, Gao J, Dong X, Zhou B, Vangapandu SN, Zhou J, Joshi HC. Multidrug resistance-associated protein-overexpressing teniposide-resistant human lymphomas undergo apoptosis by a tubulin-binding agent. Cancer Res. 2008;68:1495–1503. doi: 10.1158/0008-5472.CAN-07-1874. [DOI] [PubMed] [Google Scholar]
- 11.Becker JC, Kampgen E, Brocker E. Classical chemotherapy for metastatic melanoma. Clin Exp Dermatol. 2000;25:503–508. doi: 10.1046/j.1365-2230.2000.00690.x. [DOI] [PubMed] [Google Scholar]
- 12.O’Day SJ, Kim CJ, Reintgen DS. Metastatic melanoma: chemotherapy to biochemotherapy. Cancer Control. 2002;9:31–38. doi: 10.1177/107327480200900105. [DOI] [PubMed] [Google Scholar]
- Ridolfi R, Chiarion-Sileni V, Guida M, Romanini A, Labianca R, Freschi A, Lo Re G, Nortilli R, Brugnara S, Vitali P, Nanni O. Italian Melanoma Intergroup. Cisplatin, dacarbazine with or without subcutaneous interleukin-2, and interferon alpha-2b in advanced melanoma outpatients: results from an Italian multicenter phase III randomized clinical trial. J Clin Oncol. 2002;20:1600–1607. doi: 10.1200/JCO.2002.20.6.1600. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann MA, Sterry W, Trefzer U. Complex combination biochemotherapy regimen in advanced metastatic melanoma in a non-intensive care unit: toxicity or benefit? Jpn J Clin Oncol. 2007;37:224–229. doi: 10.1093/jjco/hym009. [DOI] [PubMed] [Google Scholar]
- 15.Landen JW, Lang R, McMahon SJ, Rusan NM, Yvon AM, Adams AW, Sorcinelli MD, Campbell R, Bonaccorsi P, Ansel JC, Archer DR, Wadsworth P, Armstrong CA, Joshi HC. Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer Res. 2002;62:4109–4114. [PubMed] [Google Scholar]
- 16.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 17.Aneja R, Dhiman N, Idnani J, Awasthi A, Arora SK, Chandra R, Joshi HC. Preclinical pharmacokinetics and bioavailability of noscapine, a tubulin-binding anticancer agent. Cancer Chemother Pharmacol. 2007;60:831–839. doi: 10.1007/s00280-007-0430-y. [DOI] [PubMed] [Google Scholar]
- 18.Wang MS, Wu Y, Culver DG, Glass JD. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincristine neuropathy. J Neuropathol Exp Neurol. 2000;59:599–606. doi: 10.1093/jnen/59.7.599. [DOI] [PubMed] [Google Scholar]
- 19.Wang MS, Fang G, Culver DG, Davis AA, Rich MM, Glass JD. The WldS protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann Neurol. 2001;50:773–779. doi: 10.1002/ana.10039. [DOI] [PubMed] [Google Scholar]
- 20.Wang MS, Davis AA, Culver DG, Glass JD. Wlds mice are resistant to paclitaxel (taxol) neuropathy. Ann Neurol. 2002;52:442–447. doi: 10.1002/ana.10300. [DOI] [PubMed] [Google Scholar]
- 21.Chen JG, Horwitz SB. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res. 2002;62:1935–1938. [PubMed] [Google Scholar]
- 22.Mitchison TJ, Kirschner MW. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- 23.Jordan MA, Thrower D, Wilson L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991;51:2212–2222. [PubMed] [Google Scholar]
- 24.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorbsky GJ, Sammak PJ, Borisy GG. Microtubule dynamics and chromosome motion visualized in living anaphase cells. J Cell Biol. 1988;106:1185–1192. doi: 10.1083/jcb.106.4.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrone L, Hersey P. The chemoresistance of human malignant melanoma: an update. Melanoma Res. 1999;9:51–58. doi: 10.1097/00008390-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–2182. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 28.Lee M, Yea SS, Jeon YJ. Paclitaxel causes mouse splenic lymphocytes to a state hyporesponsive to lipopolysaccharide stimulation. Int J Immunopharmacol. 2000;22:615–21. doi: 10.1016/s0192-0561(00)00024-2. [DOI] [PubMed] [Google Scholar]
- 29.Rowinsky EK. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu Rev Med. 1997;48:35–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 30.Silva A, Wang Q, Wang M, Ravula SK, Glass JD. Evidence for direct axonal toxicity in vincristine neuropathy. J Peripher Nerv Syst. 2006;11:211–216. doi: 10.1111/j.1529-8027.2006.0090.x. [DOI] [PubMed] [Google Scholar]
- 31.Ravula SK, Wang MS, McClain MA, Asress SA, Frazier B, Glass JD. Spatiotemporal localization of injury potentials in DRG neurons during vincristine-induced axonal degeneration. Neurosci Lett. 2007;415:34–39. doi: 10.1016/j.neulet.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.