

Abstract
NKX3.1 is a prostate tumor suppressor belonging to the NK-2 family of homeodomain (HD) transcription factors. NK-2 family members often possess a stretch of 10–15 residues enriched in acidic amino acids, the acidic domain (AD), in the flexible, disordered region N-terminal to the HD. Interactions between the N-terminal region of NKX3.1 and its homeodomain affect protein stability and DNA binding. CD spectroscopy measuring the thermal unfolding of NKX3.1 constructs showed a 2 °C intramolecular stabilization of the HD by the N-terminal region containing the acidic domain (residues 85–96). CD of mixtures of various N-terminal peptides with a construct containing just the HD showed that the acidic domain and the following region, the SRF interacting (SI) motif (residues 99–105), was necessary for this stabilization. Phosphorylation of the acidic domain is known to slow proteasomal degradation of NKX3.1 in prostate cells, and NMR spectroscopy was used to measure and map the interaction of the HD with phosphorylated and nonphosphorylated forms of the AD peptide. The interaction with the phosphorylated AD peptide was considerably stronger (Kd = 0.5 ± 0.2 mM), resulting in large chemical shift perturbations for residues Ser150 and Arg175 in the HD, as well as a 2 °C increase in the HD thermal stability compared to that of the nonphosphorylated form. NKX3.1 constructs with AD phosphorylation site threonine residues (89 and 93) mutated to glutamate were 4 °C more stable than HD alone. Using polymer theory, effective concentrations for interactions between domains connected by flexible linkers are predicted to be in the millimolar range, and thus, the weak intramolecular interactions observed here could conceivably modulate or compete with stronger, intermolecular interactions with the NKX3.1 HD.
NKX3.1 is a homeodomain (HD)1 protein belonging to the NK-2 family, a class of transcription factors critical for the development of many organs (1). NKX3.1 is involved in development of the prostate and continues to be expressed in adults, maintaining prostate cells in their differentiated state (2). Loss of one of the two NKX3.1 alleles is seen in more than 50% of early stage prostate tumor cells and is thought to be an initiating event in prostate tumor formation (3, 4). Both gene methylation and allelic loss lead to reduced NKX3.1 protein levels in the cell at a median level of 67% of normal, indicating the cell tries to boost the protein level arising from expression of the remaining allele (5). An inactivating mutation of the HD was identified as a genetic risk factor for early onset prostate cancer in a kindred predisposed to prostate cancer (6). Because of the prevalence of NKX3.1 haploinsufficiency, and the fact that early stage prostate cancer tissues retain reduced levels of protein expression, therapies designed to further increase NKX3.1 protein levels to normal levels could be important for suppressing prostate tumorigenesis.
NMR signals from a region of NKX3.1 preceding the HD intensify upon binding of NKX3.1 to DNA, an effect most likely due to this region becoming more flexible and mobile (7). The implication is that this region, which contains the acidic domain (AD) and SRF interacting (SI) motifs, interacts with the HD but becomes displaced when the HD binds to DNA. The NMR signals for the entire N-terminal region preceding the HD, including the AD and SI motifs, exhibit poor chemical shift dispersion, have strong intensities, and have Cα and Cβ chemical shift values typical of a flexible, disordered peptide structure (7, 8). In another study, phosphorylation of two threonines in the AD by CK2 was found to affect NKX3.1 protein half-life and blocking CK2 led to proteasomal degradation of NKX3.1 (9). Interactions with flexible, disordered regions of proteins play roles in a wide array of signaling and regulatory pathways, and phosphorylation is often involved in these processes (10). Here, CD spectroscopy is used to measure the thermal stability of several NKX3.1 constructs thereby determining the N-terminal regions that affect HD thermal stability, and NMR spectroscopy is used to determine the location of their interactions with the homeodomain. In addition, molecular modeling is performed to further explore the nature of the interaction of the homeodomain with the AD and SI regions.
Materials and Methods
Recombinant Proteins for CD and NMR
The NKX3.1 (1–184), (75–184), (96–184), and (114–184) construct sequences were cloned into pET30a vectors (Novagen), to create recombinant fusion proteins with a hexahistidine (His6) sequence at the N-terminus. The proteins were expressed in Escherichia coli BL21(DE3) cells in LB media. A full-length NKX3.1 (1–234) construct was also expressed in E. coli but showed poor solubility and was not studied further. For the NMR experiments, uniformly 15N-labeled proteins were produced by culturing the cells in minimal medium containing 15NH4Cl (Cambridge Isotope Laboratory) as the sole nitrogen source. Cells were grown at 37 °C to an optical density (A600) of ∼0.6 and then induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 3–4 h. The cells were suspended in 20 mM Tris buffer (pH 8.0), 500 mM NaCl, and 0.125% Triton X-100 and lysed with sonication. The proteins were isolated by Ni2+ affinity chromatography and subsequently purified via gel filtration column (HiLoad 16/60, Amersham Pharmacia Biotech) and cation exchange column (HiTrap SP HP, Amersham Pharmacia Biotech) chromatography by using the AKTA FPLC system (Amersham Bioscience). A portion of purified NKX3.1 (114–184) was treated with enterokinase to remove the leading His tag sequence. The homogeneity of purified proteins was confirmed by electrophoresis gel analysis and estimated to be ≥95% in each case. A synthetic AD (Ac-EEAETLAETEPE-NH2), ADSI (Ac-EEAETLAETEPERHLGSYLLDSE-NH2), and the phospho-AD (Ac-EEAEpThrLAEpThrEPE-NH2) peptides, where pThr is phosphothreonine, were purchased from Biosynthesis (Lewisville. TX).
CD Spectroscopy
CD spectra and temperature scans at 222 nM for NKX3.1 protein constructs were collected on a Jasco J-715 spectropolarimeter using a concentration of 30– 40 μM with a 1 mm path length cuvette. For protein/peptide mixtures, the peptide concentration was 1.6 mM, the protein concentration was 100 μM, and the spectra and 222 nM temperature scans were recorded on a Jasco J-710 spectropolarimeter with a 0.2 mm path length cuvette. All samples were in 20 mM MES (pH 6.5) and 50 mM NaCl. Phosphate buffer is a more typical choice for homeodomain structural studies; however, because phosphate is known to bind the closely related NK-2 homeodomain (11), and because the study here includes interactions of the homeodomain with phosphorylated peptides, a non-phosphate buffer was chosen. CD spectra were recorded from 260 to 200 nm, depending on solution conditions. The temperature scans were conducted at a rate of 1 °C/min from 10 to 70 °C. A CD spectrum of each sample at 20 °C was recorded before and after each temperature scan to ensure no loss or change of secondary structure due to heating.
Thermal unfolding population curves were calculated from the 222 nM temperature scans by first determining the slope between 10 and 15 °C and the slope between 65 and 70 °C; then a second-degree polynomial with the same value at 10 °C and the same slopes at 10 and 70 °C as the temperature scan was subtracted from the temperature scan, and the resulting curve was normalized to the value at 70 °C.
NMR Spectroscopy
NMR spectra were recorded on a Bruker DRX 600 instrument with a triple-axis gradient probe using standard two-dimensional (2D) 15N HSQC experiments. The purity (> 95%) of the synthetic peptides was confirmed by mass spectrometry and the concentration confirmed by one-dimensional (1D) NMR, and the protein purity (> 95%) was confirmed by NuPage gel electrophoresis and 2D 15N HSQC spectra. The sample buffer was the same as that for CD with 5% D2O. All spectra were recorded at 12 °C, except where noted otherwise. Stock solutions of NKX3.1 (114–184) and peptide were dialyzed together, using a Slide-a-Lyzer (Thermo Scientific) cassette (MWCO of 3500) and a Spectra/Por (Spectrum Laboratories) cassette (MWCO of 500) for protein and peptide, respectively. All spectra were processed and analyzed using the nmrPipe software package (12). Chemical shift perturbations were calculated as [ΔδH2 + (ΔδN/10)2]1/2, where ΔδH is the proton shift change and ΔδN is the nitrogen shift change.
For the determination of the phospho-AD binding (dissociation) constant, the formula Kd = [A-x][B-x]/[x], where [x] is the concentration of the A•B complex and [A-x] and [B-x] are the concentrations of the unbound homeodomain and phosphorylated AD peptide, respectively, is solved as a function of [B] for the fraction of bound homeodomain (f = [x]/[A]), yielding
The chemical shift changes upon titration of the phosphorylated AD peptide were assumed to be linearly proportional to f; that is, f = (Δδ)/(Δδmax), where Δδ is the observed shift change as a function of [B] and Δδmax is the maximum shift change upon complete saturation. The chemical shift changes of Ser150 and Arg176 were scaled, dividing by their average ratios relative to Arg175 Hε. Both Kd and Δδmax were determined by minimizing the sum of the squared differences between the scaled chemical shift values and their values predicted by formula.
The error range for Kd was estimated by using just two of the three sets of shift changes, recalculating Kd three times corresponding to the three possible choices of the two sets. The same procedure was repeated, except that data corresponding to one of the four phospho-AD concentrations were removed instead, each in turn, yielding four more Kd values. The predicted error range is taken as the root-mean-square difference of these Kd values compared to the Kd calculated with the full data set.
Molecular Modeling
For both the AD and phospho-AD peptides, 10 peptides with random ϕ and ψ angles were generated and minimized; short molecular dynamics were performed (50 ps), and the peptides were reminimized using Maestro and MacroModel (Schrödinger Inc., New York, NY). A homology model structure of the NKX3.1 HD was generated using Prime (Schrödinger Inc.), using the NK-2 structure [Protein Data Bank (PDB) entry 1NK3] as a template (13). Each peptide structure was docked to the NKX3.1 homeodomain using EMAP of CHARMM (14, 15). Of the 676 docked structures generated by EMAP for each initial peptide, two were chosen, with either glutamate side chains or phosphothreonine side chains within 10 Å of Ser150 HN or Arg175 Hε. One of the two was chosen with its peptide N-terminus closer to Ser150 and the other with the N-terminus closer to Arg175.
The EMAP program maps the molecular structures being docked onto coarse grids (with 2 Å spacing), and the docked structures can be moderately overlapped; therefore, minimization and molecular dynamics of the docked peptide were first performed with the protein held fixed. Distance restraints (< 5 Å) between the Cγ atom of glutamate or the phosphorus of phosphothreonine and Ser150 HN or Arg175 Hε were also employed at this stage. After restrained MD and reminimization for 50 ps, MD for an additional 50 ps were performed with the distance restraints removed, the protein side chains free, and the backbone tethered, and a final minimization was performed. The linker residues (96–124) between the docked peptides and HD were generated using Prime (Schrödinger Inc.).
Results
Thermal Stability of NKX3.1 Constructs As Determined by CD Spectroscopy
The thermal stabilities of the NKX3.1 homeodomain-containing constructs with increasing amounts of the N-terminal sequence were determined by measuring the CD signal at 222 nm to monitor the changes in α-helix content as a function of temperature. The CD spectra of the NKX3.1 (1–184), (75–184), (97–184), and (114–184) constructs are consistent with the homeodomain containing significant α-helical content and the N-terminal region consisting of flexible, disordered structure (Figure 1D), as already shown in previous NMR studies (6, 7). Panels E and F of Figure 1 show the 222 nm CD signal and estimated unfolded population as a function of temperature. NKX3.1 (114–184) constructs with and without the His tag sequence (see Materials and Methods) yielded similar spectra and the same unfolding transition temperature within error (±1 °C), and Figure 1 shows the results for all constructs with the His tag retained. To mimic the phosphorylation of acidic domain residues Thr89 and Thr93, NKX3.1 (75–184) constructs with these residues either singly or doubly mutated to glutamate were also assessed. The CD spectra for the single and double mutants are nearly the same as that of the wild-type NKX3.1 (75–184) construct, and for the sake of clarity, only the curves for the double mutant are shown in Figure 1. Table 1 summarizes the unfolding transition midpoint temperatures for all the constructs, including the single mutants.
Figure 1.
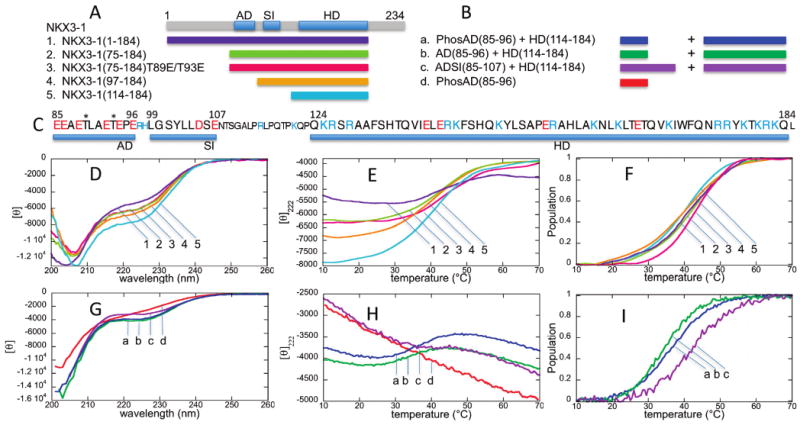
CD spectra and thermal unfolding curves. (A) NKX3.1 constructs (1–5) used for CD spectroscopy with a diagram showing the locations of the acidic domain (AD) and SRF-interacting (SI) motifs and homeodomain (HD). (B) Peptide mixtures (a–d) with NKX3.1 (114–184) used for CD spectroscopy. (C) NKX3.1 sequence highlighting the AD, SI, and HD sequences, shown in the larger font. Negatively charged residues are colored red and positively charged residues blue. The two threonine phosphorylation sites are denoted with asterisks. (D) CD spectra at 20 °C for NKX3.1 constructs (1–5). (E) Thermal unfolding curves at 222 nm for NKX3.1 constructs (1–5). (F) Unfolded population curves for NKX3.1 constructs (1–5) as a function of temperature. (G) CD spectra at 20 °C for NKX3.1/peptide mixtures (a–d). The molar ellipticity corresponds to the spectrum for the phosphorylated AD peptide; for the mixtures, the CD units are relative. (H) Thermal unfolding curves at 222 nm for NKX3.1/peptide mixtures (a–d). (I) Unfolded population curves for NKX3.1/peptide mixtures (a–c) as a function of temperature. The peptide alone (d) showed no unfolding transition.
Table 1.
Unfolding Transition Temperatures of NKX3.1 Constructsa
| NKX3.1 construct | temp (°C) | protein/peptide mixtureb | temp (°C) |
|---|---|---|---|
| 114–184 | 40 | 114–184/AD | 34 |
| 97–184 | 40 | 114–184/phospho-AD | 36 |
| 75–184 | 42 | 114–184/ADSI | 42 |
| 75–184-T89E | 43 | ||
| 75–184-T93E | 43 | ||
| 75–184-T89E/T93E | 44 | ||
| 1–184 | 41 |
All temperature values accurate to within approximately ±1 °C. The buffer for all samples consisted of 20 mM MES and 50 mM NaCl (pH 6.5).
In 100 μM NKX3.1 (114–184), 1.6 mM AD, phospho-AD, or ADSI peptide.
NKX3.1 (114–184), or the HD construct, and NKX3.1 (97–184), or the SI/HD construct, show the lowest unfolding transition temperature, 40 ± 1 °C. Addition of the AD motif to the NKX3.1 (75–184) construct provides a modest increase to 42 ± 1 °C; that of the single mutants increases to 43 ± 1 °C, and the double mutant has the highest transition temperature, 44 ± 1 °C. These increases in stability are small, barely greater than error, but they do support the notion that the AD influences the stability of the HD, and that AD phosphorylation should be expected to further influence HD stability.
To confirm the expected increase in stability due to interaction with phosphorylated AD, CD was performed on NKX3.1 (114–184) in the presence of the synthetic AD peptide with both threonines phosphorylated, and it was compared to the corresponding nonphosphorylated AD peptide. NKX3.1 has other potential CK2 phosphorylation sites beyond those in the AD (9), and in vitro phosphorylation of the NKX3.1 constructs was not pursued. The spectra and unfolding curves are shown in Figure 1G–I. The spectrum and unfolding curve for the phosphorylated AD peptide alone are also shown for reference. The CD results for the nonphosphorylated AD peptide alone were similar. The spectrum of the phosphorylated AD peptide alone, in particular how its signal at 222 nm becomes more negative as the temperature increases, is consistent with polyproline II secondary structure (16); however, additional structural characterization of the peptide, measuring HN–Hα J coupling and 1H–1H NOESY for instance, would be required to confirm that the ensemble average secondary structure lies in the polyproline II region of the Ramachandran map. Because of the high concentrations used, 100 μM NKX3.1 (114–184) and 1.6 mM peptide, a shorter, 0.2 mm cuvette was used, and the signals contain more noise due to scattering. The results are summarized in Table 1.
The phosphorylated AD peptide does provide a 2 °C increase in HD stability compared to the nonphosphorylated peptide, in agreement with the double mutant result given above. However, the actual unfolding transition temperatures, 34 ± 1 and 36 ± 1 °C, for the nonphosphorylated and phosphorylated peptides, respectively, are significantly lower than that for NKX3.1 (114–184) alone. Since previous NMR results suggested both the AD and SI regions might interact with the homeodomain (7), CD was also measured in the presence of a synthetic peptide containing both the AD and SI sequences. The resulting unfolding transition temperature (42 ± 1 °C) agrees with the value for the NKX3.1 (75–184) construct, indicating that both the AD and SI motifs are required for the stabilizing interactions with the homeodomain.
Interaction of the AD and SI Motifs with the Homeodomain by NMR
Figure 2A shows the overlaid 15N HSQC spectra of the 15N-labeled NKX3.1 (114–184) construct alone and in the presence of the phosphorylated AD peptide. Large chemical shift perturbations of up to 0.32 ppm are seen, evidence of specific interactions with the homeodomain. Figure 3A summarizes these chemical shift changes. Three of the largest changes, those of Ser150 and Arg176 backbone amide signals and the Arg175 Hε side chain signal, were monitored as a function of peptide concentration (Figure 2B), and a binding constant (Kd) of 0.5 ± 0.2 mM was determined by a least-squares fit of the data to the predicted binding curves (see Materials and Methods). Including additional residues did not alter the results or quality of the fit. Figure 2C shows the locations on the homeodomain of the resonances most highly perturbed by addition of the phosphorylated AD peptide. A spectrum was also acquired with 1.6 mM phosphorylated AD (data not shown) and displayed a pattern of additional perturbations not evident at lower concentrations, so this spectrum was not used for analysis. The cause of the anomalous perturbations is unclear; perhaps some additional mode of interaction occurs at higher peptide concentrations, though additional study would be needed to confirm this.
Figure 2.

NMR spectra titrated with the phosphorylated AD peptide. (A) Overlaid 2D 15N HSQC spectra of 100 μM NKX3.1 (114–184) without (red) and with 0.8 mM phosphorylated AD peptide (black). The resonances most perturbed by addition of phosphorylated AD are denoted with ovals. (B) Three largest chemical shift perturbations of the backbone amides of Ser150 (S150) and Arg176 (R176) and the side chain Arg175 Hε (R175Hε) as a function of phosphorylated AD peptide concentration. (C) Homeodomain locations of the amides most perturbed by the phosphorylated AD peptide.
Figure 3.
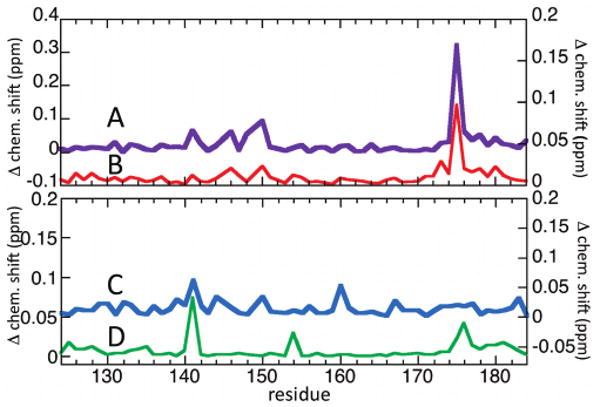
NKX3.1 HD amide chemical shift perturbations due to AD interactions. (A) NKX3.1 (114–184) with 0.8 mM phosphorylated AD peptide, with the y-axis on the left. (B) NKX3.1 (114–184) with 1.6 mM nonphosphorylated AD peptide, with the y-axis on the right. (C) NKX3.1 (114–184) plus 1.6 mM ADSI peptide, with the y-axis on the right. (D) HD perturbations in NKX3.1 (75–184) versus (114–184), with the y-axis on the left.
The experiment was repeated in the presence of the nonphosphorylated AD peptide (Figure 3B). The observed perturbations, while weaker (< 0.1 ppm), have a pattern quite similar to that of the phosphorylated AD perturbations, suggesting nonphosphorylated AD interacts specifically with the homeodomain in a similar manner, albeit with a weaker binding constant. No saturation of the chemical shift changes was apparent up to the maximum peptide concentration used, 1.6 mM, and therefore, the binding constant for nonphosphorylated AD must be weaker than this value.
The 2D 15N HSQC NMR spectra of two constructs, NKX3.1 (75–184) and NKX3.1 (114–184), were compared to identify the intramolecular interactions of the AD and SI motifs with the homeodomain (Figure 3C). The observed chemical shift changes are small, with only a few stronger than noise (∼0.02 ppm), implying either predominantly nonspecific interactions or weak specific interaction. The spectrum of the ADSI peptide with NKX3.1 (114–184) was also obtained (Figure 3D). Though not identical, some of the shift changes resemble the intramolecular changes (Figure 3C); they both show the largest change for the Arg141 side chain Hε signal, for example. The difference in the intramolecular and ADSI-induced shift perturbations compared to the AD-induced perturbations supports the CD result above showing different stabilization properties for the AD and ADSI peptides.
Modeling of the Interaction of the AD with the NKX3.1 HD
Model structures of possible complexes of the AD and phosphorylated AD peptides with the NKX3.1 HD were docked using the EMAP module of CHARMM (17), and molecular dynamics and minimization were performed using MacroModel (Schrödinger Inc.). The purpose of the models was twofold: to explore whether specific interactions, such as hydrogen bonds, might explain some of the observed chemical shift changes and to test whether the linker between the HD and AD is sufficiently long to accommodate the putative specific interactions.
Ten random peptide conformations of both the AD and the phosphorylated AD were docked individually in two orientations to the HD using EMAP (see Materials and Methods). The largest amide chemical shift changes due to AD interactions were observed for Arg175 side chain and Ser150 backbone amides, and in most of the docked complexes, hydrogen-bonded conformations between the peptide and Arg175 side chain were maintained during the unrestrained dynamics and minimization. They were not maintained for the backbone amide of Ser150, though hydrogen bonds with the side chain OH group were not uncommon. Panels A and B of Figure 4 show representative hypothetical models of the AD–HD and phosphorylated AD–HD complexes, respectively. Perhaps the significant chemical shift perturbation of the Ser150 backbone amide could be due to interaction with its side chain; another possibility is that the peptide disrupts transient N-capping interactions between Glu153 and Ser150 at the beginning of HD helix II (18).
Figure 4.
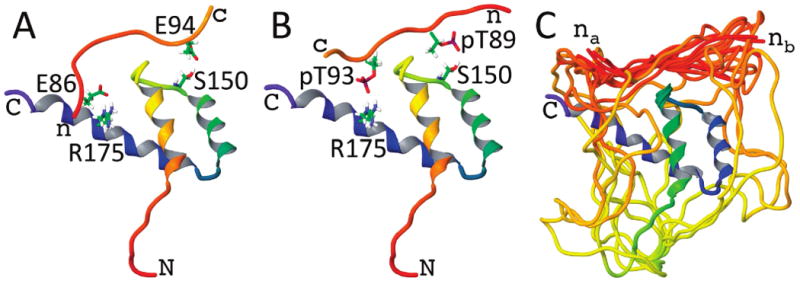
Example model structures of (A) the AD peptide and (B) the phospho-AD peptide docked to the NKX3.1 HD. The HD ribbons are shaded from red at the N-terminus to blue at the C-terminus. These two examples highlight possible hydrogen-bonded contacts between either glutamate or phosphothreonine side chains of the peptides and the Ser150 and Arg175 side chains of the HD. (C) The reconstructed linker residues between the AD and HD are shown for an ensemble of docked peptide model structures. The linker can accommodate the peptide docked with either the peptide N-terminus closer to Arg175 (nA) or closer to Ser150 (nB).
Figure 4C shows the ensemble of phosphorylated AD complexes with AD–HD linkers included. Regardless of whether the peptide N-terminus was closer to Ser150 or Arg175, there was no difficulty in generating the linker for any of the docked complexes. Were the linker region too short, this might help to explain the difference in the observed intra- versus intermolecular AD–HD chemical shift perturbations; however, the modeling clearly indicates that the linker is sufficiently long to accommodate the putative specific interactions.
Another possibility is that the peptide concentrations used in the NMR experiments (up to 1.6 mM) are not representative of the intramolecular interaction. Using polymer theory for flexible, disordered structure, an effective concentration of the covalently linked AD peptide relative to the HD can be approximated. The effective concentration is related to the probability of a polymer having zero end-to-end separation, given here using the wormlike chain model (19) in the long chain limit:
where n is the number of monomers in the chain (29 residues between the AD and HD), d is the monomer length (3.85 Å), and p is the persistence length, which is a measure reflecting how quickly the chain direction becomes randomized, and is taken here to be four monomer lengths. This effective intramolecular concentration is comparable to the peptide concentrations used for NMR. Neither linker length nor effective concentration can explain the observed differences between intra- and intermolecular AD–HD interactions, which further supports the CD and NMR results suggesting SI motif interactions directly influence AD–HD interactions.
Discussion
Biological Role of AD and SI Motifs
The CD measurements on NKX3.1 indicate that constructs containing the AD motif (residues 85–96) are stabilized a modest amount, by 2 °C relative to the HD alone. In contrast, mixing the AD peptide with the HD alone destabilizes the HD structure, but a peptide including both the AD and the following SI motif (residues 99–105) reproduces the 2 °C stabilization. Phosphorylation of the AD threonines or mutating them to glutamate further stabilizes the homeodomain an additional 2 °C. Other systems in which comparably small differences in thermal stability lead to biological consequences have been described. For instance, the R52H HD mutation found in the temperature sensitive eveID19 allele of the Drosophila even-skipped gene (20) is expected to yield a HD destabilization of 4 °C (18). Another example is the Y42H allele of human medium-chain acyl-CoA dehydrogenase, which shows lower thermal stability and activity that drops significantly for changes in temperature as small as 3 °C (21). The midpoint of the NKX3.1 HD unfolding transition, 40 °C, is not much higher than the human physiological temperature. Though conditions in cells differ from those in the CD samples, one might hypothesize that without additional stabilization, a significant population of unfolded HD could exist in cells, subject to the same ubiquitination and degradation processes that occur for misfolded and damaged proteins.
Another possible role of the AD and SI motif interactions is that they could hinder NKX3.1 HD ubiquitination by E3 ligases with specific affinity for the folded homeodomain. The E3 interactions could be hindered through steric interference, or perhaps through some mechanism involving charge neutralization of the positive HD (+12 total charge) by the negative AD (−6 and −8 for the nonphosphorylated and phosphorylated forms, respectively). In fact, six of the nine HD lysine side chains (lysines 142,147,169,178,180, and 182) lie at or near the site of AD peptide interaction identified by NMR. A potential candidate for such an E3 ligase has already been identified, TOPORS, which has been shown to ubiquitinate NKX3.1 in cells (22). In pull-down assays, TOPORS interacts with the NKX3.1 HD, and the sequences of NKX3.1 before and after the HD enhance this interaction. Whether AD phosphorylation interferes with TOPORS ubiquitination of NKX3.1 has not yet been tested.
The AD and SI motifs might also play a role in synergistic DNA binding along with other transcription factors. The N-terminal region is known to modulate the DNA binding behavior of NKX3.1 (23), and similar modulation has also been seen for the ultrabithorax homeodomain, which also contains a long, disordered N-terminal region (24). Gel shift retardation (EMSA) and ITC binding studies (Supporting Information) suggest that the AD and SI motif interactions might be responsible for the weaker DNA binding previously observed for the N-terminal NKX3.1 (1–184) construct (6). Physical interaction of the SI motif with SRF, a MADS box-containing transcription factor, contributes to synergistic activation of a reporter gene by SRF and NKX3.1 (7). The implication is that transcription factors could recruit NKX3.1 via favorable AD or SI interactions, competing with their homeodomain interactions, and thus could increase the strength of binding of NKX3.1 to adjacent DNA sites.
AD and SI Motifs in Other NK-2 Class Proteins
The NK-2 family acidic domain was first identified as a functional motif when it was observed that its deletion in Drosophila NK-2 led to a reduced level of inhibition of two downstream gene targets in transgenic fly embryos (25). Seven of the nine NK-2 class proteins in humans contain putative acidic domains (NKX2–1 and NKX2–4 do not), and four (NKX2–1, NKX2–4, NKX2–6, and NKX3.1) appear to have SI motifs. The putative AD regions occur anywhere from seven to 40 residues before the homeodomain and lack obvious sequence homology, which begs the question of how such variability can be reconciled with the apparent conservation of the AD motif in vertebrates and invertebrates. Part of the answer might lie in the intramolecular interaction and stabilization observed for NKX3.1. The negatively charged side chains of the AD could interact with the positively charged HD side chains, weakening their intra-HD mutual repulsion and thereby stabilizing the HD structure. A similar effect has been observed for the related NKX2–5 homeodomain with an increasing salt concentration, presumably due to screening of intra-HD charge repulsion (26). For charge screening, sequence conservation is not required as long as a region with a sufficient number of negatively charged residues is maintained near the HD. Perhaps the AD is evolutionarily maintained N-terminal to the HD so that the AD is already available to stabilize the HD as soon as it emerges from the ribosome.
It is not known whether the acidic domains of other NK-2 class proteins are phosphorylated like NKX3.1 is by CK2; however, the acidic domains of six of the seven human AD-containing NK-2 class proteins (excluding NKX2–6) do contain consensus CK2 phosphorylation sequences (S/TXXE/D) (27), as does Drosophila NK-2, so it is possible that CK2 phosphorylation might play a regulatory role for these proteins as well.
In conclusion, the results here suggest that the biological role of phosphorylation of the acidic domain, namely, its correlation with NKX3.1 protein levels (9), could be related to the observed physical interaction of the AD and SI motifs with the homeodomain. The AD and SI regions appear to be fully flexible by NMR, even when interacting with the homeodomain (7), a behavior similar to that observed for phosphorylated CDK inhibitor Sic1 interacting with Cdc4 (28). The observed interactions were weak, with effective binding constants no stronger than 0.5 mM; however, because the interacting domains are covalently linked, their effective concentration for interaction is predicted to be in the millimolar range. Because of their high effective concentrations, these weak intramolecular interactions could effectively compete with submicromolar binding constants and concentrations typical of intermolecular interactions inside cells. Of course, the biological mechanisms suggested in the discussion above are hypothetical, and additional experimental studies will be needed to establish which, if any, actually play significant biological roles. The percentage of eukaryotic proteins with intrinsically disordered regions has been predicted to range from 36 to 63% (29); thus, weak intraprotein interactions involving flexible motifs of the type observed for NKX3.1 could possibly play important roles in a significant percentage of all eukaryotic proteins.
Supplementary Material
Footnotes
This work was supported in part by the Intramural Research Program of the National Heart, Lung and Blood Institute and by National Institute of Environmental Health Sciences Grant ES09888 to E.P.G.
Abbreviations: CD, circular dichroism; NMR, nuclear magnetic resonance; HD, homeodomain; AD, acidic domain; phospho-AD, phosphorylated AD peptide; SRF, serum response factor; SI motif, SRF interacting motif; CK2, casein kinase II; ITC, isothermal calorimetry.
Supporting Information Available Additional analysis of previously published EMSA data and ITC data of NKX3.1 DNA binding. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Harvey RP. NK-2 homeobox genes and heart development. Dev Biol. 1996;178:203–216. doi: 10.1006/dbio.1996.0212. [DOI] [PubMed] [Google Scholar]
- 2.Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Young P, Norton CR, Gridley T, Cardiff RD, Cunha GR, Abate-Shen C, Shen MM. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966–977. doi: 10.1101/gad.13.8.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vocke CD, Pozzatti RO, Bostwick DG, Florence CD, Jennings SB, Strup SE, Duray PH, Liotta LA, EmmertBuck MR, Linehan WM. Analysis of 99 microdissected prostate carcinomas reveals a high frequency of allelic loss on chromosome 8p21-22. Cancer Res. 1996;56:2411–2416. [PubMed] [Google Scholar]
- 4.Swalwell JI, Vocke CD, Yang Y, Walker JR, Grouse L, Myers SH, Gillespie JW, Bostwick DG, Duray PH, Linehan WM, Emmert-Buck MR. Determination of a minimal deletion interval on chromosome band 8p21 in sporadic prostate cancer. Genes, Chromosomes Cancer. 2002;33:201–205. doi: 10.1002/gcc.10015. [DOI] [PubMed] [Google Scholar]
- 5.Asatiani E, Huang WX, Wang A, Ortner ER, Cavalli LR, Haddad BR, Gelmann EP. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005;65:1164–1173. doi: 10.1158/0008-5472.CAN-04-2688. [DOI] [PubMed] [Google Scholar]
- 6.Zheng SL, Ju JH, Chang BL, Ortner E, Sun JL, Isaacs SD, Sun JS, Wiley KE, Liu WN, Zemedkun M, Walsh PC, Ferretti J, Gruschus J, Isaacs WB, Gelmann EP, Xu JF. Germ-line mutation of NKX3.1 cosegregates with hereditary prostate cancer and alters the homeodomain structure and function. Cancer Res. 2006;66:69–77. doi: 10.1158/0008-5472.CAN-05-1550. [DOI] [PubMed] [Google Scholar]
- 7.Ju JH, Maeng JS, Zemedkun M, Ahronovitz N, Mack JW, Ferretti JA, Gelmann EP, Gruschus JM. Physical and functional interactions between the prostate suppressor homeoprotein NKX3.1 and serum response factor. J Mol Biol. 2006;360:989–999. doi: 10.1016/j.jmb.2006.05.064. [DOI] [PubMed] [Google Scholar]
- 8.Schwarzinger S, Kroon GJA, Foss TR, Wright PE, Dyson HJ. Random coil chemical shifts in acidic 8 M urea: Implementation of random coil shift data in NMRView. J Biomol NMR. 2000;18:43–48. doi: 10.1023/a:1008386816521. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Guan B, Maghami S, Bieberich CJ. NKX3.1 is regulated by protein kinase CK2 in prostate tumor cells. Mol Cell Biol. 2006;26:3008–3017. doi: 10.1128/MCB.26.8.3008-3017.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugasi K, Dyson JH, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez M, Weiler S, Ferretti JA, Ginsburg A. The vnd/NK-2 homeodomain: Thermodynamics of reversible unfolding and DNA binding for wild-type and with residue replacements H52R and H52R/T56W in helix III. Biochemistry. 2001;40:4923–4931. doi: 10.1021/bi0022341. [DOI] [PubMed] [Google Scholar]
- 12.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 13.Gruschus JM, Tsao DH, Wang LH, Nirenberg M, Ferretti JA. Interactions of the vnd/NK-2 homeodomain with DNA by nuclear magnetic resonance spectroscopy: Basis of binding specificity. Biochemistry. 1997;36:5372–5380. doi: 10.1021/bi9620060. [DOI] [PubMed] [Google Scholar]
- 14.Wu XW, Milne JL, Borgnia MJ, Rostapshov AV, Subramaniam S, Brooks BR. A core-weighted fitting method for docking atomic structures into low-resolution maps: Application to cryo-electron microscopy. J Struct Biol. 2003;141:63–76. doi: 10.1016/s1047-8477(02)00570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu XW, Brooks BR. Modeling of Macromolecular assemblies with map objects. Proceedings of 2007 BIOCOMP; Las Vegas. 2007. pp. 411–417. [Google Scholar]
- 16.Chen K, Liu ZG, Zhou CH, Shi ZS, Kallenbach NR. Neighbor effect on PPII conformation in alanine peptides. J Am Chem Soc. 2005;127:10146–10147. doi: 10.1021/ja052094o. [DOI] [PubMed] [Google Scholar]
- 17.Lee DY, Park SJ, Jeong W, Sung HJ, Oho T, Wu XW, Rhee SG, Gruschus JM. Mutagenesis and modeling of the peroxiredoxin (Prx) complex with the NMR structure of ATP-bound human sulfiredoxin implicate aspartate 187 of Prx I as the catalytic residue in ATP hydrolysis. Biochemistry. 2006;45:15301–15309. doi: 10.1021/bi061824h. [DOI] [PubMed] [Google Scholar]
- 18.Weiler S, Gruschus JM, Tsao DHH, Yu L, Wang LH, Nirenberg M, Ferretti JA. Site-directed mutations in the vnd/NK-2 homeodomain: Basis of variations in structure and sequence-specific DNA binding. J Biol Chem. 1998;273:10994–11000. doi: 10.1074/jbc.273.18.10994. [DOI] [PubMed] [Google Scholar]
- 19.Gobush W, Yamakawa H, Stockmayer WH, Magee WS. Statistical mechanics of wormlike chains. I. Asymptotic behavior. J Chem Phys. 1972;57:2839–2843. [Google Scholar]
- 20.Doe CQ, Smouse D, Goodman CS. Control of Neuronal Fate by the Drosophila Segmentation Gene Even-Skipped. Nature. 1988;333:376–378. doi: 10.1038/333376a0. [DOI] [PubMed] [Google Scholar]
- 21.O'Reilly L, Bross P, Corydon TJ, Olpin SE, Hansen J, Kenney JM, McCandless SE, Frazier DM, Winter V, Gregersen N, Engel PC, Andresen BS. The Y42H mutation in medium-chain acyl-CoA dehydrogenase, which is prevalent in babies identified by MS/MS-based newborn screening, is temperature sensitive. Eur J Biochem. 2004;271:4053–4063. doi: 10.1111/j.1432-1033.2004.04343.x. [DOI] [PubMed] [Google Scholar]
- 22.Guan B, Pungaliya P, Li X, Uquillas C, Mutton LN, Rubin EH, Bieberich CJ. Ubiquitination by TOPORS regulates the prostate tumor suppressor NKX3.1. J Biol Chem. 2008;238:4834–4840. doi: 10.1074/jbc.M708630200. [DOI] [PubMed] [Google Scholar]
- 23.Gelmann EP, Steadman DJ, Ma J, Ahronovitz N, Voeller HJ, Swope S, Abbaszadegan M, Brown KM, Strand K, Hayes RB, Stampfer MJ. Occurrence of NKX3.1 C154T polymorphism in men with and without prostate cancer and studies of its effect on protein function. Cancer Res. 2002;62:2654–2659. [PubMed] [Google Scholar]
- 24.Liu Y, Matthews KS, Bondos SE. Multiple intrinsically disordered sequences alter DNA binding by the homeodomain of the Drosophila Hox protein ultrabithorax. J Biol Chem. 2008;283:20874–20887. doi: 10.1074/jbc.M800375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koizumi K, Lintas C, Nirenberg M, Maeng JS, Ju JH, Mack JW, Gruschus JM, Odenwald WF, Ferretti JA. Mutations that affect the ability of the vnd/NK-2 homeoprotein to regulate gene expression: Transgenic alterations and tertiary structure. Proc Natl Acad Sci U S A. 2003;100:3119–3124. doi: 10.1073/pnas.0438043100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fodor E, Mack JW, Maeng JS, Ju JH, Lee HS, Gruschus JM, Ferretti JA, Ginsburg A. Cardiac-specific Nkx2.5 homeodomain: Conformational stability and specific DNA binding of Nkx2.5(C56S) Biochemistry. 2005;44:12480–12490. doi: 10.1021/bi050835s. [DOI] [PubMed] [Google Scholar]
- 27.Pearson RB, Kemp BE. Protein Kinase Phosphorylation Site Sequences and Consensus Specificity Motifs: Tabulations. Methods Enzymol. 1991;200:62–81. doi: 10.1016/0076-6879(91)00127-i. [DOI] [PubMed] [Google Scholar]
- 28.Mittag T, Orlicky S, Choy WY, Tang XJ, Lin H, Sicheri F, Kay LE, Tyers M, Forman-Kay JD. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci U S A. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.