

Abstract
Tumor associated macrophages mediate the link between inflammation and cancer progression. Here we showed that macrophage-derived soluble factors induce canonical Wnt signaling in colon cancer cells and promote their growth. Tumor cells induced the release of IL-1β from macrophages, which induced phosphorylation of GSK3β, stabilized β-catenin, enhanced TCF-dependent gene activation, and induced the expression of Wnt target genes in tumor cells. Neutralization experiments using anti IL-1β specific antibodies, or silencing of IL-1β in THP1 macrophages, revealed that IL-1β was required for macrophages to induce Wnt signaling and to support the growth of tumor cells. Constitutive activation of STAT1 in THP1 macrophages was essential for the induction of IL-1β and thus for the activation of β–catenin signaling in tumor cells.
Vitamin D3, an effective chemopreventive agent, interrupted this crosstalk by blocking the constitutive activation of STAT1 and the production of IL-1β in macrophages, and therefore- in a vitamin D receptor dependent manner- inhibited the ability of macrophages to activate Wnt signaling in colon carcinoma cells. Our data therefore established that vitamin D3 exerts its chemopreventive activity by interrupting a cross-talk between tumor epithelial cells and the tumor microenvironment.
Introduction
Tumors are organ like structures that include malignant cells, fibroblasts, myofibroblasts, mast cells, inflammatory cells, endothelial cells and resident macrophages, along with components of the extracellular matrix. Normal stroma can keep premalignant cells in check and can therefore delay or prevent tumor formation while abnormal, reactive stroma, contributes to, or may be required for, tumor formation, by providing growth factors, blood supply and components of the extracellular matrix (Bissell and Labarge, 2005). For example, selective deletion of SMAD4 in T cells resulted in spontaneous development of intestinal tumors (Kim et al., 2006), and depletion of mast cells (Gounaris et al., 2007) or macrophages (Oguma et al., 2008) resulted in a profound remission of Apc-initiated intestinal polyps, confirming the role of stroma and stroma-derived factors in both initiation and progression of intestinal carcinomas.
Macrophages are often the most abundant immune cells in the tumor microenvironment and are key regulators of the link between inflammation and cancer. Tumor associated macrophages (TAMs) are derived from circulating monocytes and upon recruitment to the tumor microenvironment acquire several properties of a polarized M2 phenotype. The tumor microenvironment therefore “educates” macrophages to orchestrate conditions that support tumor progression and promote metastasis and angiogenesis (Pollard, 2004). Although increased density of TAMs is associated with poor prognosis in a variety of human cancers, there are contrasting reports regarding the prognostic significance of macrophage infiltration in colon cancer (Etoh et al., 2000; Forssell et al., 2007; Oosterling et al., 2005).
Stroma-derived factors regulate tumor growth by acting in a paracrine manner to alter signaling pathways in tumor cells, such as activation of NF-κB by TNF and IL-1, and activation of STAT3 by IL-6 (Lin and Karin, 2007). Recently, TNF (Oguma et al., 2008), Hepatocyte Growth Factor (Rasola et al., 2007), PDGF (Yang et al., 2006) and FGF19 (Pai et al., 2008) were shown to activate Wnt/β–catenin signaling, the oncogenic pathway activated in the majority of colorectal cancers. Signaling through the canonical Wnt pathway results in inhibition of GSK3β activity, stabilization and accumulation of β-catenin in the cytoplasm, followed by its nuclear translocation (Moon et al., 2002).
Vitamin D3 dietary supplementation has been shown to lower the incidence of colorectal cancer by 50%, consistent with the inverse correlation between dietary vitamin D intake or sunlight exposure and human colorectal cancer (Garland et al., 1989; Kampman et al., 2000; Newmark and Lipkin, 1992; Robsahm et al., 2004). The chemopreventive properties of the active metabolite of vitamin D, 1α25(OH)2D3, stem from its ability to inhibit proliferation and angiogenesis, to induce apoptosis and differentiation, and involve its anti-inflammatory properties (Lamprecht and Lipkin, 2003; Lipkin and Lamprecht, 2006). The Wnt/β-catenin signaling oncogenic pathway appears to be an important target of the chemopreventive action of vitamin D3 (Palmer et al., 2001; Pendas-Franco et al., 2008; Shah et al., 2003; Shah et al., 2006). However, a number of cancer cell lines (Kumagai et al., 2003), including colon cancer cell lines tested in our laboratory, display limited response to vitamin D3 in vitro, and VDR expression is downregulated in late stages of colon cancer (Palmer et al., 2004), implying that vitamin D exerts some of its biological activities in a VDR independent manner, or that it targets cells in the tumor microenvironment. This is consistent with the concept that tumor stromal cells, which in general remain genetically stable (Qiu et al., 2008) may therefore be a preferred target for chemopreventive agents (Albini and Sporn, 2007).
Here we demonstrated that colon cancer cells stimulate macrophages to release IL-1β, and showed that IL-1β is sufficient to induce canonical Wnt signaling and growth of tumor cells through inactivation of GSK3β. Treatment of THP1 macrophages with 1,25(OH)2D3 - in a VDR dependent manner- abolished their ability to induce Wnt signaling in tumor cells through inhibition of constitutive STAT1 activation and the release of IL-1β.
Results
1. Factors secreted by macrophages induce canonical Wnt signaling in colon tumor cells and promote their proliferation
We developed an in vitro model to evaluate crosstalk between macrophages and colon cancer cells. Experiments were performed using HCT116 and Hke-3 cells, isogenic colon cancer cell lines that differ only by the presence of the mutant kRas allele (Shirasawa et al., 1993), and THP1 cells which display several characteristics of tumor associated macrophages (TAMs), such as defective activation of NF-κB, lack of nitric oxide production in response to LPS/IFNγ (not shown) and high constitutive STAT1 signaling (see below).
Both colon cell lines secrete factors that induce differentiation of THP1 cells into cells that exhibit characteristics of mature macrophages. Tumor cell - derived factors inhibited growth of THP1 macrophages (Supplemental Fig. 1 A, B, C, D) and induced morphological changes comparable to those induced by TPA, a known inducer of differentiation of the THP1 monocytic line into macrophages (Tsuchiya et al., 1982) (Supplemental Fig. 1E). THP1 cells grown in the presence of conditioned medium from tumor cells, like TPA- treated cells, displayed enlarged cytoplasm and reorganized actin cytoskeleton, and became irregular in shape, flat and adherent (Supplemental Fig. 1E). Tumor derived factors also induced similar changes in primary human monocytes, the precursors of tumor associated macrophages (not shown), consistent with the concept that infiltration of monocytes into the tumor microenvironment is followed by their differentiation into mature macrophages (Allavena et al., 2008).
To identify pathways altered in tumor cells exposed to macrophages, we transfected HCT116 cells with luciferase (LUC) – labeled reporter vectors that measure the activity of major signaling pathways known to be perturbed in colon cancer. We compared the activity of NF-κB, Notch, AP1, Wnt and STAT1 in HCT116 cells that were either maintained as monocultures, or were co-cultured with THP1 cells. At conditions tested, the presence of THP1 macrophages did not modulate the activity of the Notch, AP1 or STAT1 signaling, marginally affected NFκB activity (Supplemental Fig. 2), and significantly increased canonical Wnt signaling, measured by activity of the TOP-FLASH reporter. The THP1 macrophages increased Wnt signaling by 40% in HCT116 cells and by 39% in Hke-3 cells (Fig. 1A). The activity of TOP-FOP, a reporter gene with a mutated TCF4 binding site, was not affected by factors secreted by the macrophages (not shown). Peripheral blood monocytes, precursors of the tumor associated macrophages, also increased Wnt signaling in both HCT116 and Hke-3 cells (Fig. 1B).
Fig. 1. Macrophages activate Wnt signaling in colon cancer cells and promote their clonogenic growth.
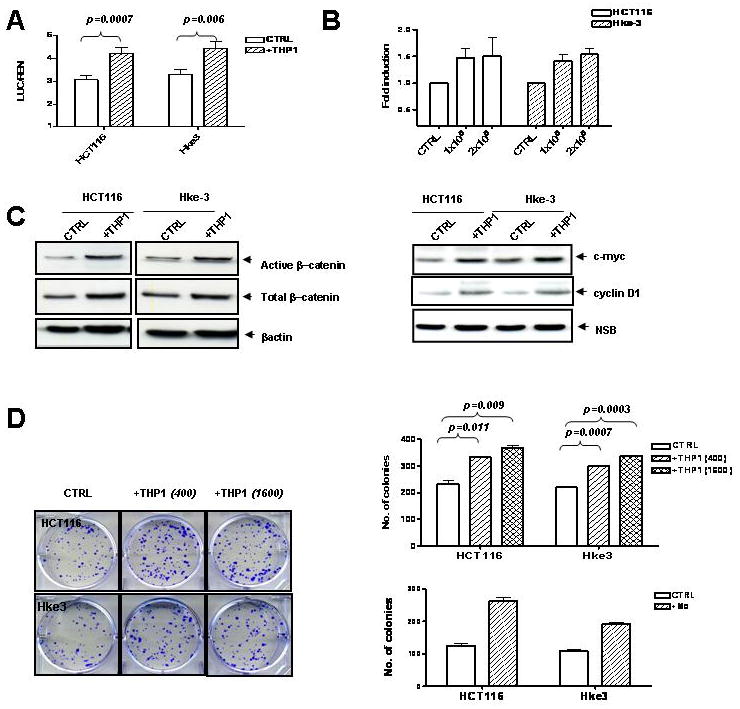
HCT116 and Hke-3 cells were transfected with TOP-FLASH reporter plasmid and were cultured in the absence or the presence of THP1 macrophages (A) or normal human peripheral blood monocytes (B). C: The levels of active β–catenin, total β–catenin, c-Myc and cyclin D1 were determined in HCT116 and Hke-3 cells that were grown alone, or were co-cultured with THP1 macrophages. D: HCT116 and Hke-3 cells were plated alone or together with THP1 macrophages or peripheral blood monocytes (Mo) as described in Material and Methods.
Consistent with increased TCF4 transcriptional activity, the levels of both unphosphorylated (active) β–catenin and total β–catenin, and the expression of c-myc and cyclin D1, two key target genes of Wnt signaling, were elevated in tumor cells that were cultured in the presence of macrophages (Fig. 1C).
To determine whether macrophages alter the clonogenic potential of tumor cells, HCT116 and HKe-3 cells were plated at a low density alone or together with an increasing number of macrophages. We showed that the presence of THP1 macrophages (Fig. 1D) or primary peripheral blood monocytes (Fig. 1D, lower panel) significantly increased the clonogenic growth of colon cancer cells.
To determine whether direct contact between macrophages and tumor cells is required for enhanced proliferation of tumor cells, we co-cultured HCT116 and THP1 cells in transwells that allowed the exchange of soluble factors, but were impermeable for cells themselves (0.4 μM pore membrane, schematically shown in Supplemental Fig. 3A). THP1 macrophages enhanced proliferation of both HCT116 and Hke-3 cells and increased the fraction of tumor cells in the S phase of the cell cycle (not shown). Consistently, both HCT116 and Hke-3 cells co-cultured with THP1 cells displayed increased expression of cyclin D1, cyclin E and cyclin A (Supplemental Fig. 3B).
Together, these data demonstrated that macrophage-derived factors increased Wnt signaling and enhanced clonogenic potential and proliferation of colonic epithelial cells and showed that the presence of activated Ras in the epithelial cells does not affect the interaction between tumor cells and macrophages.
2. IL-1β is sufficient to activate Wnt signaling and is required for increased Wnt signaling in tumor cells
Because the crosstalk between the tumor cells and macrophages was mediated by soluble factors, we compared the levels of chemokines and cytokines in conditioned medium collected from monocultures and cocultures of macrophages with HCT116 cells. Using a cytokine antibody array that assessed 36 soluble mediators (R&D Systems) we showed that the most abundant chemokine secreted by THP1 macrophages was CCL5 (Rantes), while HCT116 and Hke-3 cells expressed detectable levels of MIF and PAI-1 (Fig. 2A). Interaction of THP1 macrophages with HCT116 cells provoked a marked elevation in the expression of several chemokines including CCL5, CXCL1, CCL3, IL-8, and IL-1β, a major proinflammatory cytokine (Fig. 2A). IL-1β was also induced by co-culturing the primary peripheral blood monocytes, the precursors of tumor associated macrophages, with tumor cells (Fig. 2A/a).
Figure 2. IL-1β is sufficient and required to activate Wnt signaling.
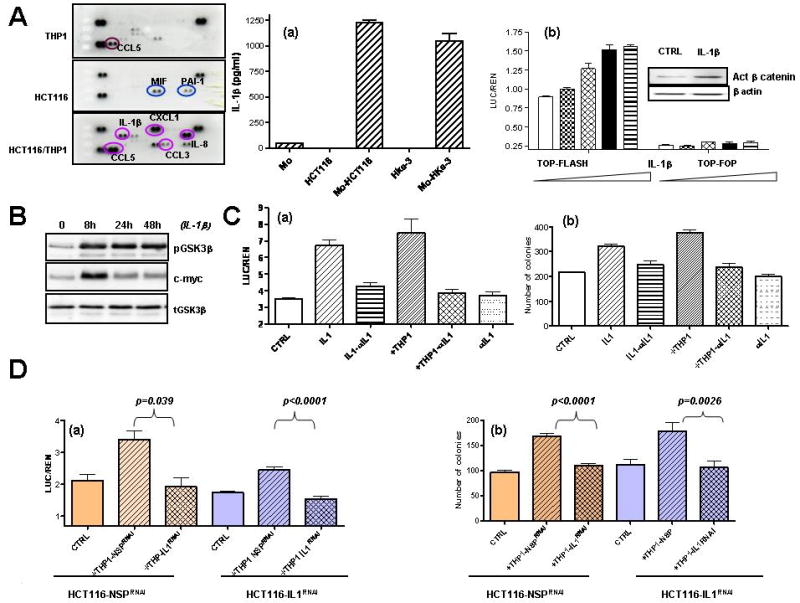
A: HCT116 and THP1 cells were cultured alone or together as indicated for 48 hours, and the expression of soluble mediators was determined by Human cytokine array (R&D Systems). (a): The amount of IL-1β was determined by ELISA in supernatants of HCT116 and Hke-3 cells, normal human monocytes (Mo) and in co-cultures. (b): HCT116 cells were transfected with TOP-FLASH or TOP-FOP reporter genes and were treated with increasing concentrations of IL-1β (0.5-10 ng/ml). Inset: The amount of active β-catenin in IL-1β treated cells. B:The levels of pGSK3 (Ser9), total GSK3 and c-myc in control and IL-1β treated HCT116 cells. C: (a) HCT116 cells were transfected with TOP-FLASH reporter gene and were treated as indicated. (b) HCT116 cells were treated as indicated and the clonogenic assay was performed as described in Material and methods. D: HCT116 were transfected with NSP (nontargeting) siRNA or siRNA specific for IL-1β. They were cultured with nontransfected THP1 cells or THP1 cells transfected with NSP siRNA or siRNA specific for IL-1β. Wnt signaling (a) and clonogenic assay were (b) were performed as indicated.
Which of these soluble factors mediated increased Wnt signaling in the tumor cells? We tested the activity of Rantes, IL-1β, and TNF, a macrophage-derived cytokine recently shown to activate Wnt signaling in gastric cancer (Oguma et al., 2008). However, unlike in gastric cells, IL-1β was a more potent inducer of TCF4/β-catenin transcriptional activity than TNF, and CCL5, a predominant cytokine secreted by THP1 cells, did not modulate Wnt signaling in HCT116 cells (not shown). IL-1β induced TCF4/β-catenin dependent reporter gene activity in a concentration dependent manner and did not affect the activity of the TOP-FOP reporter which harbors a mutant TCF4 binding site (Fig. 2A/b). Consistent with increased Wnt signaling, IL-1β increased the levels of active β–catenin in HCT116 cells (Fig. 2A/b, the inset). Treatment of HCT116 cells with IL-1β increased phosphorylation of GSK3β (Fig. 2B), which is known to inhibit its activity (Cross et al., 1995). Concomitant with inactivation of GSK3β and resulting stabilization of β-catenin, the levels of c-myc were induced by IL-1β (Fig. 2B).
The ability of IL-1β to activate Wnt signaling was inhibited by neutralizing IL-1β antibody, confirming efficacy and specificity of the antibody (Fig. 2C/a). Neutralizing IL-1β also reversed the ability of THP1 macrophages to induce Wnt signaling in HCT116 cells, demonstrating that IL-1β is required for macrophage-mediated increased Wnt signaling in the tumor cells. The neutralizing antibody against IL-1β did not affect basal Wnt signaling in HCT116 cells (Fig. 2C/a)
The ability of THP1 macrophages to increase the clonogenic growth of tumor cells was inhibited by neutralization of IL-1β (Fig. 2C/b), demonstrating that macrophages increase the growth of tumor cells mostly through IL-1β. Consistently, IL-1β was sufficient to increase the clonogenic growth of HCT116 cells (Fig. 2C/b).
To confirm that macrophages are the source of IL-1β in cocultures, the IL-1β gene was silenced in either the tumor cells or the THP1 macrophages. Silencing of IL-1β in macrophages, but not in HCT116 cells completely inhibited the ability of macrophages to induce Wnt signaling (Fig. 2D/a) and to induce the clonogenic growth of tumor cells (Fig. 2D/b). Thus, inhibition of the IL-1β release from macrophages attenuates the protumorigenic activity of macrophages.
3. 1,25(OH)2D3 inhibits the release of IL-1β and interrupts the crosstalk between macrophages and tumor cells
Next we examined whether vitamin D3, a potent chemopreventive agent for colorectal cancer, can modulate the cross talk between macrophages and tumor cells. Consistent with published results (Kumagai et al., 2003), vitamin D3 did not inhibit the growth of HCT116 cells (Supplemental Fig. 4A). Although oncogenic kRas alters the responsiveness of cells to a variety of chemopreventive and chemotherapeutic agents (Klampfer et al., 2003; Klampfer et al., 2004; Klampfer et al., 2007; Klampfer et al., 2005), the isogenic Hke-3 cells, which lack the mutant kras allele, also displayed resistance to 1,25(OH)2D3 (Supplemental Fig. 4A). In contrast, proliferation of THP1 macrophages was markedly inhibited by 1,25(OH)2D3, demonstrated by the MTT assay and BrdU incorporation (Supplemental Fig. 4B).
Because vitamin D3 has anti-inflammatory properties, we tested whether 1,25(OH)2D3 modulates the release of IL-1β from macrophages. Co-culturing of HCT116 cells and THP1 macrophages induced significant secretion of IL-1β and 1,25(OH)2D3 inhibited the release of IL-1β from macrophages induced by tumor cells, and also in response to LPS stimulation (Supplemental Fig. 4C). Surprisingly, the release of IL-8 induced by the interaction of tumor cells with macrophages was enhanced by 1,25(OH)2D3(Supplemental Fig. 4C). This suggested that 1,25(OH)2D3 may regulate the cross-talk between macrophages and tumor cells.
To test whether 1,25(OH)2D3 alters the ability of THP1 cells to activate Wnt signaling in tumor cells, HCT116 cells transfected with the TOP-FLASH reporter gene were cultured alone or together with THP1 cells, and were treated with vitamin D3. 1,25(OH)2D3 did not affect endogenous Wnt signaling in HCT116 cells, but completely prevented the ability of THP1 macrophages to enhance Wnt signaling in HCT116 cells (Fig. 3A) and inhibited the increased levels of active β–catenin and total β–catenin in HCT116 cells cultured in the presence of THP1 macrophages (Fig. 3B). Coculture of tumor cells with THP1 macrophages, like treatment with IL-1β, resulted in phosphorylation of GSK3β and treatment with 1,25(OH)2D3 prevented THP-1 mediated phosphorylation of GSK3β and induction of c-myc (Fig. 3B, right panel). THP-1 macrophages transfected with IL-1β specific siRNA failed to inactivate GSK3β and to induce c-myc (Fig. 3B, right panel), confirming the crucial role of IL-1β in the crosstalk between tumor cells and macrophages. 1,25(OH)2D3 did not inhibit butyrate-driven Wnt signaling in HCT116 cells (Supplemental Fig. 5), demonstrating that it does not act as a general inhibitor of Wnt signaling.
Fig. 3. Vitamin D3 inhibits macrophage-mediated increase in Wnt signaling.
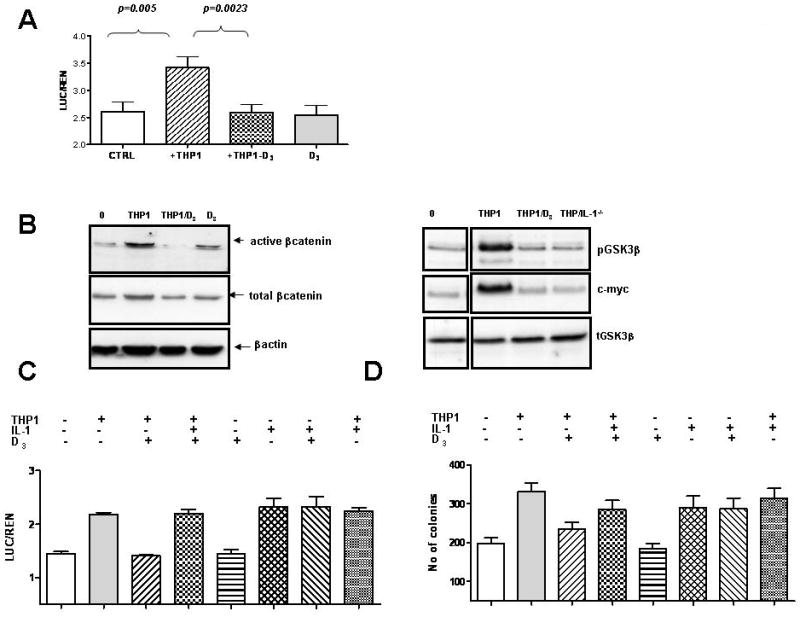
A: HCT116 cells transfected with TOP-FLASH reporter plasmid were incubated with THP1 cells as indicated. B: The levels of active β–catenin, total β–catenin, pGSK3β, total GSK3β and c-Myc were determined in colon cancer cells cultured alone or together with THP1. HCT116 cells were also cultured with THP1 cells transfected with IL-1β specific siRNA (THP1/IL1-/-) as indicated. C: HCT116 cells were transfected with TOP-FLASH reporter and were cultured in the presence of THP1 cells, IL-1β and vitamin D3 as indicated. E: The effect of vitamin D3 and IL-1β on clonogenic potential of HCT116 cells.
The ability of vitamin D3 to inhibit macrophage-mediated Wnt signaling in tumor cells was rescued by exogenous IL-1β (Fig. 3C), suggesting that 1,25(OH)2D3 acts predominantly by inhibiting the release of IL-1β from THP1 macrophages induced by tumor cells. Indeed, 1,25(OH)2D3 did not interfere with the ability of IL-1β to increase Wnt signaling in tumor cells (Fig. 3C, Supplemental Fig. 6).
Consistent with the ability to abrogate Wnt signaling, 1,25(OH)2D3 blocked the ability of THP1 macrophages to promote the clonogenic growth of HCT116 cells (Fig. 3D). Exogenous IL-1β partially restored the ability of vitamin D3 treated THP1 macrophages to promote clonogenic growth of tumor cells (Fig. 3D). Vitamin D3 did not affect the ability of IL-1β or TNF to promote the clonogenic growth of HCT116 cells (Fig. 3D, Supplemental Fig. 6), confirming that 1,25(OH)2D3 does not inhibit signaling by IL-1β. These data therefore demonstrate that 1,25(OH)2D3 interrupts the crosstalk between tumor cells and the tumor microenvironment through its ability to inhibit the release of IL-1β from macrophages.
To confirm that 1,25(OH)2D3 regulates the interactions between tumor cells and macrophages, we showed that untreated THP1 cells, but not 1,25(OH)2D3 treated THP1 macrophages, promoted proliferation of HCT116 cells (Supplementary Fig. 7A). Consistently, the increased expression of cyclin D1, cyclin A and cyclin E in HCT116 cultured with macrophages, returned to basal level upon treatment of cultures with 1,25(OH)2D3 (Supplementary Fig. 7B).
4. 1,25(OH)2D3 inhibits THP1 driven Wnt signaling and THP1-driven proliferation of tumor cells in a VDR dependent manner
To examine whether vitamin D3 exerts its biological activity in a VDR dependent manner, we silenced VDR expression in THP1 macrophages (inset, Fig. 4C). VDR deficient macrophages retained the ability to increase Wnt signaling upon 1,25(OH)2D3treatment, in contrast to nontransfected THP1 cells or THP1 cells transfected with nontargeting siRNA (NSP) (Fig. 4A). Accordingly, 1,25(OH)2D3 failed to inhibit macrophage mediated increased expression of c-myc and cyclin D1 in tumor cells upon silencing of VDR in THP1 cells (Fig. 4B). Finally, we showed that VDR deficient THP1 macrophages failed to respond to vitamin D3 and continued to secrete factors that promote proliferation of HCT116 cells upon treatment with vitamin D3 (Fig. 4C). 1,25(OH)2D3 also failed to inhibit the growth promoting activity of VDR deficient THP1 macrophages in co-culture experiments (Fig. 4D). These data established that the ability of vitamin D3 to regulate the cross-talk between THP1 macrophages and colon cancer cells requires the expression of VDR on the THP1 cells, confirming that macrophages, and not epithelial cells, were targeted by 1,25(OH)2D3. Consistent with this notion, silencing of VDR expression in HCT116 cells did not impact the interplay between epithelial cells and macrophages, or influence the ability of 1,25(OH)2D3 to interfere with this crosstalk (data not shown).
Fig. 4. Inhibition of Wnt signaling by vitamin D3 requires VDR expression on macrophages.
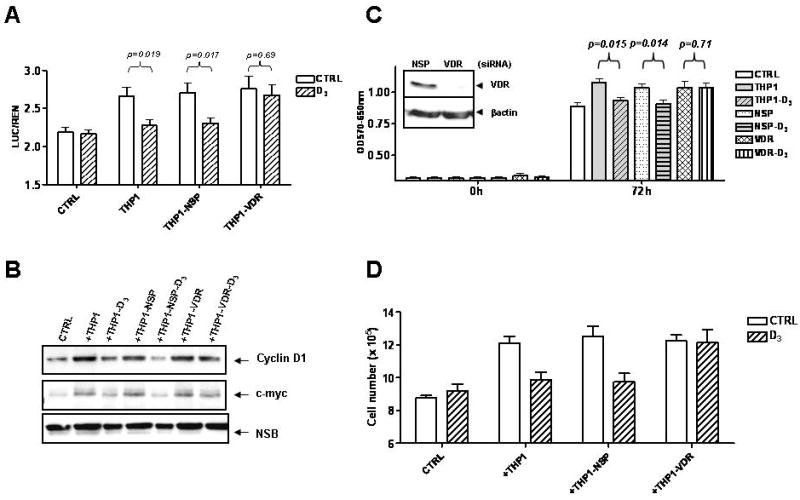
A: HCT116 cells transfected with TOP-FLASH reporter gene were cultured with nontransfected THP1 cells, or THP1 macrophages transfected with nontargeting (NSP) siRNA or VDR specific siRNA. B: Proteins were isolated from HCT116 cells co-cultured with THP1 cells as described above and the expression of cyclin D1 and c-myc was determined by immunoblotting. C: Growth of HCT116 cells with conditioned medium collected from untreated or vitamin D3 treated THP1 cells transfected with nontrageting siRNA or VDR specific siRNA, as determined by MTT assay, D: HCT116 and THP1 cells were co-cultured as indicated and the number of HCT116 cells determined after 48 hours.
5. p-STAT1 mediates the ability of THP1 macrophages to induce Wnt signaling and to promote proliferation of colon cancer cells
Tumor associated macrophages often express constitutively activated STAT1 (Biswas et al., 2006; Kusmartsev and Gabrilovich, 2005), as we demonstrated for the THP1 cells (Fig. 5A) and tumor cells have been recently shown to induce STAT1 phoshorylation on Y701 in primary macrophages (Hagemann et al., 2008). Since the expression of IL-1β is regulated by STAT1 (Lee et al., 2006; Tsukada et al., 1996), we investigated whether STAT1 was required for the release of IL-1β from macrophages. HCT116 and HKe-3 cells were co-cultured with THP1 cells transfected with nonspecific (NSP) or STAT1 specific siRNA (Fig. 5A inset). As shown in Fig. 5A, STAT1 deficient THP1 cells failed to release IL-1β in response to tumor cells or in response to stimulation with LPS. In contrast, induction of IL-8 in response to LPS was STAT1 independent (Fig. 5B).
Fig. 5. Secretion of IL-1β, but not IL-8, from THP1 macrophages, requires STAT1.
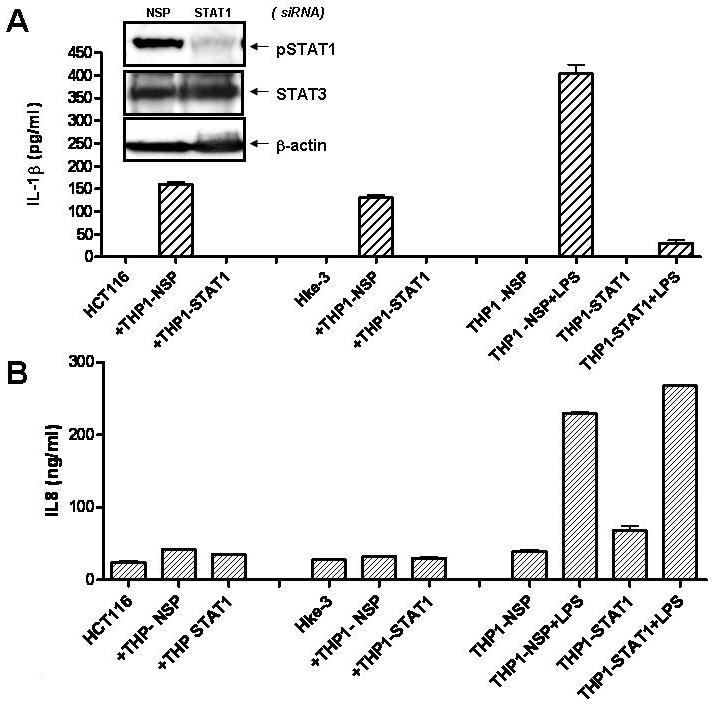
HCT116 and Hke-3 cells were cultured with THP1 cells transfected with nontargeting siRNA (THP1-NSP) or siRNA specific for STAT1 (THP1-STAT1) (A, inset). Cultures were treated with LPS (1 μg/ml) and the levels of IL-1β (A) and IL-8 (B) were determined by ELISA.
Consistently, STAT1 deficient THP1 macrophages failed to promote proliferation of the tumor cells (Fig. 6A), did not activate Wnt signaling in tumor cells (Fig. 6A, right panel) and failed to stabilize β–catenin and to increase the expression of c-myc and cyclin D1 in tumor cells (Fig. 6B).
Fig. 6. Activated STAT1 is required for pro-tumorigenic activity of THP1 macrophages.
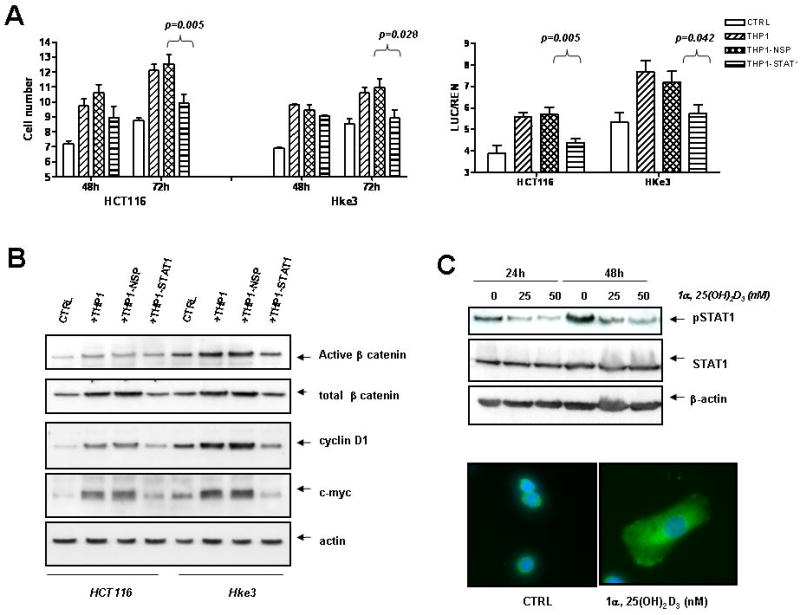
A: The number of tumor cells was determined upon coculture with nontransfected THP1 cells, THP1 cells transfected with NSP (nontargeting) siRNA or STAT1 specific siRNA. Right panel: HCT116 and Hke-3 cells were transfected with TOP-FLASH and cultured with THP1 cells as indicated. B: The levels of active β-catenin, total β-catenin, cyclin D1 and c-myc were determined by immunoblotting, C: THP1 cells were treated with vitamin D3 as indicated and the levels of pSTAT1 and total STAT1 were determined by immunoblotting. Subcellular localization of STAT1 was determined by immunofluorescence (green); nuclei were stained by DAPI. THP1 cells were left untreated or were treated with vitamin D3 (25nM) for 16 hours.
To examine whether 1,25(OH)2D3 regulates STAT1 in macrophages, THP1 cells were treated with vitamin D3 and tested for the expression of both p-STAT1 and total STAT1. As shown in Figure 6C, 1,25(OH)2D3 inhibited the expression of constitutively active STAT1 (pSTAT1Y701), without a significant and reproducible effect on the expression of total STAT1. Consistently, while most of the STAT1 in untreated THP1 cells appeared to be nuclear, treatment of cells with vitamin D3 resulted in translocation of STAT1 from the nucleus to the cytoplasm (Fig. 6C).
Therefore, our data established that tumor associated macrophages secrete IL-1β in a pSTAT1 dependent manner, activate Wnt signaling and thereby promote proliferation of the tumor cells. 1,25(OH)2D3, through its ability to inhibit pSTAT1 in macrophages and thus IL-1β production, inhibits their capability to induce Wnt signaling and to promote proliferation of tumor cells.
Discussion
The tumor microenvironment has a profound effect on the progression of colorectal cancer. Although cells in the tumor microenvironment remain genetically stable (Qiu et al., 2008), during the process of inflammation they secrete factors that target tumor cells and affect their survival, proliferation, migration and differentiation. For example, mast cells have been shown to be required for Apc-driven intestinal tumorigenesis in APCΔ468 mice (Gounaris et al., 2007), and depletion of macrophages resulted in an attenuated phenotype in APCΔ716 mice (Oguma et al., 2008). Thus, the Apc-initiated epithelium requires a supportive microenvironment in order to give rise to malignant tumors.
Macrophages have been shown to be required for the maintenance of progenitor cells in the intestinal crypt (Pull et al., 2005) and for the growth of Wnt/β-catenin activated intestinal epithelial cells (Oguma et al., 2008). Here we demonstrate that macrophages upon interaction with tumor cells secrete IL-1β in a pSTAT1 dependent manner, which provokes phosphorylation of GSK3β, activates Wnt signaling and thereby promote proliferation of the tumor cells. Alveolar macrophages from patients with lung cancer were found to secrete significantly higher amount of IL-1β than either peripheral blood monocytes from the same patient, or control subjects (Siziopikou et al., 1991), demonstrating an in vivo role of IL-1β in tumor progression.
Genome-wide expression analysis revealed that several Wnt target genes are upregulated in colon cancer cells grown in the presence of macrophages, including c-jun, uPAR, CD44, VEGF, Met, ID2, DKK3, FGF9, DLL3, FZZ9, fibronectin and Jagged 1 (data not shown). This is significant, as it has recently been reported that in vivo progression from microadenoma to macroscopic tumors in Apc Min mice is associated with augmentation of canonical Wnt signaling and increased expression of Wnt target genes (Oyama et al., 2008), demonstrating that the enhancement of Wnt signaling beyond a threshold level is required for tumor progression. In addition, β–catenin translocation is often detected at the invasive front between the tumor and surrounding tissue (Brabletz et al., 1998; Brabletz et al., 2001), consistent with the interpretation that surrounding tissue at the invasion front provides signals to the tumor cells that promote nuclear translocation of β-catenin and thus drive tumor progression. Although HCT116 cells carry a Ser45 mutation in β-catenin, expression of WT or mutant β-catenin can further increase Wnt dependent transcription, suggesting that β-catenin is only partially stabilized in HCT116 cells (Suzuki et al., 2004; Taketo, 2004). Data presented here demonstrate that Wnt signaling can be further boosted in β-catenin mutant cells by the proinflammatory cytokines. IL1 promoted Wnt signaling in RKO colon cancer cells (data not shown), which carry WT Apc and WT βcatenin, demonstrating that IL1 can promote βcatenin/TCF signaling in cells that do not harbor mutant βcatenin.
We demonstrated that IL-1β secreted by macrophages is both required and sufficient to activate Wnt signaling in tumor cells and to promote their growth. IL-1β therefore represents an important and direct link between inflammation and promotion of Wnt signaling during tumorigenesis. The nature of tumor-induced IL-1β release from macrophages and the mechanism whereby IL-1β inactivates GSK3β and leads to enhanced β-catenin/TCF4 transcriptional activity remain, for now, unknown. Polymorphism in the IL-1β gene that results in increased levels of IL-1β, has been shown to be associated with increased risk of colon cancer (Gunter et al., 2006). Consistently, mutations in NOD2 that have been linked to Crohn's disease, and therefore to increased risk of colorectal cancer, resulted in increased production of IL-1β and greater colonic inflammation (Maeda et al., 2005) and gastric specific overexpression of IL-1β was shown to be sufficient to induce spontaneous gastric inflammation and dysplasia, and to accelerate development of carcinomas in the setting of H. felis infection (Tu et al., 2008).
Finally, we demonstrated that the crosstalk between the macrophages and tumor cells can be disrupted by 1,25(OH)2D3, a potent chemopreventive agent for colorectal cancer. We showed that 1,25(OH)2D3 inhibited pSTAT1 in macrophages, prevented IL-1β production, and inhibited the ability of macrophages to induce Wnt signaling in tumor cells and to promote their proliferation. Our data therefore establish that vitamin D3 does not necessarily target epithelial cells themselves, but can inhibit the growth of tumor cells by acting on tumor associated macrophages. Of note, 1,25(OH)2D3 inhibited increased expression of c-myc and COX-2, two Wnt target genes, in the AOM/DSS model of colon cancer (Fichera et al., 2007).
Our findings describe a previously unknown link between inflammation, IL-1β, Wnt signaling and growth of colon cancer cells. We established that 1,25(OH)2D3, by acting on the cells in the tumor microenvironment, imposes growth regulation on tumor cells that display intrinsic resistance to this important chemopreventive agent. Our data therefore suggest that 1,25(OH)2D3 may have a potential to re-educate tumor associated macrophages and thereby reverse a tumor promoting microenvironment. It will be important to assess whether this crosstalk occurs in human tumors.
Material and Methods
Cell lines and co-culture experiments
The HCT116 and Hke-3 colorectal carcinoma cell lines, which differ only by the presence of the mutant k-Ras allele (Shirasawa et al., 1993), were cultured in MEM and the human monocytic cell line, THP1, was cultured in RPMI. Normal human monocytes, >90% CD14 and CD11c positive and less than 1% anti T cell receptor positive, were purchased from Astarte Biologics (Redmond, WA). Transwell Permeable Supports (Corning Incorporated, Lowell, MA were used in co-culture experiments.
Cell proliferation was assessed by the MTT assay and by BrdU incorporation (BrdU cell proliferation Assay kit, Calbiochem, Gibbstown, NJ). The assays were performed according to the manufacturer's instructions.
For clonogenic assay, HCT116 and Hke-3 cells were seeded at a density of 200 cells per well of a six well plate alone or together with THP1 macrophages or peripheral blood monocytes for 7 days. Tumor cells were cultured with THP1 monocytes directly (400 or 1600/ 6 well), as THP1 cells alone did not attach and form colonies. In contrast, experiments with primary monocytes were done using transwells, as these cells are adherent, which could potentially obscure the number of colonies. For these experiments, 3000 monocytes were added into the top chamber. Colonies were fixed and stained with 6% glutaraldehyde and 0.5% crystal violet and counted using Total Lab 1.1 software (Nonlinear Dynamics, Durham, NC, USA).
Transient transfection and Reporter gene assay
HCT116 and Hke-3 cells were transiently transfected with the TOP-FLASH or TOP-FOP luciferase reporter plasmids using the calcium phosphate method. Transfection efficiency was normalized by co-transfection with pTK-Renilla and luciferase activity was determined according to the vendor's protocol (Dual Luciferase reporter assay, Promega, Madison, WI).
THP1 cells were transfected with 20 nM of non specific siRNA (NSP) or siRNAs specific for VDR, IL-1β or STAT1 (Dharmacon, Lafayette, CO) using Lipofectamin LTX (Invitrogen, Carlsbad, CA).
Immunofluorescence
For detection of F-actin, macrophages were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 and stained with Phalloidin for 30 min. For detection of STAT1, THP1 cells were fixed in ice-cold methanol/acetic acid (95:5v/v) for 20 min at -20°C. The cells were incubated with anti-STAT1 antibody (1:100) for 1h at 37°C and with secondary anti-rabbit antibody conjugated to FITC for 45 min at 37°C. Images were acquired with a SPOT CCD camera and analyzed by SPOT software.
Western Blot
Western blots were performed using standard procedures. Membranes were blocked with 5% milk in TBS containing 0.1% Tween 20, and incubated with antibodies specific for cyclin D1, cyclin E, cyclin A, c-myc (Santa Cruz Biotechnology, Inc. Santa Cruz, CA.), active β-catenin, pSTAT1, STAT1, STAT3, pGSK3 (Millipore, Billerica, MA), total β-catenin (BD Biosciences, San Jose, CA), vitamin D receptor (Calbiochem, Gibbstown, NJ), and β-actin (Sigma Aldrich, St. Louis, MO). Immunoreactive bands were visualized by chemiluminescence (Amersham ECL™ western blotting detection kit, Piscataway, NJ).
Human Cytokine Array and ELISA
Supernatants were collected from macrophages, HCT116 cells or from cocultures of HCT116 cells with macrophages for 48 hours. Relative cytokines levels were determined using Human Cytokine Array kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Supernatants collected from monoculters or cocultures were also used for detection of IL-1β and IL-8 by human IL-1β and IL-8 ELISA kits (BD Biosciences, San Jose, CA).
Acknowledgments
We thank Dr. Paolo Norio for reading the manuscript. This work was supported by CA 111361 (to LK), U54 CA 100926 (to LA) and P30-13330 from NCI.
References
- Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–47. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–22. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Hermann K, Gunther K, Hohenberger W, Kirchner T. Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract. 1998;194:701–4. doi: 10.1016/s0344-0338(98)80129-5. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98:10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Etoh T, Shibuta K, Barnard GF, Kitano S, Mori M. Angiogenin expression in human colorectal cancer: the role of focal macrophage infiltration. Clin Cancer Res. 2000;6:3545–51. [PubMed] [Google Scholar]
- Fichera A, Little N, Dougherty U, Mustafi R, Cerda S, Li YC, et al. A vitamin D analogue inhibits colonic carcinogenesis in the AOM/DSS model. J Surg Res. 2007;142:239–45. doi: 10.1016/j.jss.2007.02.038. [DOI] [PubMed] [Google Scholar]
- Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13:1472–9. doi: 10.1158/1078-0432.CCR-06-2073. [DOI] [PubMed] [Google Scholar]
- Garland CF, Comstock GW, Garland FC, Helsing KJ, Shaw EK, Gorham ED. Serum 25-hydroxyvitamin D and colon cancer: eight-year prospective study. Lancet. 1989;2:1176–8. doi: 10.1016/s0140-6736(89)91789-3. [DOI] [PubMed] [Google Scholar]
- Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A. 2007;104:19977–82. doi: 10.1073/pnas.0704620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter MJ, Canzian F, Landi S, Chanock SJ, Sinha R, Rothman N. Inflammation-related gene polymorphisms and colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1126–31. doi: 10.1158/1055-9965.EPI-06-0042. [DOI] [PubMed] [Google Scholar]
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–8. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampman E, Slattery ML, Caan B, Potter JD. Calcium, vitamin D, sunshine exposure, dairy products and colon cancer risk (United States) Cancer Causes Control. 2000;11:459–66. doi: 10.1023/a:1008914108739. [DOI] [PubMed] [Google Scholar]
- Kim BG, Li C, Qiao W, Mamura M, Kasprzak B, Anver M, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441:1015–9. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Inhibition of Interferon gamma Signaling by the Short Chain Fatty Acid Butyrate. Mol Cancer Res. 2003;1:855–62. [PubMed] [Google Scholar]
- Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Oncogenic Ras promotes butyrate-induced apoptosis through inhibition of gelsolin expression. J Biol Chem. 2004;279:36680–8. doi: 10.1074/jbc.M405197200. [DOI] [PubMed] [Google Scholar]
- Klampfer L, Huang J, Shirasawa S, Sasazuki T, Augenlicht L. Histone Deacetylase Inhibitors Induce Cell Death Selectively in Cells That Harbor Activated kRasV12: The Role of Signal Transducers and Activators of Transcription 1 and p21. Cancer Res. 2007;67:8477–85. doi: 10.1158/0008-5472.CAN-07-0210. [DOI] [PubMed] [Google Scholar]
- Klampfer L, Swaby LA, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Oncogenic Ras increases sensitivity of colon cancer cells to 5-FU-induced apoptosis. Oncogene. 2005;24:3932–41. doi: 10.1038/sj.onc.1208552. [DOI] [PubMed] [Google Scholar]
- Kumagai T, O'Kelly J, Said JW, Koeffler HP. Vitamin D2 analog 19-nor-1,25-dihydroxyvitamin D2: antitumor activity against leukemia, myeloma, and colon cancer cells. J Natl Cancer Inst. 2003;95:896–905. doi: 10.1093/jnci/95.12.896. [DOI] [PubMed] [Google Scholar]
- Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–91. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- Lamprecht SA, Lipkin M. Chemoprevention of colon cancer by calcium, vitamin D and folate: molecular mechanisms. Nat Rev Cancer. 2003;3:601–14. doi: 10.1038/nrc1144. [DOI] [PubMed] [Google Scholar]
- Lee C, Lim HK, Sakong J, Lee YS, Kim JR, Baek SH. Janus kinase-signal transducer and activator of transcription mediates phosphatidic acid-induced interleukin (IL)-1beta and IL-6 production. Mol Pharmacol. 2006;69:1041–7. doi: 10.1124/mol.105.018481. [DOI] [PubMed] [Google Scholar]
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin M, Lamprecht SA. Mechanisms of action of vitamin D: recent findings and new questions. J Med Food. 2006;9:135–7. doi: 10.1089/jmf.2006.9.135. [DOI] [PubMed] [Google Scholar]
- Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–8. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–6. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- Newmark HL, Lipkin M. Calcium, vitamin D, and colon cancer. Cancer Res. 1992;52:2067s–2070s. [PubMed] [Google Scholar]
- Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. Embo J. 2008;27:1671–81. doi: 10.1038/emboj.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterling SJ, van der Bij GJ, Meijer GA, Tuk CW, van Garderen E, van Rooijen N, et al. Macrophages direct tumour histology and clinical outcome in a colon cancer model. J Pathol. 2005;207:147–55. doi: 10.1002/path.1830. [DOI] [PubMed] [Google Scholar]
- Oyama T, Yamada Y, Hata K, Tomita H, Hirata A, Sheng H, et al. Further upregulation of {beta}-catenin/Tcf transcription is involved in the development of macroscopic tumors in the colon of Apc Min/+ mice. Carcinogenesis. 2008 doi: 10.1093/carcin/bgn001. [DOI] [PubMed] [Google Scholar]
- Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res. 2008;68:5086–95. doi: 10.1158/0008-5472.CAN-07-2325. [DOI] [PubMed] [Google Scholar]
- Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–87. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer HG, Larriba MJ, Garcia JM, Ordonez-Moran P, Pena C, Peiro S, et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med. 2004;10:917–9. doi: 10.1038/nm1095. [DOI] [PubMed] [Google Scholar]
- Pendas-Franco N, Aguilera O, Pereira F, Gonzalez-Sancho JM, Munoz A. Vitamin D and Wnt/beta-catenin pathway in colon cancer: role and regulation of DICKKOPF genes. Anticancer Res. 2008;28:2613–23. [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40:650–5. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Fassetta M, De Bacco F, D'Alessandro L, Gramaglia D, Di Renzo MF, et al. A positive feedback loop between hepatocyte growth factor receptor and beta-catenin sustains colorectal cancer cell invasive growth. Oncogene. 2007;26:1078–87. doi: 10.1038/sj.onc.1209859. [DOI] [PubMed] [Google Scholar]
- Robsahm TE, Tretli S, Dahlback A, Moan J. Vitamin D3 from sunlight may improve the prognosis of breast-, colon- and prostate cancer (Norway) Cancer Causes Control. 2004;15:149–58. doi: 10.1023/B:CACO.0000019494.34403.09. [DOI] [PubMed] [Google Scholar]
- Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem. 2003;278:48137–45. doi: 10.1074/jbc.M307154200. [DOI] [PubMed] [Google Scholar]
- Shah S, Islam MN, Dakshanamurthy S, Rizvi I, Rao M, Herrell R, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol Cell. 2006;21:799–809. doi: 10.1016/j.molcel.2006.01.037. [DOI] [PubMed] [Google Scholar]
- Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–8. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- Siziopikou KP, Harris JE, Casey L, Nawas Y, Braun DP. Impaired tumoricidal function of alveolar macrophages from patients with non-small cell lung cancer. Cancer. 1991;68:1035–44. doi: 10.1002/1097-0142(19910901)68:5<1035::aid-cncr2820680522>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–22. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- Taketo MM. Shutting down Wnt signal-activated cancer. Nat Genet. 2004;36:320–2. doi: 10.1038/ng0404-320. [DOI] [PubMed] [Google Scholar]
- Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982;42:1530–6. [PubMed] [Google Scholar]
- Tsukada J, Waterman WR, Koyama Y, Webb AC, Auron PE. A novel STAT-like factor mediates lipopolysaccharide, interleukin 1 (IL-1), and IL-6 signaling and recognizes a gamma interferon activation site-like element in the IL1B gene. Mol Cell Biol. 1996;16:2183–94. doi: 10.1128/mcb.16.5.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–19. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell. 2006;127:139–55. doi: 10.1016/j.cell.2006.08.036. [DOI] [PubMed] [Google Scholar]