

Abstract
Purpose
While tyrosine kinase inhibitors (TKI) have improved survival in advanced GIST, complete response is rare and most patients eventually fail the first line treatment with imatinib. Sunitinib malate is the only approved 2nd line therapy for patients with imatinib-resistant or -intolerant GIST. The clinical benefit of sunitinib is genotype-dependent in regards to both primary and secondary mutations, with GIST patients harboring the KITAY502-3ins exon 9 mutation being the most sensitive.
Experimental Design
As sunitinib resistance is now emerging, our goal was to investigate mechanisms of progression and to test the efficacy of novel TKI on these resistant mutants in vitro. ENU mutagenesis of Ba/F3 cells expressing the KITAY502-3ins mutant was used to investigate novel patterns of resistant mutations evolving in the presence of sunitinib.
Results
Tumors from patients who developed sunitinib resistance after at least 1-year of radiographic response were analyzed, showing similar findings of a primary KITAY502-3ins mutation and a secondary mutation in the KIT activation loop. Ba/F3 cells expressing these sunitinib-resistant double mutants showed sensitivity to both dasatinib and nilotinib.
Conclusions
Sunitinib resistance in GIST shares similar pathogenetic mechanisms identified in imatinib failure, with acquisition of secondary mutations in the activation domain after an extended initial response to the drug. Moreover, in vitro mutagenesis with or without ENU of Ba/F3 cells expressing KITAY502-3ins showed acquisition of secondary mutations restricted to the 2nd kinase domain of KIT. In contrast, in vitro resistance to imatinib produces a broader spectrum of secondary mutations including mutations in both KIT kinase domains.
Keywords: KIT, exon 9, GIST, imatinib, sunitinib resistance
Introduction
Although imatinib achieves a partial response or stable disease in most GIST patients, with lasting responses over 5-year period in more than 20% of patients, complete responses are rare (1). Clinical responses to imatinib in GIST depend on the presence and type of KIT or platelet derived growth factor receptor, PDGFRA gain-of-function mutations. Thus patients with exon 11 mutations show a partial response rate of 84%, while patients with tumors harboring a KIT exon 9 or no detectable mutation had a partial response rate of 48% and 0%, respectively (2). It is now clear that a majority of patients who initially benefit from imatinib, eventually become resistant. The most common mechanism of acquired resistance is through a secondary KIT mutation, usually located either in the N-terminal or C-terminal kinase domain, which disrupts imatinib binding by stabilizing the receptor in a more active conformation. The mechanism for the development of secondary KIT mutations remains unclear, but resistant patients with identifiable second site mutations had been treated with imatinib longer than resistant patients lacking second site mutations (3). The only FDA approved second line TKI for patients with advanced GIST who have progressed on or are intolerant to imatinib is sunitinib malate (Sutent, Pfizer, New York, NY). The clinical benefit from sunitinib following imatinib failure is influenced by the genomic location of both the primary and secondary mutations of the activated kinase. Thus, progression-free and overall survivals are significantly longer for patients with either KIT exon 9 mutation or KIT/PDGFRA wild-type tumors.
Furthermore, imatinib-resistant secondary mutations within the ATP-binding pocket (KIT exon 13 and 14) appear to be sensitive to sunitinib inhibition (4). However, after an initial response patients developing sunitinib resistance are being diagnosed in the clinic. It remains unclear if similar mechanisms identified in imatinib failure are also responsible for the development of sunitinib resistance. Since sunitinib activity encompasses a broader spectrum of targeted kinases as compared to imatinib, including anti-vascular endothelial growth factor receptor (VEGFR) activity, it is possible that additional mechanisms play a role in the acquisition of resistance. The goal of our study was first to investigate the clinicopathologic and genomic characteristics associated with patients failing sunitinib therapy. Second, using an in vitro model we tested the efficacy of novel TKI on the sunitinib-resistant mutants. Furthermore, in order to predict patterns of mutations arising during sunitinib therapy, we used a cell-based screen to identify mutations giving rise to drug-resistance, the results of which can be used to generate a genotype-dependent algorithm for drug selection. Utilizing N-Ethyl-N-nitrosourea (ENU), a DNA alkylating agent which is a highly potent mutagen in mice (5), we established a robust, unbiased mutagenesis system.
ENU mutagenesis alters predominantly AT base pairs and produces A/T->T/A transversions, A/T->G/C transitions and with much lower frequency G/C ->A/T transitions, G/C->C/G transversions, A/T->C/G transitions and % G/C->T/A transitions thus producing a broad spectrum of missense mutations, which either may be loss- or gain-of-function mutations. ENU mutagenesis was used to compare incidence and types of BCR-ABL kinase domain (KD) mutants emerging in the presence of imatinib, dasatinib, and nilotinib, alone and in dual combinations in Ba/F3 cells. We have used this approach to investigate the development of drug resistant mutations in KIT. As the pattern of imatinib-induced resistant mutations has been described in-depth, we focused on identifying mutations conferring sunitinib resistance and acquired secondary mutations associated with KIT502-3AYins primary mutations. By transforming the Ba/F3 murine pro-B cell line with KIT502-3AYins, with or without ENU mutagenesis, we were able to reproduce the pattern and relative abundance of sunitinib- and imatinib-resistant secondary mutations previously identified in drug-resistant GIST patients with the exon 9 mutation.
Materials and Methods
Clinicopathologic Features
Patients with a diagnosis of GIST who developed imatinib resistance and subsequently developed progression on sunitinib after at least one year on therapy, were identified from our prospective sarcoma database at Memorial Sloan-Kettering Cancer Center. Those who underwent surgical resection of their resistant tumors were included in the study. Based on the previous experience with imatinib-resistance, we selected a 1-year cutoff of clinical response to sunitinib therapy (defined as failure to progress radiographically) in order to exclude patients with primary resistance and to allow sufficient time for clonal selection of second-site mutations under drug pressure. Clinical information was obtained from the prospective sarcoma database and review of medical charts, including extent of disease at the start of each TKI treatment, duration of imatinib and sunitinib therapy prior to development of resistance and/or surgical resection and the best clinical response obtained on each TKI. This study was approved by the Institutional Review Board.
The histologic slides from the surgical specimens were reviewed and the diagnosis of GIST was confirmed based on the morphology and immunostaining for the KIT antibody. Tissue from both imatinib and sunitinib-resistant GIST resection was available in most cases for analysis of tumor response, such as degree of necrosis, mitotic activity, and expression of KIT.
KIT/PDGFRA Genotyping
Mutation analysis was performed as described previously (6). Genomic DNA was isolated from snap-frozen tumor tissue samples stored at -70°C, using a standard phenol-chloroform organic extraction protocol. All cases were tested for the known sites of KIT (exons 9, 11, 13, 14, and 17) and PDGFRA (exons 12, 14 and 18) mutations. One μg of genomic DNA was subjected to PCR using Platinum TaqDNA Polymerase High Fidelity (Life Technologies, Inc). Primer sequences and annealing temperatures were as described (6, 7). Direct sequencing of PCR products was performed for all exons tested and each ABI sequence was compared to the NCBI human KIT and PDGFRA gene sequences.
DNA constructs
The retroviral vector plasmid containing WT human KIT cDNA (GNNK- isoform), pMSCV-WTKIT-IRES-GFP, was generously provided by Dr. Gary Gilliland (Harvard Medical School). KIT mutations recapitulating the genotype found in sunitinib malate-resistant GIST patients were generated by site-directed mutagenesis PCR, using QuickChange II XL site-directed Mutagenesis Kit (Qiagen, Inc). KIT double mutant isoforms hosting primary exon 9 mutation and an acquired secondary mutations in exon 13 or 17 were generated as follows: KIT502-3AYins/V654A, KIT502-3AYins/D820Y, and KIT503-3AYins/N822K. KIT502-3AYins single mutant was used as a control. All constructs were verified with direct sequencing.
Establishing Ba/F3 KIT mutants stable transformant cell lines
The IL3-dependent Ba/F3 murine pro-B cell line was obtained from the German Collection of Microorganisms and Cell Culture. Ba/F3 cells were transfected and selected as previously described (8). Briefly, Ba/F3 cells were co-transfected with retroviral vector plasmids containing KIT cDNA mutant isoforms and linear hygromycin resistant DNA via electroporation. The electroporated cells were first grown in the presence of Hygromycin for 10 days followed by sorting according to GFP fluorescence. GFP positive cells were further grown for another 2 weeks in the presence of IL3, then transformed to IL3-independent upon IL3 withdrawal. Ba/F3 KIT502-3AYins cells were supplemented with 20 ng/ml of KIT ligand, all the double mutants described above did not require KIT ligand supplement. Cells were stained with PE-conjugated anti-CD117 antibody (BD Biosciences) and monitored by flow cytometry. Cell lysates were subjected to Western Blotting using anti-KIT antibody (Calbiochem).
Inhibitors and in vitro drug testing
The following TKI were used: imatinib, sunitinib, sorafenib, dasatinib, and nilotinib. Nilotinib and dasatinib were synthesized in-house and kindly provided by Dr. Bayard Clarkson's lab at MSKCC. Imatinib, sunitinib and sorafenib were purchased commercially and purified with column chromatography by Dr. Bayard Clarkson's lab at MSKCC. The kinases inhibitors were dissolved at 10mM in dimethyl sulfoxide (DMSO) as stocks and working dilutions were freshly made prior to experiments.
Cell proliferation assays
Ba/F3 cells (0.5 × 105/well) expressing KIT mutants were incubated with 10, 100, 1000, 5000 and 10,000 nM of imatinib, sunitinib, nilotinib, dasatinib and sorafenib in 96-well plates at 37°C for 48 hours in triplicate. Cells were incubated for 4 hours with 3H-thymidine (1 μCi [0.037 MBq]) before harvesting and 3H-thymidine incorporation was determined. Growth inhibition was plotted as the ratio of the average 3H-thymidine incorporation in drug-treated wells relative to no-drug controls. IC50s for tested inhibitors were calculated by GraphPad Prism software, version 5.00.
Apoptosis assays
Cells at a density of 1 × 106 were cultured in 24-well plates and incubated with imatinib, sunitinib, nilotinib, dasatinib, and sorafenib at concentrations to a range of 10, 100, 1000 and 5000 nM for 48 hours. Cells were harvested and stained with anti-AnnexinV-PE antibody and 7AAD (BD Biosciences, San Jose, CA). A minimum of 20,000 events were analyzed by FACScan (Becton-Dickison) within an hour after staining. Data were analyzed by FlowJo 7.1.3.
Immunoprecipitation and Western Blotting
Ba/F3 cells expressing KIT mutant isoforms were starved from serum and treated with the indicated concentrations of each inhibitor for 90 minutes. Cell lysis, immunoprecipitations, and immunoblotting were performed as previously described (8). Cell lysates were incubated with anti-KIT antibody (Assay Designs, Inc.) and Magna beads (Pierce Biotechnology) overnight at 4°C. The precipitated KIT was separated by electrophoresis and transferred to nitrocellulose. Blots were probed with anti-phospho-tyrosine antibodies PY20 and PY99 (Santa Cruz Biotechnology), stripped, and reprobed with rabbit polyclonal anti-KIT antibody (Calbiochem).
N-ethyl-N-nitrosourea (ENU) mutagenesis
Ba/F3 KIT502-3AYins cells were maintained in complete culture media, RPMI 1640 containing 10% serum, 1% pen/strep, and 20ng/ml of KIT ligand at an exponential growth rate. Cells were incubated with ENU (50 μg/ml) at a density of 5 × 106 cells/ml for 24 hours, and then washed 3 times with RPMI, replated in complete media, and expanded for a week under exponential growth conditions.
In vitro mutagenesis screen
ENU-exposed Ba/F3 KIT502-3AYins cells were cultured in 96-well plates (1.0 × 105 cells/well) in 150 μl of complete media, in the presence of various concentrations of sunitinib and imatinib. Sunitinib was supplemented at 0.05, 0.1, 0.25, 0.5 and 1 μM, and imatinib at 0.5, 1, 2.5, 5 and 10 μM, which corresponded to 1, 2, 5, 10 and 20 times their IC50s (sunitinib=54 nM, imatinib=517 nM), respectively. The number of resistant colonies was counted by visual inspection every 2-3 days, for at least 4-6 weeks, and. Single colonies were picked and expanded for analysis in 1 ml complete media in the presence of the corresponding concentration of inhibitors used in the screen. Genomic DNA was extracted and subjected to PCR for possible mutations in the main hot spots (KIT exons 13, 14 and 17) (3). In parallel, Ba/F3 KITAY502-3ins cells without ENU exposure were incubated with the same concentrations for each inhibitor. Resistant colonies were inspected and analyzed with the same protocol as the ENU-exposed cells. Ba/F3 KITWK557-8del cells with or without ENU treatment were grown in complete media supplemented with 0.05, 0.1, 0.25, 0.5 and 1.0 μM of imatinib, according to 1, 2, 5, 10 and 20 times its IC50, respectively. Resistant colony selection and analysis were conducted as described above as an isoform control.
Fluorescence in situ Hybridization (FISH)
Cells were treated with colcemid (Gibco) at 50ng/ml for 45minutes before harvesting. KITAY502-3ins DNA was labeled with Digoxingenin-11-dUTP (Roche) by nick translation. The slides and the probes were co-denatured at 70°C for 5 min, then incubated at 37°C overnight in a dark moist chamber. Post-hybridization wash was carried at 45°C with 50% formamide/2 × SSC. Mouse anti-digoxigenin IgG (5μg/ml) (Roche) and FITC-conjugated goat anti-mouse IgG (5μg/ml) (Invitrogen) were used for detection. The cells were then stained with DAPI (Molecular Probes). Slides were viewed with a Nikon E600 epifluorescence microscope.
Results
Secondary KIT mutations in the activation loop are associated with sunitinib resistance
We identified three imatinib resistant patients who showed a clinical response to sunitinib for more than a year (mean 23 months) before progressing and subsequently referred for surgical management. Tissue from the sunitinib resistant nodules was available for genotyping and transcriptional profiling. The sunitinib-resistant patients shared similar clinicopathologic and molecular findings: all had their primary GIST located in the small bowel, harbored a primary KIT exon 9 mutation and showed a partial response to imatinib therapy for a mean of 24 months (range 18-28), before developing progression. Furthermore, both patients with tissue available pre-sunitinib therapy lacked second-site imatinib-resistant mutations.
On microscopic evaluation, all sunitinib resistant tumors showed increased cellularity, brisk mitotic activity and strong and diffuse KIT immunoreactivity. The genotype analysis of the 8 nodules from the three sunitinib resistant patients revealed three different amino acid substitutions in the KIT activation loop (exon 17): D820Y, D820E, and N822K. In one patient only one of the 6 resistant nodules analyzed showed the presence of a second-site KIT mutation (D820E).
Ba/F3 cells expressing KIT Exon 9/Exon 17 Double Mutants are Sensitive to Dasatinib and Nilotinib Inhibition
To investigate the sensitivity of sunitinib-resistant KIT mutants to other TKIs, we established stable Ba/F3 cell lines expressing primary KIT exon 9 (KIT502-3AYins) and secondary mutations in KIT exon 13 (KITV654A) and exon 17 (KITD820Y, KITN822K). The KIT502-3AYins/D820Y and KIT502-3AYins/N822K were identified in sunitinib resistant GIST patients. The 3rd secondary mutation identified in a sunitinib-resistant patient, exon 17 KITD820E, failed transfection. The KIT502-3AYins/V654A mutant previously described in an imatinib-resistant, sunitinib sensitive patient (4) was used as a control.
The efficacy of five TKIs with anti-KIT activity, including imatinib, sunitinib, nilotinib, dasatinib and sorafenib, was tested in Ba/F3 KIT transfectants expressing the sunitinib-resistant double mutatants, KIT502-3AYins/D820Y and KIT502-3AYins/N822K, recapitulating mutations identified in patients, and compared to the imatinib- and sunitinib-sensitive KIT ectodomain mutant, KIT502-3AYins, as well as to the imatinib-resistant and sunitinib-sensitive double mutant, KIT502-3AYins/V654A. Each cell line was treated with similar escalating doses of the 5 inhibitors. Inhibition of KIT kinase activity was monitored by immunoblotting and the biological consequences were evaluated by determining proliferation inhibition and induction of apoptosis. Growth-inhibitory effects were determined by 3H incorporation, while induction of apoptosis was evaluated by flow cytometry using AnnexinV-PE Apoptosis Detection kit (Pharmingen).
Ba/F3 cells expressing exon 17 secondary mutations, KIT502-3AYins/D820Y, were resistant to either imatinib or sunitinib inhibition. Imatinib did not inhibit KIT phosphorylation at concentrations <5000 nM, nor did it show effects in biological assays. Sunitinib did not show any cellular effects below 1000 nM, inhibiting cell growth with an IC50 of 1486 nM, and inducing mild apoptosis at 1000 nM (Fig 1), although it abrogated KIT kinase activity between 100 to 1000 nM. However, dasatinib completely abolished KIT phosphorylation at a low dose, 10 to 100 nM. Dasatinib also showed an excellent efficacy inhibiting cell growth, with an IC50 of 40.6 nM and inducing apoptosis between 100 and 1000 nM. Nilotinib treatment resulted in marked decrease of KIT kinase activity below 100 nM and showed potent proliferation inhibition with an IC50 of 248 nM. Induction of apoptosis was achieved with a higher dose of nilotinib, 1000 nM (Fig 1). Similar experiments were done with Ba/F3 cells expressing the KIT502-3AYins/N822K mutant resulting in a comparable drug sensitivity profile. Although imatinib and sunitinib partially inhibited KIT kinase activity of this double mutant, cell proliferation or induction of apoptosis was not affected below 1000 nM. In contrast, dasatinib and nilotinib inhibited the kinase activity at concentrations below 100 nM and 1000 nM, inhibited cell growth with an IC50s of 66.2 nM and 309 nM, and induced apoptosis at 100 nM and 1000 nM, respectively. In contrast Ba/F3KIT502-3AYins cells were highly sensitive to both dasatinib and sunitinib, and less sensitive to imatinib and nilotinib inhibition. Dasatinib and sunitinib inhibited cell growth with an IC50 of 2.9 and 54 nM, respectively. Dasatinib induced significant apoptosis at 10 nM, whereas sunitinib induced apoptosis between 10 and 100 nM. Furthermore, dasatinib dramatically decreased KIT kinase autophosphorylation at <10nM, while sunitinib showed marked inhibition of KIT kinase autophosphorylation between 10 to 100 nM. Imatinib produced moderate inhibition on KIT kinase activity at 1000 nM, with an IC50 of 517 nM, while nilotinib and sorafenib inhibited kinase activity at 100 nM, with an IC50s of 695 and 861 nM, respectively. These 3 inhibitors induced significant apoptosis at 1000 nM (Fig 2).
Fig 1.
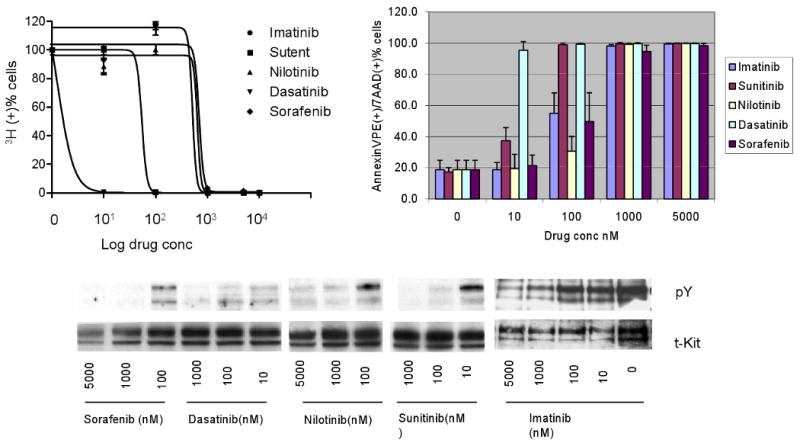
Imatinib resistant Ba/F3 KIT502-3AYins/D820Y double mutant is sensitive to dasatinib and nilotinib inhibition, but insensitive to sunitinib and imatinib. Dasatinib and nilotinib remarkably abolished the mutant KIT kinase activity at 100 nM and 1000 nM, and inhibited cell growth with IC50s of 40.6 and 248 nM, respectively. Both drugs induced more than 50% of apoptosis at 1000 nM. Sunitinib and imatinib treated cells showed reduced KIT kinase activity at 1000 and 5000 nM, respectively. Sunitinib inhibited cell proliferation with an IC50 of 1486 nM and induced significant apoptosis at 5000 nM. In contrast, imatinib did not show growth inhibition or induce apoptosis up to 5000 nM. Sorafenib inhibited cell proliferation with an IC50 of 842 nM and induced moderate apoptosis at 500 nM.
Fig 2.
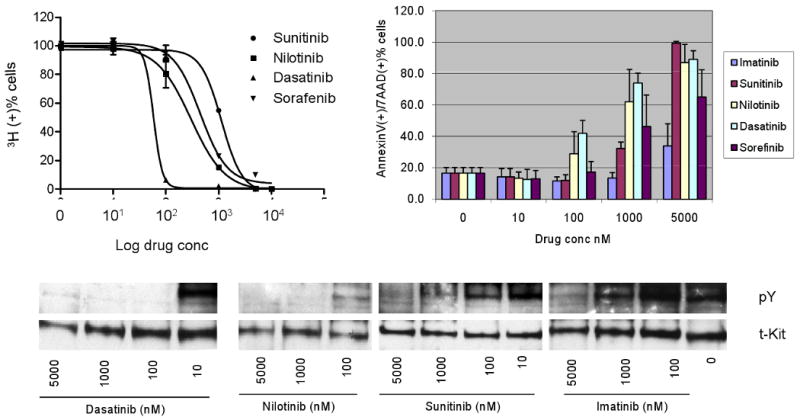
In vitro drug sensitivity of Ba/F3 KIT502-3AYins single mutant. Sunitinib and dasatinib inhibited KIT kinase activity at ≤10 nM, inhibited cell proliferation with IC50s of 54 and 2.9 nM, and induced significant apoptosis at 100 and 10 nM respectively. Imatinib, nilotinib, and sorafenib inhibited KIT phosphorylation between 100-1000 nM, inhibited cell growth with IC50s of 517, 695 and 861 nM, respectively, and induced significant apoptosis at 1000 nM.
As previously noted in the imatinib-resistant patients (9), our in vitro results validate that the secondary mutations in the ATP binding pocket remain sensitive to sunitinib inhibition. Inhibition of KIT activation of the Ba/F3KIT502-3AYins/V654A double-mutant was achieved with less than 10 nM of sunitinib, while inhibition of cell proliferation with an IC50 of 20.6 nM, concomitant with induction of apoptosis at 100 nM. Dasatinib treated cells also demonstrated dramatic decrease of KIT kinase activity at 10 nM, while the IC50 for proliferation was 334.5 nM and at 1000nM induction of apoptosis about 70%. Imatinib partially inhibited KIT phosphorylation at 1000 nM, but did not arrest the growth or induce apoptosis below 5000 nM, compared to the un treated control. Although nilotinib showed overt inhibition on KIT phosphorylation at 1000 nM and an IC50 of 1045 nM with regards to inhibition of cell proliferation, it did not induce apoptosis at a comparable dose. Table 1 summarizes these results in comparison with the IC50 of Ba/F3 KIT double mutants carrying a KIT exon 11 primary mutation (reported published in reference (8)).
Table 1.
Comparative efficacy of TKI in BA/F3 KIT double mutants, harboring either a KIT exon 9 or exon 11 primary mutation
| IC50s (nM) | |||
|---|---|---|---|
| Mutations | Nilotinib | Dasatinib | Sorafenib |
| KIT502-503AY/V654A | 1045 | 334.5 | 1672 |
| KITV560del/V654A | 192 | 585 | 1074 |
| KIT502-503AY/D820Y | 248 | 40.6 | 842 |
| KITV559D/D820Y | 297 | 432 | 944 |
| KIT502-503AY/N822K | 369 | 66.2 | 933 |
| KITV560del/N822K | NA | NA | NA |
Sorafenib Showed Moderate Inhibition on KIT502-3AYins Single Mutant, but only Mild Efficacy on KIT502-3AYins Double Mutants
In Ba/F3 cells expressing KIT502-3AYins sorafenib inhibited the mutant kinase at 100 to 1000 nM, induced moderate apoptosis at 100 nM, and inhibited cell proliferation with an IC50 of 861 nM. In contrast, it inhibited exon 9 double mutants, KIT502-3AYins/D820Y, KIT502-3AYins/N822K, and KIT502-3AYins/V654A, with IC50s of 842 nM, 933 nM, and 1672 nM, respectively, but did not induce significant apoptosis at comparable doses.
In vitro screen for sunitinib resistance revealed second-site mutations in the activation loop of KIT
To investigate the mechanisms of sunitinib-resistance and generate a comprehensive map of acquired mutations, we developed a cell-based screen for drug-resistance of KIT502-3AYins associated secondary mutations. Ba/F3 KIT502-3AYins cells were treated with the mutagen ENU. Subsequently the ENU treated Ba/F3 KIT502-3AYins cells were incubated with sunitinib or imatinib to identify drug resistant colonies. The incidence, patterns and types of mutations acquired by sunitinib selection were compared to the ones induced by imatinib treatment. Furthermore, non-ENU-exposed Ba/F3 KIT502-3AYins cells were selected with the identical doses of sunitinib and imatinib, in order to compare the resistance frequencies and mutation spectrum. To exclude the possibility that resistant colonies develop intrinsically due to genetic instability, Ba/F3 KIT502-3AYins cells were also screened without inhibitors. Cells cultured with KIT ligand, KITL, supplemented in the absence of TKI grew back in a week, without finding secondary mutations in randomly tested cells.
In concordance with the sunitinib-resistant genotypes observed in patients, the cell-based mutagenesis also showed that acquired secondary mutations in Ba/F3 KIT502-3AYins cells were only found in the KIT activation loop. Sequencing of 100 sunitinib-resistant clones resulting from the ENU treated Ba/F3 KIT502-3AYins cells and 117 clones from the untreated cells showed that all secondary mutations are single amino acid substitutions in KIT exon 17, including D816V, D816F, D816A, D816H, D816Y and D820G (Fig 3). Among these mutations, D816V was the most prevalent substitution, accounting for 6.7% of ENU-treated resistant colonies. ENU-mutagenesis did not generate distinct secondary mutations compared to the non-ENU group. However, the spectrum of sunitinib-resistant mutations was more limited compared to the imatinib selected screen. Sequencing of 78 clones obtained after ENU treatment and 158 clones from non-ENU Ba/F3 KIT502-3AYins cells, imatinib resistant mutations were observed in both KIT kinase domains, including T670I, D816V and D816F.
Fig. 3.

Distribution of point mutations in a cell-based screen for sunitinib resistance of BaF3KITAY502-3ins cells compared with the genotype in sunitinib- and imatinib-resistant patients. (A) Secondary mutations identified under the sunitinib screen were restricted to the KIT activation loop, being single amino acid substitutions at positions of D816 and D820. (B) Secondary mutations identified in sunitinib-resistant (red font) were limited to the activation loop in the C-terminal kinase domain of KIT protein, and imatinib-resistant (black) patients with a primary KIT exon 9 mutation were located in both ATP-binding and catalytic domain, with a similar amino acid substitution pattern (3, 9, 24).
The incidence of each type of mutation identified in sunitinib-resistant clones emerging from Ba/F3 KIT502-3AYins cells varied from 1-14%, according to different doses (Fig 4A); with overall mutation rates of 3.3%, 9.5% and 5.6% at 2, 5, and 10 times IC50. The rate of imatinib-resistant clones under similar conditions was higher, of 19.4%, 8% and 67%, respectively. The resistant mutations were limited to D816V at higher doses (10 × IC50) for both inhibitors tested. At more than 20 times IC50, both drugs sufficiently suppressed the emergence of resistant colonies.
Fig. 4.
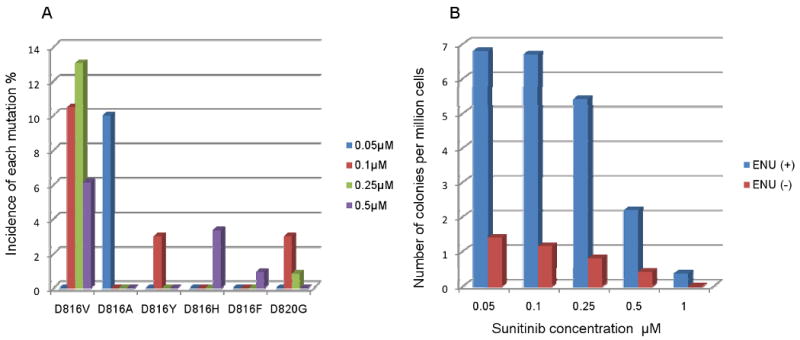
The spectrum of acquired secondary mutations and the frequency of resistant colonies in cell-based screen for sunitinib-resistance. A. The spectrum of secondary mutations was dominated by D816V, followed by D816A. Other substitutions seen at a similar lower rate included D816F, D816H, D816Y and D820G. B. The frequency of resistant colonies was dose dependent and was higher in ENU exposed Ba/F3KITAY502-3ins cells. Escalating doses reduced the incidence of resistant colonies. Exposure to ENU-mutagen significantly increased the frequency of resistant colonies.
In contrast, the mutation rates of imatinib-resistant clones selected by similar dosages in Ba/F3 KIT557-8WKdel cells were about 90%, regardless of ENU treatment, displaying a similar mutation spectrum, including D816V, D816Y, N822K, N655K, T670I (data not shown). The most prevalent mutation remained at D816, D816V with ENU-treatment (79%) and D816Y in the non-ENU group (86.5%).
The Frequency of Resistant Clones Increased with ENU-exposure and Lower Concentration of Inhibitors
To determine whether the induced drug resistance was dose-dependent, ENU-treated Ba/F3 KIT502-3AYins cells were exposed to escalating concentrations of sunitinib, of 0.05, 0.1, 0.25, 0.5 and 1 μM, corresponding to 1, 2, 5, 10 and 20 times IC50, respectively. Non-ENU treated Ba/F3 KIT502-3AYins cells were treated with similar doses of sunitinib and imatinib. The frequencies of resistant colonies were determined by visual inspection of the number of colonies relative to the total number of cells. The frequency of resistant colonies decreased with increasing sunitinib dose, with a rate of 6.8 × 10-6, 6.7 × 10-6, 5.4 × 10-6, 2.2 × 10-6 and 0.35 × 10-6 at 1, 2, 5, 10 and 20 times IC50, respectively (Fig. 4B). Imatinib selected colonies of ENU-treated Ba/F3 KIT502-3AYins cells showed similar dose-dependent resistant frequencies of 10×10-6, 5.8×10-6, 2.9×10-6, 0.35×10-6 and 0.1×10-6 at similar doses.
Cells exposed to lower doses than the IC50 of sunitinib (i.e.10 and 20 nM) grew back without individual colony formation within a week, suggesting that these concentrations did not sufficiently inhibit Ba/F3 KIT502-3AYins cells. Conversely, at more than 20 times the IC50 of sunitinib, no resistant colonies evolved within 60 days. Similar observations were made with imatinib.
Untreated Ba/F3 KIT502-3AYins cells yielded a lower frequency of resistant colonies, as compared to ENU-treated cells at similar drug concentrations (p<0.01, using pair of comparison Qui square). The frequency of resistant colonies observed in non-ENU conditions was 1.4, 1.15, 0.8, 0.4 and 0 colonies per million cells, at 1, 2, 5, 10 and 20 times IC50 of sunitinib, respectively (Fig. 4B). The incidence of imatinib-resistant colonies in non-ENU treated group was 5.1, 3.1, 1.45, 0.05 and 0 colonies per million cells, at equipotent dosages. No overt difference in the median growth time of resistant colonies was seen between the sunitinib and imatinib selected BaF3KIT502-3AYins cells.
Sunitinib-Selected Resistant Clones were Insensitive to Sunitinib and Imatinib Inhibition
To validate that clones derived from the sunitinib resistant screen were indeed refractory to sunitinib inhibition, cells were cultured in the presence of various concentrations of sunitinib, and the IC50s were determined. In parallel, the imatinib IC50s was also determined in these cells. Sunitinib-selected clones, with or without secondary mutations, were tested, showing similar insensitivity to sunitinib compared to sunitinib sensitive parental Ba/F3KITAY502-3ins cells. The imatinib IC50s in these clones was >5 μM.
KITAY502-3ins Sunitinib Resistant Clones show biochemical activation of KIT but no alteration in KIT copy number
By Western blot, protein extracts from KITAY502-3ins resistant clones showed similar levels of total and phosphorylated KIT expression to the parental sunitinib-sensitive Ba/F3-KITAY502-3ins cells. To explore the possibility of KIT copy number changes in sunitinib resistant clones without detectable secondary mutations, we investigated the genomic integration of KITAY502-3-ins cDNA plasmid in both the parental Ba/F3 KITAY502-3ins and sunitinib resistant clones. Cells were subjected to FISH using Digoxigenin-labeled KITAY502-3ins cDNA, showing a single integration site on chromosome 6C2 in both groups. Therefore, no genetic difference related to the KIT locus was noted between the parental Ba/F3 KITAY502-3-ins and the sunitinib resistant clones.
Discussion
The primary genetic event responsible for the pathogenesis of GIST is a gain-of-function mutation in the KIT proto-oncogene or, less commonly, in the platelet derived growth factor alpha (PDGFRA) gene (10, 11). More than two-thirds of all GIST patients carry mutations in KIT exon 11, which encodes the juxtamembrane domain, while approximately 10-15% of patients show mutations within the extracellular domain of KIT, in exon 9 (6, 12). The vast majority of KIT exon 9 mutations represent an identical tandem duplication of six nucleotides, encoding AY 502–503. GISTs harboring KIT exon 9 mutations define a distinct subset, characterized predominantly by small bowel location and an aggressive clinical behavior (6, 13).
Therapeutic inhibition of KIT and PDGFRA kinase activity by imatinib mesylate (Gleevec, Novartis, Basel, Switzerland) has emerged as front-line treatment for patients with metastatic or locally advanced inoperable GIST. Imatinib achieves disease control in 70-85% of patients with advanced GIST. The clinical response varies significantly according to different molecular subtypes of GIST. As such, patients with GIST carrying KIT exon 11 mutations show the best response to imatinib with a partial response rate of 84%, compared to only 48% in patients with KIT exon 9 mutations (2). KIT exon 9 mutations were the strongest adverse prognostic indicator for response to imatinib, increasing the relative risk of progression by 171% and the relative risk of death by 190% (14). Furthermore, patients whose tumors harbored KIT exon 9 mutations had a significantly superior progression-free survival when treated with the high-dose regimen, compared to patients with exon 11 mutations (14). However, in spite of the prolonged responses with escalating imatinib dose, most patients subsequently experienced disease progression.
The only FDA approved second line TKI for patients with advanced GIST, who have progressed on or are intolerant of imatinib, is sunitinib malate. Sunitinib is a small molecule TK inhibitor with potent anti-angiogenic and anti-tumor activities which has demonstrated efficacy against GIST, acceptable tolerability and safety in a double-blind placebo-controlled phase III trial (15). Patients with KIT exon 9 mutations GISTs showed the most sustained response to sunitinib inhibition. As sunitinib resistance is now emerging in the clinical practice, new therapeutic strategies are needed to treat these patients.
As learned from the imatinib-resistance experience, second site KIT mutations develop more frequently in association with primary KIT exon 11 mutations, which confer the most sustained clinical response to imatinib. This finding might reflect the long time span of exposure to the drug and not only due to the location of the primary mutations. Thus secondary mutations were found in 73-86% of imatinib-resistant patients harboring exon 11 primary mutations and only 19-33% of patients with exon 9 primary mutation developed secondary mutations (3, 9, 16). Furthermore, the pattern of the second site mutations in the setting of imatinib resistance was exclusively point mutations, evenly divided between the first and second kinase domains (3, 17, 18). In keeping with these observations, the three patients who developed sunitinib resistance after at least one year of clinical benefit had second site mutations in the KIT kinase activation domain (N822K, D820Y and D820E). Expression of these sunitinib-resistant double mutants in an in vitro Ba/F3 model showed sensitivity to both dasatinib and nilotinib, suggesting alternative therapeutic options for these patients.
To establish a comprehensive profile of acquired secondary mutations which confer drug-resistance to KIT exon 9 mutants, Ba/F3 cells expressing KIT502-3AYins were subjected to ENU mutagenesis followed by sunitinib selection and emerging resistant clones were identified and characterized. The KIT mutations identified by in vitro mutagenesis recapitulated the type and location of second site KIT mutations observed in both sunitinib-and imatinib-resistant patients. Thus all sunitinib-resistant secondary mutations identified in the Ba/F3-KIT502-3AYins cells were similarly point mutations in the KIT kinase activation loop. However, the mutations identified in the in vitro screen were dominated by D816V. The high incidence of this mutation is most likely conferred by the significant advantage in its activation rate. Substitutions at this site, D816H/V, have shown increases in activation rate of 184- and 536-fold, respectively (19). Furthermore, D816 mutations appear to negatively influence the inhibitory conformation of the JM domain of KIT (19). Several substitutions at this site have been described in TKI-resistant GIST patients, including D816H/F/A/G/E/V. However, the rarity of D816V mutation in progressing GIST patients might be an indication of its high malignancy, which may induce tumor lethality in vivo. Alternatively, the possibility of this dominant D816V genotype being intrinsic to the Ba/F3 cells system remains a consideration, since Ba/F3 cells are murine pro-B cells and D816V is the common mutation in mastocytosis.
Importantly, KIT exon 13 and 14 mutations were not detected in either the sunitinib-resistance screen or in progressing tumors of patients of sunitinib resistant patients, as sunitinib is known to be efficacious with ATP-pocket second site mutations (4, 9). In contrast, in vitro mutagenesis of Ba/F3 KIT502-3AYins cells and selection with imatinib identified secondary mutations mapped in both KIT kinase domains, i.e. mostly D816V and T670I. These findings are in agreement with results of in vitro mutagenesis of Ba/F3 Bcr-Abl cells, in which the vast majority of imatinib-resistant mutations were substitutions within the kinase domain of the fusion protein (20, 21). However, the spectrum of resistance mutations obtained with Ba/F3 KIT502-3AYins cells was narrower compared to the results obtained with Bcr-Abl. The observed difference most likely is not related to the methodology used, which was very similar in both studies, but rather intrinsic to the specific oncogenes tested. The relatively limited variety of mutations observed in the Ba/F3 KIT502-3AYins screen cannot be explained only by the lower rate of mutation detected in the resistant clones, since a significantly higher rate of mutations (i.e. 90%) noted in the Ba/F3-KIT557-8WKdel cells, was not accompanied by an enhanced mutation spectrum. In order to overcome the low yield of resistant colonies, ENU mutagenesis was applied, which indeed increased the frequency of both sunitinib- and imatinib-resistant colonies, indicating that ENU-mutagenesis is beneficial in establishing an unbiased and robust cell-based resistance screen system of KIT gene.
The results of ENU mutagenesis followed by sunitinib selection were in concordance with the findings observed with sunitinib-resistant patients, defining D816 and D820 amino acids as the most vulnerable sites for acquired mutations within the KIT kinase activation loop. Similarly, imatinib-selection identified T670, D816 and D820 as resistant mutation hot spots. The frequency of secondary mutations with sunitinib or imatinib selection with Ba/F3 KIT502-3AYins cells was quite low (Fig 4). In contrast, clones carrying a juxtamembrane KIT557-8WKdel mutation had a significantly higher mutation rate. In keeping with these differences, previous biochemical studies revealed that tumors with KIT exon 9 mutation had less AKT activation than tumors with mutations of exon 11, suggesting that these KIT mutants may recruit different downstream substrates (22). The latter hypothesis may also explain the genotype-dependent response of GIST patients to different tyrosine kinase inhibitors (TKI).
Recently the crystal structure of the KIT ectodomain was characterized, showing that KIT dimerization is driven by bivalent binding of KIT ligand and stabilized by lateral D4-D4 and D5-D5 interactions of two neighboring KIT ectodomains (23). Activating oncogenic mutations reported so far within the ectodomain of KIT map to the D5-D5 interface, which presumably stabilize receptor dimers and thus activate the kinase in the absence of ligand. In contrast, juxtamembrane domain mutations are thought to induce kinase activation by disrupting the auto-inhibitory conformation of the juxtamembrane domain. The different mechanisms of activation of constitutive KIT kinase activity seen with different mutant oncoproteins may impact on the mutation rates yielded from the mutagenesis screen. KIT exon 11 mutants largely rely on the structural changes conferred by secondary mutations to overcome inhibition of KIT kinase activity and with extra-cellular domain mutants similarly acquisition of secondary mutations which stabilize the active conformation of the KIT kinase are required. It is therefore not surprising that the spectrum of secondary mutations between the two mutant isoforms did not differ. Although both D816 and D820 do not interact directly with the inhibitors, the mutations at these sites affect the A-loop conformation as well as the overall conformation of the kinase (Fig. 5) preventing drug binding (19).
Fig. 5.
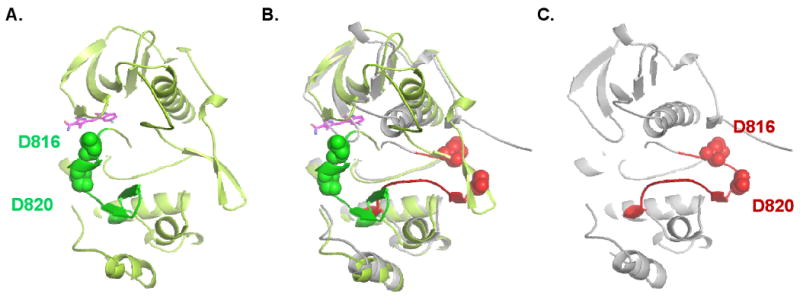
Comparison of the structures of the inactive KIT kinase in complex with sunitinib and the activated KIT kinase. A) Inactive kinase shown in light green, the activation loop in green and sunitinib in magenta; B) superposition of the inactive (A) and the active kinase structures (C); C) structure of the activated KIT kinase is shown in grey and the activation loop in red. The position of second site mutation in the activation loop, D816 and D820, are indicated. (adapted from (19)).
Unfortunately, comprehensive molecular studies are constrained by the low number of patients who are surgical candidates after failing two TKIs and thus the tissue available for investigating mechanisms of resistance is limited. Taken together, the data obtained from clinical samples as well as from the in vitro mutagenesis screens suggest that sunitinib-resistance shares similar pathogenetic mechanisms seen in imatinib-failure, with acquisition of secondary mutations in the activation loop conferring resistance to both drugs. In contrast, the imatinib-resistance screens demonstrated a slightly higher mutation rate and a wider mutation spectrum, spanning both kinase domains. In addition, the in vitro resistance screens applied to either ectodomain or juxtamembrane domain KIT mutants suggest that the location of the primary mutation may trigger different mechanisms of drug resistance, with a higher rate of resistant mutations seen with primary KIT exon 11 mutants.
Acknowledgments
The authors would like to thank Kai Xu, from the Structural Biology Program, Sloan Kettering Institute; Diann DeSantis for clinical follow-up and Milagros Soto for editorial support.
Supported in part by: ACS MRSG CCE-106841 (CRA), P01CA47179 (CRA, RGM, SS), Life Raft Group (CRA, PB), GIST Cancer Research Fund (CRA, RPD), Shuman Family Fund for GIST Research (CRA, RGM), CA102613 (RPD), CA102774 (PB), HL/DK55748 (PB).
Footnotes
Translational Relevance: Therapy with tyrosine kinase inhibitors (TKI) benefits approximately 80% of patients with advanced GIST, but most patients eventually develop drug resistance. Sunitinib malate is a broad spectrum TKI approved as 2nd line therapy for imatinib-resistant GIST patients. The clinical benefit of sunitinib is genotype-dependent, with GIST patients harboring a KIT exon 9 mutation being the most sensitive. As sunitinib resistance is now emerging, our goal was to investigate mechanisms of progression and test the efficacy of novel TKI on these resistant mutants in vitro. To establish a comprehensive profile of acquired mutations which confer drug-resistance to KIT exon 9 mutants, Ba/F3 cells expressing KIT502-3AYins were subjected to ENU mutagenesis and sunitinib selection. The KIT mutations identified by in vitro screen recapitulated the pattern of second site mutations observed in TKI-resistant patients. Our findings highlight new mechanisms of resistance to second generation TKI and provide a pre-clinical rationale for alternative therapeutic options for patients failing sunitinib therapy.
References
- 1.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 3.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 4.Prenen H, Cools J, Mentens N, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12:2622–7. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- 5.Russell WL, Kelly EM, Hunsicker PR, Bangham JW, Maddux SC, Phipps EL. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc Natl Acad Sci U S A. 1979;76:5818–9. doi: 10.1073/pnas.76.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–37. [PubMed] [Google Scholar]
- 7.Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204–15. doi: 10.1158/1078-0432.CCR-07-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo T, Agaram NP, Wong GC, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13:4874–81. doi: 10.1158/1078-0432.CCR-07-0484. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–9. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 11.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 12.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–21. [PubMed] [Google Scholar]
- 13.Lasota J, Kopczynski J, Sarlomo-Rikala M, et al. KIT 1530ins6 mutation defines a subset of predominantly malignant gastrointestinal stromal tumors of intestinal origin. Hum Pathol. 2003;34:1306–12. doi: 10.1016/s0046-8177(03)00407-6. [DOI] [PubMed] [Google Scholar]
- 14.Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 15.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 16.Nishida T, Kanda T, Nishitani A, et al. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 2008;99:799–804. doi: 10.1111/j.1349-7006.2008.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–9. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743–9. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 19.Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106:1542–7. doi: 10.1073/pnas.0812413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Bubnoff N, Veach DR, van der Kuip H, et al. A cell-based screen for resistance of Bcr-Abl-positive leukemia identifies the mutation pattern for PD166326, an alternative Abl kinase inhibitor. Blood. 2005;105:1652–9. doi: 10.1182/blood-2004-06-2445. [DOI] [PubMed] [Google Scholar]
- 21.Bradeen HA, Eide CA, O'Hare T, et al. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108:2332–8. doi: 10.1182/blood-2006-02-004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs) Oncogene. 2004;23:3999–4006. doi: 10.1038/sj.onc.1207525. [DOI] [PubMed] [Google Scholar]
- 23.Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130:323–34. doi: 10.1016/j.cell.2007.05.055. [DOI] [PubMed] [Google Scholar]
- 24.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]