

Abstract
There is a great need for the development of novel chemotherapeutic agents that overcome the emergence of multidrug resistance in cancer. We catalogued the National Cancer Institute’s Developmental Therapeutics Program (DTP) drug repository in search of compounds showing increased toxicity in multidrug resistant (MDR) cells. By comparing the sensitivity of parental cell lines with multidrug resistant derivatives, we identified 22 compounds possessing MDR-selective activity. Analysis of structural congeners led to the identification of 15 additional drugs showing increased toxicity in Pgp-expressing cells. Analysis of MDR-selective compounds led to the formulation of structure activity relationships (SAR) and pharmacophore models. This data mining coupled with experimental data points to a possible mechanism of action linked to metal chelation. Taken together, the discovery of the MDR-selective compound set demonstrates the robustness of the developing field of MDR-targeting therapy as a new strategy for resolving Pgp-mediated multidrug resistance.
Keywords: chemotherapy, multidrug resistance, P-glycoprotein, NCI-60, NSC73306
Introduction
Members of the ABC (ATP Binding Cassette) transporter superfamily are widely recognized as major contributors to controlling drug distribution and pharmacokinetics, and the acquisition of anticancer drug resistance. Expressed largely in the plasma membranes of cancer cells, ABC transporters mediate cellular resistance to anticancer agents by the ATP-dependent efflux of toxic chemotherapeutics from cells. While several ABC transporters have been shown to transport anticancer drugs in vitro, P-glycoprotein (Pgp/MDR1/ABCB1) stands out by conferring the highest level of resistance to a vast array of drugs and because of its demonstrated association with clinical multidrug resistance (MDR) and poor clinical outcome (1). Pgp transports large, hydrophobic, positively charged molecules that can have strikingly dissimilar structures, including clinically relevant compounds such as anticancer drugs, human immunodeficiency virus (HIV)-protease inhibitors, immunosuppressive agents and antiepileptics (1–3). Despite promising in vitro results obtained with several generations of Pgp inhibitors, successful modulation of clinical MDR through the chemical blockade of drug efflux from cancer cells has not been successful (1), and new strategies are required,
Intriguingly, Pgp can contribute not only to acquired resistance but also to paradoxical drug sensitivity (4–14). “Collateral sensitivity” of otherwise MDR cells represents a promising but yet unrealized strategy for targeting Pgp mediated MDR. Given the importance of drug-transporter interactions and the desire to uncover compound classes with new modalities, we recently analyzed the Developmental Therapeutics Program (DTP) drug activity dataset, and reported the prediction and validation of small molecules recognized by various ABC transporters (7). The drug database compiled by the DTP of the National Cancer Institute (NCI) contains the cytotoxicity profiles of more than 100,000 compounds across 60 human cancer cell lines (NCI-60 panel). This dataset, combined with other descriptors of the cell panel provides clues to mechanisms of chemosensitivity and resistance (7, 15, 16). Statistical correlations between the sensitivity of these cell lines to a panel of anti-cancer drugs and the expression of Pgp across the NCI-60 cell panel identified new Pgp substrates, but also suggested that cells expressing higher levels of Pgp are more sensitive to the thiosemicarbazone NSC73306. This agent was selected for in vitro evaluation (17), and we have subsequently shown that NSC73306 is truly selective for functional Pgp; cells are hypersensitive to NSC73306 in proportion to their Pgp function, and this selectivity is abrogated by functional inhibition of Pgp or down-regulation of Pgp expression by siRNA (18). Furthermore, NSC73306 itself is not an inhibitor or substrate of Pgp, suggesting that this compound does not confer its toxicity in the same fashion as the so called ‘collateral sensitivity’ agents.
We are currently undertaking pre-clinical assessment of NSC73306. In parallel, we sought to identify new ‘MDR-selective’ compounds to expand the scope of bioactive agents and to gain insight into the Pgp-specific mechanism of action. To this end, here we report the screening of 42,000 compounds from the DTP dataset in silico for ‘MDR-selective’ activity, predicted by the correlation of cytotoxicity with Pgp expression across the NCI-60 cell line panel. This prediction set was then validated in vitro, focusing on clusters of structurally related compounds. Twenty-two compounds possessing MDR-selective activity were identified and analysis of structural congeners in the DTP repository led to the identification of 15 additional compounds showing increased toxicity in Pgp-expressing cells. The discovery of these MDR-selective compounds demonstrates the robustness of the developing field of MDR-targeting therapy as a new strategy for resolving Pgp-mediated multidrug resistance.
Materials and Methods
Chemicals
Unless otherwise stated, compounds were obtained from the NCI DTP drug repository. NSC716765, NSC716766, and NSC716772 were synthesized by us (17); the octapeptide NSC633657 (D-Phe-Cys-Tyr-D-Trp-Lys-Ser-Cys-Thr-NH2) was prepared by John Stonik, NCI. NSC617961 and NSC617963 were sourced from the Auckland Cancer Society Research Centre, New Zealand (19). NSC697124 and NSC697125 were sourced from Instituto de Quimica, UNAM, Mexico. 1,10-phenanthroline, 2,2’-bipyridine and tris(1,10-phenanthroline)ruthenium(III) chloride was purchased from Sigma-Aldrich (St Louis, MO).
Drug database
The DTP Human Tumor Cell Line Screen has screened tens of thousands of compounds for growth inhibition of human cancer cell lines. Publicly available screening results of ~43,000 compounds were downloaded from the DTP website 1 (July 2007 Release). Scrutiny of the dataset indicated that it was not immediately amenable to stringent statistical analysis due to missing dose-response values or lack of activity in the dose window tested (12.1% of the cell line screen data points are missing and 44.9% of growth inhibition values indicate inactivity) (20). Since uninformative drug profiles are not expected to yield useful leads, we analyzed only those drugs that were measured inside the range of cytotoxicity with no more than 50% missing values, resulting in a higher quality dataset. Putative MDR-selective compounds were identified based on correlation of their cytotoxicity patterns to ABCB1 expression, as described in (7).
Cell lines and culture conditions
KB-3-1 is the parental human (HeLa) epidermoid carcinoma cell line of KB-V1 cells that overexpress Pgp (MDR1) as a result of selection in vinblastine (21). NIH-MDR-G185 is an isogenic sub-clone of the parental line NIH3T3 transfected with wild type pHaMDR1/A (22). MES-SA Dx5, a drug resistant cell line expressing high levels of Pgp, was derived from a human uterine sarcoma line (MES-SA) by doxorubicin selection (23). KB-3-1, KB-V1, NIH3T3 and NIH-MDR-G185 cells were grown in DMEM cell culture medium at 37 °C in 5% CO2. MES-SA and MES-SA Dx5 cells were grown in McCoy’s medium. NIH-MDR-G185, KB-V-1 and Dx5 cells were maintained in 60 ng/mL colchicine, 1µg/mL vinblastine and 500 nM doxorubicin (adriamycin) respectively to maintain Pgp expression. All cell culture media (GIBCO) was supplemented with 10% fetal bovine serum (FBS, GIBCO), 5 mM glutamine (GIBCO) and 50 unit/ml penicillin and streptomycin (GIBCO). Retroviral expression of ABCB1 in A431 cells was achieved by a method described previously (24). The human skin–derived, epidermoid carcinoma cells, A431, were maintained in alpha-MEM (Life Technologies, Grand Island, NY) supplemented as above.
Cell viability assay
Viability was assessed as described previously (7). Cytotoxicity assays were performed in triplicate, and curves were fitted by Prism software (GraphPad Software, Inc., San Diego, CA) using nonlinear least-squares regression in a normalized sigmoidal dose-response model with variable slope. Curve fit statistics were used to determine the concentration of test compound that resulted in 50% toxicity (IC50). Differences between the GI50 values were analyzed by two-sided paired Student’s t test and results were considered statistically significant at p < 0.05.
Results
Correlative information from the NCI-60 screen and qRT-PCR profiling identifies putative MDR-selective compounds that are more active in cells that overexpress the drug-resistance transporter Pgp
In an effort to identify candidate MDR-selective compounds, we used quantitative real-time PCR (qRT-PCR) expression data (7) to correlate Pgp mRNA levels and the publicly available DTP drug screening toxicity profiles across the NCI-60 panel. Our analysis included DTP’s complete public dataset, consisting of 42,657 candidate anticancer agents that had been submitted for screening. Dose-response data of the compounds screened against the NCI60 cell panel were downloaded from DTP’s website (July 2007 release) and curated to remove uninformative dose-response profiles.
97.4 % of drug-Pgp correlations fall within the range −0.4 < r < 0.4, considered to accommodate compounds with poor or no correlation with Pgp expression, indicating that for most drugs Pgp is not a key determinant of toxicity. While the analysis identified several putative substrates at r < −0.4, we focused on the positively correlated Pgp-drug correlations (r ≥ 0.4). The result of data conditioning confined the 42K set to 64 compounds (“Discovery-set”) showing a strong positive correlation (r ≥ 0.4) between Pgp expression and efficacy patterns across the NCI-60 cell line panel (Table I and Supplementary Table SI). Based on our earlier validation of the bioinformatic correlation employed here, we hypothesized that these compounds would likely possess MDR-selective activity. Indeed, NSC73306, reported previously by us as an MDR-selective agent, was again identified in this screen (Table I) (7).
Table I.
Putative MDR-selective compounds.
| Compound (NSC) |
Pearson | Selectivity ratio |
Compound (NSC) |
Pearson | Selectivity ratio |
Compound (NSC) |
Pearson | Selectivity ratio |
|---|---|---|---|---|---|---|---|---|
| 10580 | 0,41 | 1,71±0,26* | 639743 | 0,42 | NA | 697120 | 0,44 | NA |
| 43320 | 0,56 | 2,08±0,37* | 641208 | 0,43 | 2,13±0, 09*** | 697124 | 0,46 | 2,07±0,49* |
| 73306 | 0,51 | 3,64±0,93* | 647574 | 0,42 | NT | 697125 | 0,54 | 2,33±0,79* |
| 86715 | 0,53 | <=1 | 649816 | 0,47 | 1,17±0,12* | 697128 | 0,43 | NA |
| 3,82±0,85** | ||||||||
| 168468 | 0,48 | * | 651782 | 0,43 | NA | 697129 | 0,47 | NA |
| 292408 | 0,45 | 2,80±0,76* | 651859 | 0,43 | NA | 697135 | 0,43 | NA |
| 356777 | 0,4 | 1,64±0,42** | 653864 | 0,41 | NA | 697137 | 0,43 | NA |
| 403148 | 0,4 | NT | 654724 | 0,41 | NA | 697678 | 0,43 | NA |
| 617961 | 0,42 | <=1 | 669341 | 0,41 | <=1 | 697920 | 0,41 | NA |
| 617963 | 0,53 | <=1 | 672027 | 0,55 | <=1 | 705301 | 0,43 | NA |
| 621481 | 0,45 | <=1 | 672036 | 0,42 | 1,27±0,16* | 705305 | 0,43 | NA |
| 624967 | 0,41 | NA | 673999 | 0,59 | 1,99±0,32* | 709976 | 0,47 | NA |
| 626670 | 0,42 | NA | 681125 | 0,44 | NA | 710857 | 0,44 | 1,47±0,25* |
| 632591 | 0,4 | NA | 688942 | 0,4 | NA | 713048 | 0,41 | 1,71±0,34* |
| 632731 | 0,53 | NA | 692419 | 0,42 | <=1 | 716765 | 0,5 | 1,80±0,20** |
| 632733 | 0,46 | NA | 693046 | 0,43 | <=1 | 716766 | 0,48 | 1,64±0,20** |
| 632736 | 0,47 | NA | 693336 | 0,45 | NA | 716768 | 0,52 | 1,92±0,07*** |
| 632738 | 0,43 | NA | 693630 | 0,4 | NA | 716772 | 0,52 | 2,67±0,61** |
| 632955 | 0,47 | <=1 | 693871 | 0,68 | 8,48±3,15** | 720611 | 0,55 | NA |
| 633657 | 0,45 | NT | 693872 | 0,45 | 3,90±1,37** | 720612 | 0,64 | NA |
| 635977 | 0,42 | NA | 695331 | 0,44 | 2,60±0,86* | |||
| 636097 | 0,42 | <=1 | 695333 | 0,58 | 3,11±1,99* |
Compounds identified as having MDR-selective activity, listed with their Pearson’s correlation coefficients - the correlation between Pgp expression and DTP determined efficacy of each compound across the NCI-60 cell line panel. To validate predicted MDR-selective activity in vitro, compounds were sourced by a variety of methods, predominantly through the NCI DTP drug library, but also via other investigators and synthesis in our laboratories. For compounds that were available, IC50 values were determined using the MTT cytotoxicity assay on the parental KB-3-1 cell line, and its P-glycoprotein expressing derivative, KB-V1. Those unavailable for further testing are noted as such (NA). MDR1 selectivity is calculated as the ratio of a compound’s IC50 against KB-3-1 cells divided by its IC50 against KB-V1 cells. A value > 1 indicates that the compound kills Pgp-expressing cells more effectively than parental cells - so-called MDR-selective activity (bolded), as assessed by two-sided paired Student’s t test of multiple MTT assays
Significance levels: P < 0.05
P < 0.01
P < 0.001.
Compounds with a ratio≤1 inhibit growth in both cell lines with similar potency (0.5<r≤1), indicating that they are either Pgp substrates or their toxicity is not modulated by Pgp. Compounds with an IC50 > 50 µM were considered to be inactive (not toxic, NT), and as such their MDR1 selectivity could not be determined.
To characterize the structural coherence of the Discovery-set, the 64 compounds were clustered based on commonality in their structural features (chemical descriptors), revealing distinct Tanimoto clustering (Figure 1). Strikingly, 9 of the 64 compounds contain a thiosemicarbazone functional group and 5 of these are isatin-β-thiosemicarbazones: NSC73306, NSC716765, NSC716766, NSC716768, NSC716772, NSC669341, NSC693336, NSC695331, and NSC695333 (isatin-β-thiosemicarbazones in italics). This suggests that the isatin-β-thiosemicarbazone core of NSC73306, the only MDR-selective agent reported in the DTP drug database so far, is strongly associated with MDR-selective activity. Notably, the biological activity of thiosemicarbazones often involves endogenous metal ion coordination (25) (26), and the metal chelates of thiosemicarbazones are regularly reported to be more active than the compound alone (27). The 64 compounds also include eight metal complexes (Figure 1). Seven of these contain 1,10-phenanthroline (phen) or 2,2’-bipyridine (bipy) bidentate ligands, and six contain lanthanoid metal ions. Phen and bipy are well-characterized chelating agents known to demonstrate anti-proliferative activity (28). Two highly similar 8-hydroxyquinoline analogs, NSC693871 and NSC693872 are also potent chelators with known anti-proliferative activity (29) (30, 31).
Figure 1.

Dendrogram showing the average-linkage hierarchical clustering of 64 putative MDR-selective compounds. The distance matrix is derived from Tanimoto similarity indices3. Numbers represent NSC codes. Black: compound not available for testing; in bold: confirmed MDR-selective activity; italicized: lack of MDR-selective activity; asterix (*): metal complex. Clusters of structurally related compounds where one or more analogs are active are highlighted by their core common structure (structures for all compounds are shown in Supplementary Table SI).
A distinct cluster is formed by seven natural product-derived sesquiterpenic benzoquinones based on the natural product perezone (NSC697125, 2-(1,5-dimethyl-4-hexenyl)-3-hydroxymethyl-p-benzoquinone), (32). Perezone and six closely related analogs NSC697125 (aminoperezone), NSC697124 (isoaminoperezone), NSC697129 (anilineperezone), NSC697120, NSC697128, NSC697137, NSC697135 are known to display a range of pharmacological properties including redox activity and mitochondrial decoupling (33). The benzoquinone moiety of perezones responsible for redox activity can also be found in other NSC compounds such as the catechol of NSC10580 (34).
Beyond these three major classes (encompassing 24 of 64 compounds), a variety of structural motifs and known drug derivatives exist, presenting a number of promising opportunities for lead drug validation. For example, there are three podophyllotoxin (or etoposide) analogs (NSC403148, NSC651859 and NSC653864); and an olivacine analog (NSC86715) that was previously identified in a screen for MDR1-related agents (35); and a recently patented Bcl-2 inhibitor (NSC168468).
Verification of MDR-selective activity in cytotoxicity assays
The 64 compounds listed in Table I correlated with elevated toxicity in proportion to Pgp expression across the NCI60 cell lines. To validate the predicted Pgp-potentiated activity, 35 compounds that were made available by DTP were tested using a cell line pair not included in the NCI-60 panel. MTT cytotoxicity assays were performed against KB-3-1, a human adenocarcinoma cell line, and KB-V1, a multidrug resistant derivative of KB-3-1 that over-expresses MDR1/Pgp (21) (Table I). By definition, MDR-selective compounds show selective toxicity towards cells expressing Pgp, defined here as the ratio of a compound’s IC50 against KB-3-1 and KB-V1 cells. A compound that is more toxic to KB-V1 cells yields a ratio greater than 1 and indicates MDR-selective activity. Of the 35 compounds tested, 3 compounds had an IC50 > 50 µM and thus were considered non toxic (NT). Ten of the remaining 32 compounds that were tested inhibited growth in both cell lines with similar potency, suggesting that Pgp does not modulate the toxicity of these compounds.
Conversely, 22 compounds showed preferential growth inhibition in the Pgp overexpressing KB-V1 cell line. To assess whether the potentiation of cytotoxicity against Pgp-expressing cells is robust and not cell-line specific, four compounds that were available in sufficient quantities were tested on additional multidrug resistant cell lines. Irrespective of the selecting drug used to maintain Pgp expression, the tissue of origin, and in otherwise isogenic, Pgp-transfected cell lines, all four compounds showed elevated toxicity in Pgp-expressing cells relative to their parental line, demonstrating that the MDR-selective activity is not restricted to the KB-3/KB-V1 cell pair (Table II). Furthermore, PSC833, a high affinity Pgp-inhibitor, was used to establish the requirement for functional Pgp to mediate sensitization in KB-V1 cells (Table II). For each drug, inhibition of Pgp rendered the MDR cells less sensitive to the compounds, confirming that functional Pgp is required for the increased toxicity of the identified MDR-selective agents. This activity profile is shared by NSC73306 (18), and shows that the MDR1-selective activity of NSC73306 is not unique, but represents a robust modality for targeting multidrug resistance.
Table II.
MDR-selective agents show elevated toxicity in relation to Pgp expression and function in Pgp expressing cell lines.
| NSC | KB-3-1/ KB-V1 |
KB-V1 (PSC) |
NIH-3T3/ G185 |
MES-SA/ MES- SA/Dx5 |
A431/ A431 B1 |
|---|---|---|---|---|---|
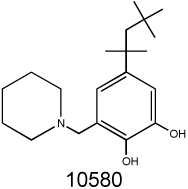 |
1.71 | 2.20 | 4.27 | 4.78 | 4.32 |
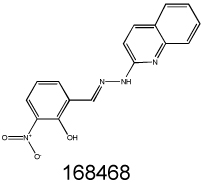 |
3.82 | 2.87 | 3,10 | 3.82 | 1.43 |
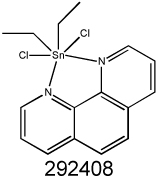 |
2.80 | 3.20 | 1.77 | 3.50 | 4.13 |
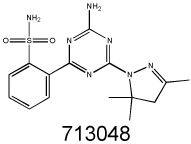 |
1.71 | 4.90 | 3.98 | 5.06 | 2.18 |
MDR1 selectivity of NSC10580, NSC168468, NSC292408 and NSC713048 comparing parental (KB-3-1, NIH-3T3, MES-SA and A431) and their respective Pgp-expressing derivatives (KB-V1, G185, MES-SA Dx5, and A431 B1) were determined by MTT cytotoxicity assays. For KB-V1, cytotoxicity was also assayed in the presence and absence of the Pgp inhibitor PSC833 (2 µM), to determine whether functional Pgp is required for MDR-selective activity.
Effect of structural diversification on MDR-selective activity
We searched the DTP database for structural analogs of the confirmed MDR-selective compounds NSC10580, NSC168468, NSC292408, NSC713048 (Table II) and NSC73306 (18). It was hypothesized that some of the close structural analogs of the 5 MDR-selective compounds used as seeds would demonstrate increased toxicity in KB-V1 cells. For each lead, structurally related compounds were identified based on respective Tanimoto coefficients, with a threshold distance of 0.6 (Supplementary Table SII). The similarity search identified 77 analogs, of which 58 were available for testing. Of these, MTT assays identified 15 additional compounds that were more toxic to Pgp-expressing KB-V1 cells (Supplementary Table SII).
In total, the approach of analyzing the correlation between gene expression and drug activity combined with structural similarity analysis yielded 37 MDR-selective agents. This set contains several structurally coherent subgroups, which were further analyzed for structure activity relationships. To identify common features that may be responsible for MDR-selective activity, we generated pharmacophore models for two prominent clusters, containing the thiosemicarbazones and the 10580-analogs (Supplementary Figure S1), using the most active compounds as templates (NSC693871 and NSC337743 for the TSC and 10580 clusters, respectively). Both models contain at least two hydrogen bond acceptors, a hydrogen bond donor as well as an aromatic ring (Supplementary Figure S1). QSAR analysis of the thiosemicarbazones and analogs of NSC168468 identified descriptors that predict MDR1-selective activity with very high accuracy (r2=0.98 and r2=0.85, respectively. (See Supplementary Figure S2 and Supplementary Table SIII for details)). Given the low number of compounds tested, these predictions are preliminary and further refinement and testing of the model is currently underway with synthetic libraries.
The role of metal binding in MDR-selective activity
Most compounds identified here are poorly characterized – 28 of the 64 putative MDR-selective compounds do not appear in any peer-reviewed publication (SciFinder 2008 structure-search). The NCI-60 screening data represent a unique, publicly available, information-rich source that has the potential to interface screening profiles with gene expression data and structural analysis, and to assign a putative mechanism of action to compounds (36). Self-organizing maps (SOM) provide a visually compelling clustering algorithm to analyze the relation of drug activity patterns to functional categories representing distinct modes of action (37). To find out if the MDR-selective compounds are associated with a common mechanism of action, the Discovery-set was projected on a SOM representing cytotoxicity measurements of the DTP tumor cell screen2. Most of the confirmed MDR-selective compounds project to a distinct region populated by metal chelation complexes and chelators whose activity is linked to nucleic acid metabolism (37) (31). A particularly strong cluster, suggestive of a shared mechanism of action, is formed by structurally diverse compounds including NSC73306 and NSC693871 (Supplementary Figure S3).
Along with metal-ion chelators, such as thiosemicarbazones or the hydroxyquinolines (NSC693871 and NSC693872), the Discovery-set contains several metal complexes.
The relationship between chelating molecules and coordination complexes is inextricable; solution equilibria results in dissociation of a complex into ligands and metal ions, and it may be either the complex or a component such as the liberated ligand, metal ion, or counter ion that is responsible for biological activity (38). To explore the role of the ligands and metal ions in the activity of MDR-selective compounds, experiments were performed with free ligands and chelated complexes. First, we examined the actual metal binding capacity of NSC73306. The two likely coordination modes of NSC73306 are the N,S bidentate and N,S,O tridentate coordination additionally through the ind-2-one oxygen (Figure 2a). Addition of Fe2+, Cu2+ and Zn2+ resulted in immediate color changes associated with rapid complexation (Figure 2b). Electrospray ionization mass spectrometry (ESI-MS) of each mixture confirmed the molecular weight of NSC73306 (326.1 + H+) and revealed a 2:1 ligand:metal complex, with each ligand singly deprotonated (M(L-H)2 + H+) indicating avid metal binding (Figure 2c).
Figure 2.

Chelation alone is not sufficient to confer MDR-selective activity A. A number of coordination modes are possible for the TSC NSC73306 (left), the most likely being bidentate (middle) or tridentate (right). B. Solution color changes of NSC73306 indicating complexation with metal sulfate salts, L to R: NSC73306, NSC73306 + Fe2+, NSC73306 + Cu2+ and NSC73306 + Zn2+. C. ESI-MS (ES+) of NSC73306 (L, mw = 326.1 gmol−1), and mixtures with metal ions (+II oxidation state) in 1:1 MeOH:H2O. The highest mass peak observed is shown, in each case corresponding to a 2:1 ligand:metal complex D. Compounds such as NSC632738 and NSC292408 that possess 1,10-phenanthroline ligands can dissociate to yield free 1,10-phenanthroline, capable of exerting its own MDR-selective activity. In contrast, the highly [Ru(phen)3] complex that does not release phen does not display MDR-selective activity.
If a stable metal-bound species was responsible for the enhanced activity of NSC73306, its pre-formed complexes should demonstrate improved activity. To address this hypothesis, metal ions were pre-mixed with NSC73306 and cytotoxicity of the complexes was determined by MTT assays (metal ions alone were not toxic). The Cu-TSC complex, which is not stable in the reducing environment of the cell, proved significantly more toxic to both cell lines (Table III). However, intracellular reduction of Cu2+ to Cu+ results in the dissociation of the complex and the liberation of NSC73306 (not shown). Therefore, the increased toxicity of the Cu-TSC complex in both KB-3-1 and KB-V1 cells is probably is due to the chaperoning of highly toxic Cu2+ into the cell as a complex with NSC73306. Complexation with iron lowered cytotoxicity and slightly diminished selectivity, probably by chelation that is relatively stable even within the cell. Zinc(II) generally forms more stable complexes than iron(II) (in accordance with the Irving-Williams series), yet zinc coordination did not influence MDR-selective activity. While it is possible that the Zn-NSC73306 complex retains its activity, it appears that a stable complexed species of NSC73306 is not the MDR-selective active species; if it were, significant improvement in activity and selectivity would be observed when using preformed complexes.
Table III.
Cytotoxicity of MDR-selective ligands and their pre-incubated metal complexes against the KB-3-1 parental adenocarcinoma cell line, and the MDR sub-line KB-V1 that expresses high levels of Pgp.
| Compound. | KB-V1, IC50 (µM) | KB-3-1, IC50 (µM) | MDR1-selectivity ratio | |
|---|---|---|---|---|
| NSC73306 | 3.3 ± 1.3 | 14.2 ± 1.2 | 4.30 | |
| 1,10-phenanthroline | 1.16 ± 0.16 | 4.04 ± 0.19 | 3.50 | |
| desferrioxamine | 43.1 ± 8.6 | 0.75 ± 0.12 | 0.020 | |
| triapine | 5.9 ± 2.6 | 1.4 ± 2.5 | 0.20 | |
| KP772 | - | - | 2.0* | |
| [Ru(phen)3]Cl2 | > 50 | > 50 | - | |
| K.SCN | > 50 | > 50 | - | |
| [NdCl2(C2H6)(phen)]Cl2 | 1.05 ± 0.66 | 7.73 ± 2.87 | 2.80 | |
| Fe(II)-NSC73306 | 10.2 ± 0.9 | 19.6 ± 2.6 | 1.90 | |
| Cu(II)-NSC73306 | 0.56 ± 0.02 | 0.57 ± 0.1 | 1.00 | |
| Zn(II)-NSC73306 | 4.8 ± 4.7 | 20.8 ± 5.4 | 4.30 | |
| Fe2+ | > 100 | > 100 | - | |
| Cu2+ | > 100 | > 100 | - | |
| Zn2+ | > 100 | > 100 | - | |
From Heffeter et al. Biochem. Pharmacol. 2007, 73, 1873–1886, against the KB-3-1 and P-gp expressing KBC-1 cell line pair.
Given that phen complexes with a range of metal ions (Nb3+, Ce3+, Sn2+ and La3+) were uncovered, we tested the MDR-selective activity of the free ligand. Following our initial paper on the expression of ABC genes and drug-gene pairs (7), a lanthanoid tris-phenathroline complex (KP772 ([tris(1,10-phenanthroline)lanthanum(III)] trithiocyanate)) was reported to possess selective toxicity in MDR cells (39). This compound was also tested by the DTP (NSC632737) and, notably, our screen identifies it as a putative MDR-selective compound (structural analog of NSC292408, see Supplementary Table SII). Phen and NSC292408 demonstrated a similar MDR-selective activity (3.5-fold and 2.8-fold, respectively), suggesting that it is the free ligand that is active. The La3+ and the SCN− counter-ion components that complete KP772, as well as the other metal ions were not toxic in the dose windows studied. The ruthenium(II) complex, ([Ru(phen)3]Cl2]) (Figure 2d) was also tested. As the rate of ligand exchange is approximately 8 orders of magnitude slower in Ru(II) complexes than La(III) complexes (40), insignificant levels of phen would be exchanged from the Ru(II) complex in a 72 h cytotoxicity experiment. The inert ruthenium complex was completely inactive. Of the NSC292408 (a tin-phenanthroline complex) analogs available for testing, only one, the platinum-phenanthroline NSC615541, showed MDR-selective activity (Table SII). The inactive NSC292408 analogs either lack the MDR-selective phen ligands or are such stable complexes that they may not be released to exert their activity. The fact that these chelating ligands alone possess cytotoxic response profiles similar to their metal chelates indicates that the active metal complexes serve as carriers for the chelators, which themselves are the active drug molecules, perhaps by forming metal chelates in situ (31).
Discussion
An ultimate goal in cancer therapy is to devise individually tailored treatment protocols targeting specific pathways of cellular proliferation or drug resistance. Pgp represents one of the best-studied mechanisms of resistance to hydrophobic anticancer drugs. Pgp-mediated drug transport of cytotoxic drugs is modulated by a wide range of agents, many of which have been tested in clinical trials. However, despite the clear rationale for the use of efflux inhibitors in combination with cytotoxic agents, the development of these compounds has been slow, and the clinical benefit of Pgp inhibition is still in question. Our aim was to explore the DTP drug database for compounds that may offer a radically different solution by exploiting, rather than suppressing, Pgp function to induce cytotoxicity. Such drugs (termed “MDR-selective compounds”) target otherwise multidrug resistant cycling cells, without inhibiting the transporter and avoiding side effects associated with the damage of resting cells that constitutively express Pgp. We based our approach on the presumption that a positive correlation between a compound’s cytotoxicity profile and the expression of Pgp in the NCI-60 cell panel may be the result of a causal interaction, where the activity of Pgp sensitizes the cell to the cytotoxicity of the compound. It may be argued that this approach has several limitations: the cytotoxicity profiles across the NCI60 panel are derived from single MTT assays, leading to variability in the quality of the primary data; the possibility that compounds are not pure as submitted; and the activity of drug metabolism and resistance pathways may confound the statistical approach, producing false negative and false positive results. Given these and other confounding factors, it is remarkable that 37 MDR-selective compounds were validated in this study. Future studies will determine if any of the compounds not available for testing (Table I) are also MDR-selective. Of particular interest is the series of perezone derivatives that appear in a structurally coherent group among the top-scoring untested compounds (Figure 1).
The new MDR-selective compounds presented here have several features reminiscent of NSC73306, the first Pgp-targeting drug identified in our initial report (7).
Since the MDR-selective toxicity is reversed in the presence of Pgp-inhibitors, we can conclude that the enhanced toxicity of the compounds shown in Table II is indeed dependent on Pgp-activity, and is not due to an off-target effect linked to the genetic drift of KB-V1 cells. Furthermore, the results obtained with an isogenic cell line pair support that functional expression of Pgp is necessary and sufficient to convey increased sensitivity to these compounds. The degree of Pgp-dependent toxicity varied: while some compounds showed a minor (albeit statistically significant) MDR-selective activity, others, such as NSC693871 were considerably more toxic in KB-V1 cells. As observed with NSC73306, expression of Pgp in MDR cell lines decreased upon exposure to the MDR-selective agents, supporting the causal link between the toxicity and Pgp function (manuscript in preparation).
Analysis of the MDR-selective molecules in the context of the DTP drug database has the potential to relate the information in the tumor cell line data set to structural feature analysis. Granted that the general toxicity of the MDR-selective compounds identified here is probably mediated by a number of mechanisms, the structural coherence of the Discovery-set may imply shared modes of MDR-selective toxicity that pertain to structurally related compound subsets. There is a significant enrichment in certain chemical features within the Discovery-set: the nine TSCs comprise 14% of the Discovery-set, while representing only 1% of the original set of the screened compounds, and there are only 25 phen and 56 bipy compounds in the original dataset (0.059 % and 0.12%, respectively). Thus, the enrichment of TSC (~12 fold), phen (over 1500 fold) and bipy (over 750 fold) reinforces that these features are associated with MDR-selective activity. In all three structural cohorts, there are two nitrogen atoms in a cis(1,4) arrangement, separated by two carbons. The initial QSAR (Table S3) is a first step toward a systematic substructure analysis coupled with statistical correlation of compound activity, to highlight structural features necessary for MDR-selectivity (41). Recently the TSC Dp44mT was reported to be more toxic to KB-V1 cells (42). Dp44mT was not submitted to the DTP, therefore it does not appear in our list of putative MDR-selective agents. Interestingly, it also contains at least two hydrogen bond acceptors, a hydrogen bond donor as well as an aromatic ring, underscoring the pharmacophore model derived from the TSCs analyzed in this study (Figure S2).
The abundance of metal chelators in the Discovery-set is striking. The eight metal-containing complexes represent 12.5% of the Discovery-set, while only 2.7% of the compounds found in the DTP library form complexes (including sodium and calcium cation formulations). Several MDR-selective compounds project to the S region of the SOM representing the DTP drug response data (Supplementary Figure S3), along with chelating agents targeting metallo enzymes, especially iron- and copper-dependent proteins (31). Typically, metal complexes projecting to the S region show some degree of similarities in both structure and cytotoxic response profiles. Interestingly, the type of the chelated metal is varied, suggesting that the cytotoxicity of S-region complexes is determined primarily by the organic component of the complex (the ligand) and the type of metal is only playing a minor role (31). Our results indicate that the MDR-selective chelators can form complexes with a range of biologically important metals ions, including iron (Figure 2). Extensive research efforts have been dedicated to the design of iron or copper chelators as anticancer agents, and several drugs in the clinic such as bleomycin, the anthraquinones, triapine and hydroxyurea demonstrate the validity and diversity of metal interaction as a viable chemotherapeutic target (38). Based on the results obtained by this correlative observational study, we speculate that metal ion interaction is key to the cytotoxicity of at least a subset of the MDR-selective compounds. Iron-chelating drugs may kill cancer cells by a number of mechanisms, including, (1) deprivation of nutritionally essential iron, (2) iron-drug associations that lead to redox cycling (6), or (3) highly specific metal-enzyme targets, such as ribonucleotide reductase (43). We speculate that the activity of Pgp may increase the susceptibility of the cells to these mechanisms. At the same time, our results also suggest that metal chelation alone is not sufficient for Pgp-potentiated activity. Since metal complexes were as active as the ligands alone (Table III), chelates may serve as chaperones facilitating free diffusion of the ligands into the cells (44). In line with this hypothesis, NSC73306 does not interact with Pgp (18), whereas triapine (a TSC ribonucleotide reductase inhibitor devoid of MDR-selective activity) is actively effluxed by Pgp (45). Thus, it appears that evasion of Pgp mediated efflux as well as the ability to chelate metals are necessary, if not sufficient requirements for the MDR-selective activity of the chelator compounds. Dissociation of the complexes within the cells leading to the release of the free ligand and MDR-selective activity may be related to reactive oxygen species generated by redox cycling, as has been demonstrated for the activity of other TSCs (46).
Taken together, it appears that the target of the MDR-selective compounds is not Pgp per se. The paradoxical vulnerability uncovered by the MDR-selective compounds may be linked to the efflux of an endogenous substrate providing e.g. redox-resistance, to the influence of Pgp on membrane lipid composition, or to other, Pgp-related metabolic changes that characterize MDR cells.
The results here indicate that MDR-selective compounds may be used against MDR cells, providing a range of active pharmacophores. According to a recent report, the TSC Dp44mT has a broad-spectrum activity against a wide range of cancer cell types in vivo, including those possessing the MDR phenotype mediated by Pgp (42). The fact that a related TSC shows strong in vivo anticancer activity in human xenografts is promising, and suggests that the MDR-selective chelators identified in this study may have potent and broad antitumor activity. Further evaluation of the compounds identified here will help to elucidate this intriguing modality, and provide the basis of a fresh therapeutic approach to resolving MDR cancers.
Supplementary Material
Acknowledgements
We thank the NCI DTP for generation of the database used in this study. The technical help of Zsuzsanna Sebestyen is gratefully acknowledged.
This work has been supported by OTKA (PF60435), EMBO-SDIG and Marie Curie Grants (046560, 041547) as well as the Intramural Research Program of the National Institutes of Health. Gergely Szakács is the recipient of a János Bolyai Scholarship and a Special Fellow Award from the Leukemia and Lymphoma Society.
Footnotes
References
- 1.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 2.Szakacs G, Varadi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) Drug Discov Today. 2008;13:379–393. doi: 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 3.Turk D, Szakacs G. Relevance of multidrug resistance in the age of targeted therapy. Curr Opin Drug Discov Devel. 2009;12:246–252. [PubMed] [Google Scholar]
- 4.Nicholson KM, Quinn DM, Kellett GL, Warr JR. Preferential killing of multidrug-resistant KB cells by inhibitors of glucosylceramide synthase. Br J Cancer. 1999;81:423–430. doi: 10.1038/sj.bjc.6690711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bentley J, Quinn DM, Pitman RS, Warr JR, Kellett GL. The human KB multidrug-resistant cell line KB-C1 is hypersensitive to inhibitors of glycosylation. Cancer Lett. 1997;115:221–227. doi: 10.1016/s0304-3835(97)04739-3. [DOI] [PubMed] [Google Scholar]
- 6.Shabbits JA, Mayer LD. P-glycoprotein modulates ceramide-mediated sensitivity of human breast cancer cells to tubulin-binding anticancer drugs. Mol Cancer Ther. 2002;1:205–213. [PubMed] [Google Scholar]
- 7.Szakacs G, Annereau JP, Lababidi S, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. 2004;6:129–137. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 8.Warr JR, Quinn D, Elend M, Fenton JA. Gain and loss of hypersensitivity to resistance modifiers in multidrug resistant Chinese hamster ovary cells. Cancer Lett. 1995;98:115–120. [PubMed] [Google Scholar]
- 9.Lehne G, De Angelis P, den Boer M, Rugstad HE. Growth inhibition, cytokinesis failure and apoptosis of multidrug-resistant leukemia cells after treatment with P-glycoprotein inhibitory agents. Leukemia. 1999;13:768–778. doi: 10.1038/sj.leu.2401392. [DOI] [PubMed] [Google Scholar]
- 10.Lehne G, Sorensen DR, Tjonnfjord GE, et al. The cyclosporin PSC 833 increases survival and delays engraftment of human multidrug-resistant leukemia cells in xenotransplanted NOD-SCID mice. Leukemia. 2002;16:2388–2394. doi: 10.1038/sj.leu.2402663. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan O, Jaroszewski JW, Clarke R, et al. The multidrug resistance phenotype: 31P nuclear magnetic resonance characterization and 2-deoxyglucose toxicity. Cancer Res. 1991;51:1638–1644. [PubMed] [Google Scholar]
- 12.Bell SE, Quinn DM, Kellett GL, Warr JR. 2-Deoxy-D-glucose preferentially kills multidrug-resistant human KB carcinoma cell lines by apoptosis. Br J Cancer. 1998;78:1464–1470. doi: 10.1038/bjc.1998.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warr JR, Bamford A, Quinn DM. The preferential induction of apoptosis in multidrug-resistant KB cells by 5-fluorouracil. Cancer Lett. 2002;175:39–44. doi: 10.1016/s0304-3835(01)00721-2. [DOI] [PubMed] [Google Scholar]
- 14.Bergman AM, Pinedo HM, Talianidis I, et al. Increased sensitivity to gemcitabine of P-glycoprotein and multidrug resistance-associated protein-overexpressing human cancer cell lines. Br J Cancer. 2003;88:1963–1970. doi: 10.1038/sj.bjc.6601011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinstein JN, Myers TG, O'Connor PM, et al. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 16.Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 17.Hall MD, Salam NK, Hellawell JL, et al. Synthesis, Activity, and Pharmacophore Development for Isatin-beta-thiosemicarbazones with Selective Activity toward Multidrug-Resistant Cells. J Med Chem. 2009 doi: 10.1021/jm800861c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludwig JA, Szakacs G, Martin SE, et al. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 2006;66:4808–4815. doi: 10.1158/0008-5472.CAN-05-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atwell GJ, Rewcastle GW, Baguley BC, Denny WA. Potential antitumor agents. 50. In vivo solid-tumor activity of derivatives of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J Med Chem. 1987;30:664–669. doi: 10.1021/jm00387a014. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Klinginsmith J, Dong X, et al. Chemical data mining of the NCI human tumor cell line database. J Chem Inf Model. 2007;47:2063–2076. doi: 10.1021/ci700141x. [DOI] [PubMed] [Google Scholar]
- 21.Shen DW, Cardarelli C, Hwang J, et al. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J Biol Chem. 1986;261:7762–7770. [PubMed] [Google Scholar]
- 22.Cardarelli CO, Aksentijevich I, Pastan I, Gottesman MM. Differential effects of P-glycoprotein inhibitors on NIH3T3 cells transfected with wild-type (G185) or mutant (V185) multidrug transporters. Cancer Res. 1995;55:1086–1091. [PubMed] [Google Scholar]
- 23.Wang E, Lee MD, Dunn KW. Lysosomal accumulation of drugs in drug-sensitive MES-SA but not multidrug-resistant MES-SA/Dx5 uterine sarcoma cells. J Cell Physiol. 2000;184:263–274. doi: 10.1002/1097-4652(200008)184:2<263::AID-JCP15>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Ujhelly O, Ozvegy C, Varady G, et al. Application of a human multidrug transporter (ABCG2) variant as selectable marker in gene transfer to progenitor cells. Hum Gene Ther. 2003;14:403–412. doi: 10.1089/104303403321209005. [DOI] [PubMed] [Google Scholar]
- 25.Antholine W, Knight J, Whelan H, Petering DH. Studies of the reaction of 2-formylpyridine thiosemicarbazone and its iron and copper complexes with biological systems. Mol Pharmacol. 1977;13:89–98. [PubMed] [Google Scholar]
- 26.Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of ribonucleotide reductase with antineoplastic activity. Adv Enzyme Regul. 1999;39:3–12. doi: 10.1016/s0065-2571(98)00017-x. [DOI] [PubMed] [Google Scholar]
- 27.Hall IH, Lackey CB, Kistler TD, et al. Cytotoxicity of copper and cobalt complexes of furfural semicarbazone and thiosemicarbazone derivatives in murine and human tumor cell lines. Pharmazie. 2000;55:937–941. [PubMed] [Google Scholar]
- 28.Byrnes RW, Antholine WE, Petering DH. Interactions of 1,10-phenanthroline and its copper complex with Ehrlich cells. Free Radic Biol Med. 1992;12:457–469. doi: 10.1016/0891-5849(92)90099-3. [DOI] [PubMed] [Google Scholar]
- 29.Shen AY, Wu SN, Chiu CT. Synthesis and cytotoxicity evaluation of some 8-hydroxyquinoline derivatives. J Pharm Pharmacol. 1999;51:543–548. doi: 10.1211/0022357991772826. [DOI] [PubMed] [Google Scholar]
- 30.Pierre JL, Baret P, Serratrice G. Hydroxyquinolines as iron chelators. Curr Med Chem. 2003;10:1077–1084. doi: 10.2174/0929867033457584. [DOI] [PubMed] [Google Scholar]
- 31.Huang R, Wallqvist A, Covell DG. Anticancer metal compounds in NCI's tumor-screening database: putative mode of action. Biochem Pharmacol. 2005;69:1009–1039. doi: 10.1016/j.bcp.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Enriquez RG, Fernandez-G JM, Gnecco D, Penicaud A, Reynolds WF. The crystal and molecular structures of isoperezone, aminoperezone and isoaminoperezone: a comparative study of their crystal packing. Journal of Chemical Crystallography. 1998;28:529–537. [Google Scholar]
- 33.Cuellar A, Carabez A, Chavez E. Ca2+ releasing effect of perezone on adrenal cortex mitochondria. Life Sci. 1987;41:2047–2054. doi: 10.1016/0024-3205(87)90479-6. [DOI] [PubMed] [Google Scholar]
- 34.Kovacic P. Unifying mechanism for anticancer agents involving electron transfer and oxidative stress: clinical implications. Med Hypotheses. 2007;69:510–516. doi: 10.1016/j.mehy.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y, Blower PE, Yang C, et al. Correlating gene expression with chemical scaffolds of cytotoxic agents: ellipticines as substrates and inhibitors of MDR1. Pharmacogenomics J. 2005;5:112–125. doi: 10.1038/sj.tpj.6500297. [DOI] [PubMed] [Google Scholar]
- 36.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 37.Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Mining the National Cancer Institute's tumor-screening database: identification of compounds with similar cellular activities. J Med Chem. 2002;45:818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Wong J, Lovejoy DB, Kalinowski DS, Richardson DR. Chelators at the cancer coalface: desferrioxamine to Triapine and beyond. Clin Cancer Res. 2006;12:6876–6883. doi: 10.1158/1078-0432.CCR-06-1954. [DOI] [PubMed] [Google Scholar]
- 39.Heffeter P, Jakupec MA, Korner W, et al. Multidrug-resistant cancer cells are preferential targets of the new antineoplastic lanthanum compound KP772 (FFC24) Biochem Pharmacol. 2007;73:1873–1886. doi: 10.1016/j.bcp.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Draper MP, Martell RL, Levy SB. Indomethacin-mediated reversal of multidrug resistance and drug efflux in human and murine cell lines overexpressing MRP, but not P-glycoprotein. Br J Cancer. 1997;75:810–815. doi: 10.1038/bjc.1997.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blower PE, Yang C, Fligner MA, et al. Pharmacogenomic analysis: correlating molecular substructure classes with microarray gene expression data. Pharmacogenomics J. 2002;2:259–271. doi: 10.1038/sj.tpj.6500116. [DOI] [PubMed] [Google Scholar]
- 42.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol. 1992;19:1–10. [PubMed] [Google Scholar]
- 44.Hall MD, Failes TW, Yamamoto N, Hambley TW. Bioreductive activation and drug chaperoning in cobalt pharmaceuticals. Dalton Trans. 2007:3983–3990. doi: 10.1039/b707121c. [DOI] [PubMed] [Google Scholar]
- 45.Rappa G, Lorico A, Liu MC, et al. Overexpression of the multidrug resistance genes mdr1, mdr3, and mrp in L1210 leukemia cells resistant to inhibitors of ribonucleotide reductase. Biochem Pharmacol. 1997;54:649–655. doi: 10.1016/s0006-2952(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 46.Bernhardt PV, Sharpe PC, Islam M, Lovejoy DB, Kalinowski DS, Richardson DR. Iron chelators of the dipyridylketone thiosemicarbazone class: precomplexation and transmetalation effects on anticancer activity. J Med Chem. 2009;52:407–415. doi: 10.1021/jm801012z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.