

Abstract
We have recently identified the secreted protein IGFBP7 as a factor required for an activated BRAF oncogene to induce senescence or apoptosis in primary human cells. In human melanomas containing an activating BRAF mutation (BRAF-positive melanomas), IGFBP7 is epigenetically silenced, which appears to be a critical step in melanoma genesis. Restoration of IGFBP7 function by addition of recombinant IGFBP7 (rIGFBP7) induces apoptosis in BRAF-positive human melanoma cell lines, and systemically administered rIGFBP7 markedly suppresses growth of BRAF-positive primary tumors in xenografted mice. Here we further evaluate the role of IGFBP7 in treatment of BRAF-positive melanoma and other malignancies. We find that in human metastatic melanoma samples IGFBP7 is epigenetically silenced and at an even higher frequency than that found in primary melanomas. Using a murine experimental metastasis assay, we show that systemic administration of rIGFBP7 markedly suppresses growth of metastatic disease and prolongs survival. Analysis of the NCI60 panel of human cancer cell lines reveals that in addition to melanoma, IGFBP7 induces apoptosis in several other cancer types, in particular colorectal cancer cell lines. In general, IGFBP7 induced apoptosis in human cancer cell lines that had an activating mutation in BRAF or RAS, and that were sensitive to chemical inhibition of BRAF-MEK-ERK signaling. Significantly, systemically administered rIGFBP7 blocks growth of colorectal tumors containing an activating RAS or BRAF mutation in mouse xenografts. The results presented here, in conjunction with those from previous studies, justify the further development of IGFBP7 as an anti-cancer agent.
Keywords: IGFBP7, metastatic melanoma, colorectal cancer, BRAFV600E
Introduction
The proto-oncogene RAF encodes a serine-threonine protein kinase that functions as an immediate downstream effector of RAS (reviewed in (1)). RAF activates the MAP kinase extracellular signal regulated kinase (MEK), which in turn phosphorylates and activates extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2). Activating mutations in BRAF promote cell proliferation and transformation by constitutively activating the RAF-MEK-ERK signaling pathway. Activating BRAF mutations are found at high frequency in human cancers and are particularly prevalent in melanoma where they occur at a frequency of 50-70% (2).
Paradoxically, when expressed in primary cells, an activated BRAF mutant can block cellular proliferation by inducing senescence or apoptosis (3, 4). Recently, we identified 17 genes required for activated BRAF-mediated apoptosis and senescence, one of which encodes the secreted protein IGFBP7 (4). Analysis of human tissue samples indicates that loss of IGFBP7 expression is a critical step in melanoma development. Most importantly, we found that recombinant IGFBP7 (rIGFBP7) induces apoptosis in BRAF-positive human melanoma cell lines, and systemically administered rIGFBP7 markedly suppresses growth of BRAF-positive melanoma in xenografted mice. Growth suppression results both from inhibition of BRAF-MEK-ERK signaling and activation of an apoptotic pathway that culminates in the upregulation of BNIP3L, a pro-apoptotic BCL2 family protein.
The selective sensitivity of activated BRAF-containing human cancer cell lines to IGFBP7, and the ability of IGFBP7 to suppress BRAF-positive tumor growth in mouse xenografts, suggests a possible role for IGFBP7 in treating BRAF-positive malignancies. Here we further evaluate the potential role of IGFBP7 for treatment of melanoma and other cancers.
Materials and Methods
Immunohistochemistry
The study was approved by the UMass Medical Center institutional review board (IRB #12543). Archival materials from metastatic melanoma were retrieved from the pathology files of Boston University School of Medicine, Boston, MA. The histologic sections of all cases were re-reviewed and the diagnoses confirmed by a dermatopathologist (MM). All patient data were de-identified. Immunohistochemical analysis was performed as previously described (4). BRAF genotyping was performed using mutant allele-specific amplification (MASA)-PCR as previously described (5). The PCR reaction was performed using forward primers 5'-TAGGTGATTTTGGTCTAGCTACAGT-3' (to amplify wild-type BRAF) and 5’-GGTGATTTTGGTCTAGCTACAAA-3' (to amplify the mutant BRAFV600E allele) and reverse primer 5'-GGCCAAAATTTAATCAGTGGA-3' using the following conditions: denaturation for 2 min at 94°C, followed by 40 cycles of denaturation for 30 s at 94°C, annealing for 45 s at 52°C, and extension for 45 s at 72°C.
Bisulfite Sequencing
Bisulfite modification was carried out essentially as previously described (4). Six clones were sequenced for each human tissue sample using nested primers BisulBP7-For1 (5’-AGAAGTTTAAATATATTGAT-3’), BisulBP7-For2 (5’-GGAAATGGGGAGAAATTAGA-3’) and BisulBP7-Rev2 (5’-GTTGGGTTGTTGTTTTTGTT-‘3).
Tumor Formation Assays
Recombinant IGFBP7 (rIGFBP7) was produced and purified from baculovirus-infected cells as previously described (4). In the experiments of Fig. 2A, rIGFBP7 (100 μg in 100 μl) or PBS was injected into the tail vein of athymic Balb/c (nu/nu) mice (Taconic) (n=5 mice per group). One day later, mice were injected through the tail vein with 7×105 A375M-Fluc cells (a kind gift of Sanjiv Gambhir, Stanford University, in June 2007; (6)), and 3 and 6 days later with rIGFBP7 (20 μg) or PBS. On day 7 mice were injected with D-Luciferin and imaged using a Xenogen IVIS imaging system. Survival probability was calculated using Kaplan-Meier analysis. In the experiments of Fig. 2B, 7×105 A375M-Fluc cells were injected through the tail vein and 5, 8 and 11 days later rIGFBP7 (20 μg in 100 μl total volume) was injected (n=5 mice per group). Animal experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
Figure 2.

IGFBP7 suppresses tumor growth and increases survival in an experimental metastasis assay. A, On day 1, rIGFBP7 or PBS was delivered by tail vein injection followed by introduction of A375M-Fluc cells on day 2 (n=5 mice per group). Two additional doses of rIGFBP7 were administered by tail vein injection on days 3 and 6. Survival probability was calculated using Kaplan-Meier analysis. (Right) Bioluminescent imaging on day 7. B, On day 1, A375M-Fluc cells were introduced by tail vein injection followed by administration of rIGFBP7 or PBS by tail vein injection on days 5, 8 and 11 (n=5 mice per group). (Right) Bioluminescent imaging on day 7.
For colorectal cancer cell experiments, 5×106 HT29 or SW-620 cells were injected subcutaneously into the right flank of athymic Balb/c (nu/nu) mice (n=5 mice per group). When tumors reached a size of 100 mm3, 100 μg rIGFBP7 was delivered by tail vein injection at days 6, 9 and 12. Tumor dimensions were measured every three days and tumor volume was calculated using the formula π/6 × (length) × (width)2.
Analysis of NCI60 Cell Lines
The NCI60 panel of human cancer cell lines were obtained from National Cancer Institute (NCI) in September 2006, and grown in RPMI with 10% FCS. Cells were plated and treated with 10 μg rIGFBP7 for 24 hrs and analyzed for apoptosis induction by annexin V staining, or treated with U0126 (10 μM; Cell Signaling Technology Inc.) for 24 hrs and analyzed for proliferation by trypan blue exclusion. All experiments were performed in triplicate.
Results
IGFBP7 is Epigenetically Silenced at High Frequency in Metastatic Melanoma
Our previous study focused exclusively on human primary melanoma samples and mouse models of primary melanoma (4). However, metastatic disease represents the major unmet need for melanoma treatment (7). To evaluate the potential role of IGFBP7 in metastatic melanoma, we examined IGFBP7 expression by immunohistochemistry in a series of human metastatic melanoma samples. Each sample was also analyzed for the presence of the activating BRAFV600E mutation. The results (representative examples are shown in Fig. 1A and the results are summarized in Fig. 1B) reveal that all 20 metastatic melanomas analyzed failed to express detectable levels of IGFBP7, regardless of BRAF status.
Figure 1.

Analysis of IGFBP7 expression in human metastatic melanoma samples. A, Immunohistochemical analysis of IGFBP7 expression in representative human metastatic melanoma tissue samples. As a positive control, IGFBP7 expression is shown in a primary melanoma sample. Samples were stained with hematoxylin and eosin (H&E). Images are shown at 2X and/or 20X. B, Summary of IGFBP7 expression in human metastatic melanoma samples. The BRAF status is shown; all samples were negative for the NRASQ161R mutation. C, Bisulfite sequence analysis of the IGFBP7 promoter in human metastatic melanoma tissue samples.
To investigate whether the loss of IGFBP7 expression in metastatic melanomas resulted from epigenetic silencing, we performed bisulfite sequencing analysis on eight of the samples, which either contained or lacked the activating BRAFV600E mutation. Significantly, all eight samples contained dense hypermethylation of the IGFBP7 promoter (Fig. 1C), indicative of epigenetic silencing and explaining the lack of detectable IGFBP7 expression.
IGFBP7 Suppresses Tumor Growth and Increases Survival in a Mouse Model of Metastatic Melanoma
We previously showed that IGFBP7 suppressed tumor growth in a mouse xenograft model of primary melanoma (4). We were therefore interested in determining whether IGFBP7 could also be used to treat metastatic disease, which would be the most important clinical application. For these experiments, we used an established murine experimental metastasis assay in which human melanoma cells form pulmonary metastases following tail vein injection (see, for example, (6, 8)). The experiments used A375M-Fluc cells, which are a highly metastatic, BRAFV600E-positive human melanoma cell line expressing the firefly luciferase (Fluc) gene (6), which enables bioluminescent optical imaging.
In the first set of experiments, in which we asked whether IGFPB7 could function prophylactically, rIGFBP7, or as a control PBS, was delivered by tail vein injection on day 1 followed by introduction of A375-Fluc cells on day 2. Two additional doses of rIGFBP7 were administered by tail vein injection on days 3 and 6. Bioluminescent imaging on day 7 revealed substantial pulmonary metastasis in all untreated animals, whereas pulmonary metastasis was undetectable in animals receiving rIGFBP7 treatment (Fig. 2A). More importantly, all untreated animals died by day 20, whereas rIGFBP7-treated animals survived through day 30, when the experiment was terminated.
In a second set of experiments, A375-Fluc cells were first introduced by tail vein injection (day 1). Subsequently, rIGFBP7, or PBS, was administered by tail vein injection on days 5, 8 and 11. Bioluminescent imaging on day 5, prior to IGFBP7 administration, revealed that measurable pulmonary tumors were present prior to initiation of therapy (Supplementary Fig. 1). On day 7, substantial pulmonary metastasis was observed in all untreated animals, whereas pulmonary metastasis was undetectable in all animals receiving rIGFBP7 (Fig. 2B). None of the untreated animals survived beyond day 24, whereas all the animals receiving rIGFBP7 treatment survived through day 50, when the experiment was terminated.
Susceptibility of NCI60 Human Cancer Cell Lines to IGFBP7 Treatment
Activating BRAF mutations are also found in a number of other solid tumors including colorectal, ovarian and non-small cell lung cancers (2). In addition, up to 30% of solid tumors contain activating RAS mutations, which can also increase BRAF-MEK-ERK signaling (reviewed in (9)). We therefore investigated the potential use of IGFBP7 in treatment of other cancers. Toward this goal, we analyzed the NCI60 panel of human cancer cell lines for sensitivity to apoptosis induced by rIGFBP7. In parallel, we tested the ability of the chemical MEK inhibitor, U0126, to block proliferation of the NCI60 cell lines. The BRAF or RAS mutational status in each of the NCI60 cell lines was derived from previously published data (10). The results, shown in Figure 3, enable us to draw several conclusions.
Figure 3.

Susceptibility of NCI60 human cancer cell lines to treatment with IGFBP7 or a MEK inhibitor. A, Cell lines were treated with 10 μg rIGFBP7 for 24 hrs and analyzed for apoptosis by annexin V staining. B, Cell lines were treated with U0126 (10 μM) for 24 hrs and analyzed for proliferation by trypan blue exclusion. All experiments were performed in triplicate. Error bars represent SEM.
First, consistent with our previous findings (4), we found that human melanoma cell lines were highly sensitive to IGFBP7-mediated apoptosis. Second, the vast majority of the NCI60 cell lines were unaffected by rIGFBP7, as expected for a targeted therapeutic as opposed to a general cytotoxic agent. Third, several breast, ovarian, lung and, in particular, colorectal cancer cell lines underwent significant apoptosis following addition of rIGFBP7. Finally, in general the human cancer lines that were sensitive to IGFBP7-mediated apoptosis contained an activating BRAF or RAS mutation and were also sensitive to U0126-mediated growth arrest. We note, however, there were several exceptions to this general trend. For example, several breast cancer (BT-549 and MCF7) and colorectal cancer (HCC-2998 and KM-12) cell lines lacked an activating BRAF or RAS mutation and were sensitive to IGFBP7 but not to U0126. Conversely, several human cancer cell lines such as HS 578T (breast), A549 (lung) and CCRF-CEM (hematopoietic) contained activating RAS mutations and were sensitive to U0126 but not to IGFBP7.
IGFBP7 Suppresses Tumor Growth in Human Colorectal Cancer Mouse Xenografts
The ability of rIGFBP7 to induce apoptosis in human colorectal cancer cell lines raised the possibility that rIGFBP7 could suppress growth of colorectal tumors. To test this possibility we chose two human colorectal cancer cell lines, one of which contained an activating BRAF mutation (HT29) and the second of which contained an activating KRAS mutation (SW-620). Each cell line was injected subcutaneously into the flanks of nude mice and when tumors reached a size of 100 mm3, 100 μg rIGFBP7, or PBS, was delivered by tail vein injection at days 6, 9 and 12. Figure 4 shows that systemic administration of rIGFBP7 completely suppressed growth of both HT29 and SW-620 tumors.
Figure 4.
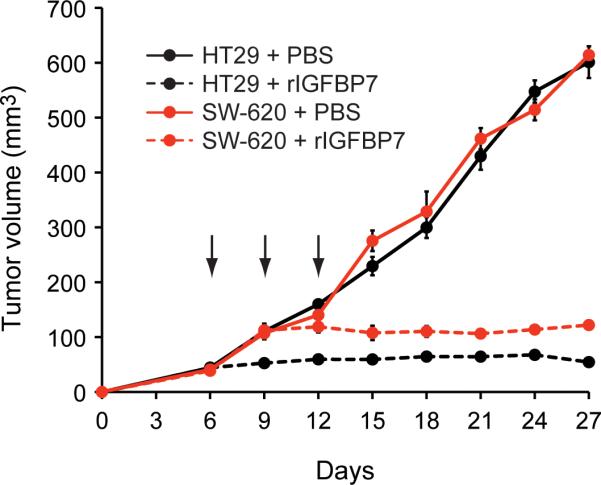
IGFBP7 suppresses tumor growth in mouse xenografts of human colorectal cancer cell lines. HT29 (containing a BRAFV600E mutation) or SW-640 (KRASG12V) cells were injected into the flanks of nude mice (n=5 mice per group). When tumors reached 100 mm3, 100 μg rIGFBP7 was administered by tail vein injection at days 6, 9 and 12 (indicated by arrows). Error bars represent SEM.
Discussion
Here we have performed a series of experiments to further investigate the potential role of IGFBP7 in treatment of melanoma and other cancers. Of particular interest was metastatic melanoma, an aggressive disease that is refractory to conventional chemotherapeutic agents and lacks adequate treatment options (reviewed in (7)). Similar to our previous results using a primary melanoma model (4), we found that systemically administered rIGFBP7 suppressed tumor growth and prolonged survival in a murine experimental metastasis assay. Analysis of IGFBP7 in the recently developed mouse models of BRAF-positive melanoma (11, 12) remains an important future objective.
We had previously shown that IGFBP7 expression is lost in primary melanomas bearing an activating BRAF mutation but not in primary melanomas with wild type BRAF (4). However, here we found that IGFBP7 expression was undetectable in all metastatic melanomas analyzed, regardless of BRAF status. The higher rate of loss of IGFBP7 expression in metastatic samples suggests that during melanoma development there is a strong selection against IGFBP7 expression, providing further evidence that IGFBP7 is a melanoma tumor suppressor gene.
Analysis of the NCI60 panel of cell lines revealed that IGFBP7 induced apoptosis in several cancer types in addition to melanoma. In general, IGFBP7 induced apoptosis in human cancer cell lines that had an activating mutation in BRAF or RAS, and that were sensitive to chemical inhibition of BRAF-MEK-ERK signaling. Previous studies have shown that cancer cells harboring an activated BRAF mutation are highly dependent on BRAF-MEK-ERK signaling for proliferation and survival (13), and that BRAF mutation predicts sensitivity to MEK inhibition (14). These findings provide the rationale for developing therapeutic strategies that target the BRAF-MEK-ERK signaling pathway for treatment of melanoma and other cancers in which BRAF is mutated (13). Inhibitors of BRAF have been developed but unfortunately have performed poorly in clinical trials (15, 16).
We have previously shown that IGFBP7 inhibits BRAF-MEK-ERK signaling and efficiently induces apoptosis in BRAF-positive melanoma cell lines (4). The ability of IGFBP7 to both inhibit BRAF-MEK-ERK signaling and irreversibly induce apoptosis following transient exposure may make it particularly efficacious for treating malignancies that are dependent upon BRAF-MEK-ERK signaling.
We found that colorectal cancer cell lines were also highly susceptible to IGFBP7-mediated apoptosis, consistent with the high frequency of activating BRAF or RAS mutations, and presumably increased BRAF-MEK-ERK signaling, in colorectal cancers (1, 9). Significantly, previous studies have found that IGFBP7 expression is lost in human colorectal cancers (17, 18), consistent with the possibility that IGFBP7 is a colorectal cancer tumor suppressor (19, 20). Most importantly, we found that systemically administered rIGFBP7 markedly suppressed growth of colorectal tumors containing an activating RAS or BRAF mutation in mouse xenografts. These collective results support the encouraging possibility that IGFBP7 may also have a role in treatment of colorectal cancer.
Supplementary Material
Acknowledgments
We thank Sanjiv Gambhir for providing A375M-Fluc cells; the UMMS Diabetes and Endocrine Research Center (DERC) for immunohistochemical staining; Susan Griggs for assistance with the animal experiments; Xiaochun Zhu for rIGFBP7 protein production; and Sara Evans for editorial assistance.
Grant support: Our Danny Cancer Fund (N. Wajapeyee), Howard Hughes Medical Institute (M.R. Green).
References
- 1.Dhomen N, Marais R. New insight into BRAF mutations in cancer. Curr Opin Genet Dev. 2007;17:31–9. doi: 10.1016/j.gde.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 4.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu X, Quiros RM, Gattuso P, Ain KB, Prinz RA. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003;63:4561–7. [PubMed] [Google Scholar]
- 6.Collisson EA, De A, Suzuki H, Gambhir SS, Kolodney MS. Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Cancer Res. 2003;63:5669–73. [PubMed] [Google Scholar]
- 7.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 8.Hoeflich KP, Gray DC, Eby MT, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- 9.Saxena N, Lahiri SS, Hambarde S, Tripathi RP. RAS: target for cancer therapy. Cancer Invest. 2008;26:948–55. doi: 10.1080/07357900802087275. [DOI] [PubMed] [Google Scholar]
- 10.Ikediobi ON, Davies H, Bignell G, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–12. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Dankort D, Curley DP, Cartlidge RA, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–31. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 14.Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fecher LA, Amaravadi RK, Flaherty KT. The MAPK pathway in melanoma. Curr Opin Oncol. 2008;20:183–9. doi: 10.1097/CCO.0b013e3282f5271c. [DOI] [PubMed] [Google Scholar]
- 16.Madhunapantula SV, Robertson GP. Is B-Raf a good therapeutic target for melanoma and other malignancies? Cancer Res. 2008;68:5–8. doi: 10.1158/0008-5472.CAN-07-2038. [DOI] [PubMed] [Google Scholar]
- 17.Luo MJ, Lai MD. Identification of differentially expressed genes in normal mucosa, adenoma and adenocarcinoma of colon by SSH. World J Gastroenterol. 2001;7:726–31. doi: 10.3748/wjg.v7.i5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shao L, Huang Q, Luo M, Lai M. Detection of the differentially expressed gene IGF-binding protein-related protein-1 and analysis of its relationship to fasting glucose in Chinese colorectal cancer patients. Endocr Relat Cancer. 2004;11:141–8. doi: 10.1677/erc.0.0110141. [DOI] [PubMed] [Google Scholar]
- 19.Ruan W, Xu E, Xu F, et al. IGFBP7 plays a potential tumor suppressor role in colorectal carcinogenesis. Cancer Biol Ther. 2007;6:354–9. doi: 10.4161/cbt.6.3.3702. [DOI] [PubMed] [Google Scholar]
- 20.Ruan WJ, Lin J, Xu EP, et al. IGFBP7 plays a potential tumor suppressor role against colorectal carcinogenesis with its expression associated with DNA hypomethylation of exon 1. J Zhejiang Univ Sci B. 2006;7:929–32. doi: 10.1631/jzus.2006.B0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.