

Abstract
C-1027 is a chromoprotein antitumor antibiotic consisting of an apo-protein and the C-1027 chromophore. The C-1027 chromophore possesses four distinct structural moieties – an enediyne core, a deoxy aminosugar, a benzoxazolinate, and an (S)-3-chloro-5-hydroxy-β-tyrosine – the latter two of which are proposed to be appended to the enediyne core via a convergent biosynthetic strategy. Here we report the in vitro characterization of SgcF, an epoxide hydrolase from the C-1027 biosynthetic gene cluster that catalyzes regio- and stereospecific hydrolysis of styrene oxide, serving as an enediyne core epoxide intermediate mimic, to form a vicinal diol. Abolishment of C-1027 production in the ΔsgcF mutant strain Streptomyces globisporus SB1010 unambiguously establishes that sgcF plays an indispensable role in C-1027 biosynthesis. SgcF efficiently hydrolyzes (S)-styrene oxide, displaying an apparent Km of 0.6 ± 0.1 mM and kcat of 48 ± 1 min−1, via attack at the α-position to exclusively generate the (R)-phenyl vicinal diol, consistent with the stereochemistry of the C-1027 chromophore. These findings support the role of SgcF in the proposed convergent pathway for C-1027 biosynthesis, unveiling an (R)-vicinal diol as a key intermediate. Interestingly, SgcF can also hydrolyze (R)-styrene oxide to afford preferentially the (R)-phenyl vicinal diol via attack at the β-position, albeit with significantly reduced efficiency (apparent Km of 2.0 ± 0.4 mM and kcat = 4.3 ± 0.3 min−1). Although the latter activity unlikely contributes to C-1027 biosynthesis in vivo, such enantioconvergence arising from complementary regioselective hydrolysis of a racemic substrate could be exploited to engineer epoxide hydrolases with improved regio- and/or enantiospecificity.
Keywords: C-1027, enediyne, epoxide hydrolase, SgcF, Streptomyces globisporus
Introduction
The enediynes are some of the most cytotoxic molecules known to date, and two members of this family, calicheamicin and neocarzinostatin (NCS), are currently in clinical use as anticancer drugs.1, 2 The enediyne cores of molecules such as C-1027 and NCS possess multiple vicinal diols (Figure 1), to which the various peripheral moieties are attached to impart the distinct characteristics of each enediyne antitumor antibiotic. These functional groups may be envisaged to originate from epoxide precursors that undergo hydrolytic ring-opening catalyzed by epoxide hydrolases (EHs).
Figure 1.

The structures of C-1027 and neocarzinostatin, with vicinal diol moieties (shaded) predicted to be derived by epoxide hydrolases from enediyne core epoxide precursors.
The ubiquity of EHs in nature is evidenced by their widespread occurrence in mammals, plants, insects, and various microorganisms such yeast, fungi, and bacteria.3–6 Most EHs belong to the α/β-hydrolase superfamily,7–9 and crystallographic studies have confirmed the presence of this structural fold in several EHs.10–17 Detailed analysis of available sequence and structural data has revealed that EHs possess a modular architecture consisting of three conserved and three variable regions (Figure S1, Supporting Information).18, 19 The conserved regions include the N- and C-terminal catalytic regions that together make up the core catalytic domain of the α/β-hydrolase fold, as well as the lid or cap domain. The variable regions include: (i) an N-terminal region that may include a membrane anchor or may be missing altogether as in most bacterial EHs; (ii) an NC-loop linking the lid and the N-terminal catalytic domain; and (iii) a variable loop in the lid domain (cap-loop). The active site is located at the interface between the core and lid domains and contains five residues that are absolutely conserved among EHs – the nucleophile (Asp or Glu) and His of the nucleophile-His-acid triad (the acid residue of which is variable in its position), the 2 residues of the oxyanion hole (X of the HGXP motif, and the residue following the catalytic nucleophile), and a conserved Tyr in the 5th helix of the lid domain (i.e., α8 in Figure S2). Although most EHs possess a second Tyr in the 1st helix of the lid domain (i.e., α4 in Figure S2), this residue is not always conserved.19
The conserved structure and active site residues reflect a common mechanism for epoxide hydrolysis (Figure 2). The two tyrosine residues in the lid domain form hydrogen bonds to the oxirane oxygen atom,10, 20–22 thereby activating the epoxide to be attacked by the nucleophilic Asp/Glu residue of the catalytic triad to yield a covalent alkyl-enzyme intermediate.23–25 A water molecule, activated by a conserved His-Asp charge relay system, hydrolyzes the monoester intermediate to afford the vicinal diol as a product. The transient oxyanion tetrahedral intermediate is stabilized via hydrogen bonds to the backbone amide hydrogens of the oxyanion hole residues.15–17, 26
Figure 2.

The proposed mechanism of epoxide hydrolases. The catalytic triad and the oxygen atom from water are shown in red, and the active site residues reflect those of AnEH from Aspergillus niger.17
Although mammalian EHs have been studied for decades for their roles in xenobiotic detoxification27, 28 and other physiological functions,29 microbial EHs have recently become attractive for biocatalytic applications due to their regio- and enantio-selectivity and solubility,3, 4, 30, 31 and little is known about their roles in natural product biosynthesis. For an asymmetric epoxide such as styrene oxide, microbial EHs with various regio- and enantioselectivity have been reported. Most microbial EHs preferentially hydrolyze one enantiomer of an epoxide racemate, and thereby may be utilized to generate enantiopure epoxides or diols via kinetic resolution. Interestingly, a few EHs have been reported to catalyze the hydrolysis of both enantiomers to form enantiomerically-enriched diol products.32 This enantioconvergent process occurs by means of complementary enantio- and regioselective activities, whereby nucleophilic attack may occur at either epoxide carbon.
We have been studying the biosynthesis of the 9-membered enediynes C-1027, NCS, and maduropeptin as model systems to gain fundamental insight into the biochemical logic of enediyne biosynthesis. The biosynthetic gene clusters of each have been cloned, the involvement of which in enediyne biosynthesis has been confirmed by in vivo inactivation of selected genes within each cluster,33–35 and in vivo and in vitro characterizations have unveiled numerous examples of unprecedented biochemistry and enzymology,36–41 including a convergent biosynthetic strategy for this family of metabolites featuring vicinal diol intermediates (Figure 3). Sequence analysis indeed revealed one gene, sgcF, from the C-1027 biosynthetic gene cluster and two genes, ncsF1 and ncsF2, from the NCS biosynthetic gene clusters, respectively, whose deduced gene products show high amino acid homology to known microbial EHs, including the Asp-His-Asp catalytic triad (Figure S2). These findings are consistent with the proposed biosynthetic pathways, presenting outstanding opportunities to study the regio- and enantioselectivity of EHs involved in natural product biosynthesis.
Figure 3.

The proposed biosynthetic pathway for C-1027 involving an SgcF-catalyzed formation of a (R)-vicinal diol intermediate from a (S)-enediyne core epoxide precursor. The oxygen atom from water is shown in red.
Here we report the in vitro characterization of SgcF as an EH, supporting an (R)-vicinal diol as a key intermediate for C-1027 biosynthesis. SgcF efficiently catalyzes regio- and enantiospecific hydrolysis of (S)-styrene oxide, which serves as an enediyne core epoxide mimic, forming a covalent alkyl-enzyme intermediate at C-1 followed by hydrolysis to afford the (R)-phenyl vicinal diol exclusively. Interestingly, SgcF also hydrolyzes (R)-styrene oxide, albeit with significantly reduced efficiency, and regioselective formation of the monoester intermediate at the C-2 position also yields the (R)-phenyl vicinal diol as a preferred product. These data showed that SgcF catalyzes the net trans-addition of H2O to (S)-styrene oxide in a Markovnikov manner but followed the opposite stereochemical course for (R)-styrene oxide in an anti-Markovnikov manner. Given the critical role EHs play in the biosynthesis of various natural products, including the enediynes, such enantioconvergence arising from complementary regioselective hydrolysis of a racemic substrate could be exploited to engineer epoxide hydrolases with improved regio- and/or enantiospecificity to increase natural product structural diversity.
Experimental
Strains, Plasmids and Cosmids
Escherichia coli DH5α and E. coli BL21(DE3) (Novagen, Madison, WI) were used as hosts for subcloning and heterologous expression, respectively, E. coli ET12567/pUZ8002 was used as the donor strain for conjugation42, and Streptomyces globisporus wild-type strain (C-1027-producing) has been described previously43. Plasmid pCDF-2 Ek/LIC was from Novagen, and pBS1005 was described previously43. SuperCos1 was from Stratagene (La Jolla, CA).
DNA Isolation and Manipulation
Plasmid preparation was carried out using commercial kits (Qiagen, Santa Clarita, CA). General procedures for DNA restriction digests, ligations and genetic manipulations in E. coli were performed according to standard protocols. E. coli-Streptomyces conjugation to introduce DNA into S. globisporus was performed according to the standard procedure44 with the following modifications. S. globisporus spores were suspended in TSB medium and heat-shocked at 50 °C for 10 min, followed by incubation at 30 °C for 6 hr. Germinated spores were mixed with E. coli ET12567/pUZ8002 cells and spread onto ISP4 plates freshly supplemented with 20 mM MgCl2.
Chemicals and Instruments
Substrates including racemic styrene oxide, (R)-styrene oxide, and (S)-styrene oxide, products including racemic 1-phenyl-1,2-ethanediol, (R)-1-phenyl-1,2-ethanediol, and (S)-1-phenyl-1,2-ethanediol, and [18O]-H2O were purchased from Sigma-Aldrich (St. Louis, MO). Dithiothreitol (DTT) was purchased from Research Products International Corp (Mt. Prospect, IL). Complete protease inhibitor tablet, EDTA-free, was from Roche Applied Science (Indianapolis, IN). Medium components and buffers were from Fisher Scientific (Pittsburgh, PA). Synthetic DNA oligonucleotides were purchased from the University of Wisconsin-Madison Biotechnology Center (Madison, WI). PCR was performed with a PerkinElmer GeneAmp 2400. Electrospray ionization-mass spectroscopy (ESI-MS) was performed with an Agilent 1100 MSD SL ion trap mass spectrometer (Agilent Technologies, Inc. Santa Clara, CA). NMR spectra were recorded using a Varian UI-500 spectrometer (Varian, Inc., Palo Alto, CA). High performance liquid chromatography (HPLC) analyses were carried out on a Varian HPLC system equipped with Prostar 210 pumps, a photodiode array (PDA) detector, and an Alltech Appolo C18 reverse phase column (5 μm, 4.6 × 250 mm, Grace Davison Discovery Sciences, Deerfield, IL), using a 20 min linear gradient from 0 to 60% acetonitrile in H2O. The enantiomeric separation was performed on a Waters HPLC system equipped with 600 pumps, a 996 PDA detector, and a Chiralcel OD-H chiral column (5 μm, 4.6 × 250 mm, Grace Davison Discovery Sciences) using a 60 min isocratic elution with 2.5% isopropanol in n-hexane.
Inactivation of SGCF
The sgcF gene was inactivated by the λ-red mediated PCR-targeting method according to the literature protocol.45 Since the backbone of pBS1005 is pOJ446,43 which carries the replication origin of plasmid SCP2*, a 19-kb XbaI fragment from pBS1005 was subcloned into the same site of SuperCos1 to afford pBS1094. The apramycin (Apr) resistance gene aac(3)IV/oriT cassette was amplified by using the following pair of primers: sgcFF1: 5 ′-CGCGCCGCCCCGCTCCCACAATCACGAGGGTGGATTCACTATTCCGGGGATCCGTCGACC -3′/sgcFR1: 5′-TCCGGCCGACGGGCGCTCCACTTCGTATGTGCCCTACTGGTTGTAGGCTGGAGCTGCTTC - 3′ (underlined letters represent the oligonucleotide homologous to the flanking DNA regions of sgcF). The resulting PCR product was introduced into E. coli BW25113/pIJ790 harboring pBS1094 by electroporation, yielding the ΔsgcF mutated cosmid pBS1095 through λ-red mediated homologous recombination. Cosmid pBS1095 was then introduced into S. globisporus by E. coli-Streptomyces conjugation. Exconjugants that were apramycin resistant and kanamycin sensitive were selected as double crossover mutants and named SB1010, the genotype of which was confirmed by PCR using the following pair of primers: sgcFF1: 5′-CCGGGGACGGTCAATCTAG- 3′/sgcFR1: 5′-GCTCCCACAATCACGAGG- 3′.
C-1027 Production, Isolation and Analysis
Fermentation of S. globisporus wild-type and the ΔsgcF mutant strain SB1010 for C-1027 production was carried out using A9 medium as described previously.46 Briefly, a spore suspension of the S. globisporus wild-type or recombinant strain was inoculated into 50 mL A9 medium in a 250 mL baffled flask and incubated at 28 °C, 250 rpm for 2 days. Five mL of the resultant seed culture was then used to inoculate 50 mL A9 medium in 250 mL baffled flasks, and incubation continued at 28 °C, 250 rpm for 5 days. The culture was centrifuged (4,000 rpm, 4 °C, 10 min) to collect the supernatant. This supernatant was first adjusted to pH 4.0 with 1 N HCl followed by centrifugation (4,000 rpm, 4 °C, 8 min). The resultant supernatant was then added (NH4)2SO4 to 70% saturation and re-adjusted to pH 4.0 to precipitate the C-1027 chromoprotein complex. The precipitated C-1027 chromoprotein complex was finally collected by centrifugation (4,150 rpm, 4 °C, 20 min) and was extracted with acetone to isolate the C-1027 chromophore, which was subjected to HPLC analysis immediately.
The HPLC analysis was carried out on a C18 column (5 μm, 250 mm × 4.6 mm, Alltech, Lexington, KY). The column was equilibrated with 25% solvent A-75% solvent B and eluted with 25% solvent A-75% solvent B for 10 min followed by a linear gradient to 10% solvent A-90% solvent B in 15 min at a flow rate of 1 mL/min and with UV detection at 350 nm (solvent A, 10 mM phosphate buffer, pH 7.2 and solvent B, acetonitrile). Bioassay against Micrococcus luteus ATCC 9431 was performed as described.43
Cloning, Overproduction, and Purification of SgcF
The sgcF gene was amplified by PCR from cosmid pBS100533, 43 using Platinum Pfx polymerase (Invitrogen, Carlsbad, CA) with the following primers: forward 5′-GAC GAC GAC AAG ATG CGT CCC TTC CGT ATC GA -3′ and reverse 5′-GAG GAG AAG CCC GG TCA GCG GAG CGG AGG GT -3′ (starting and stop codons underlined). The gel-purified PCR products were cloned into the pCDF-2 Ek/LIC vector using ligation-independent cloning as described by Novagen (Madison, WI) to give pBS1096 for E. coli expression. Overproduction in E. coli BL21 (DE3) and purification of SgcF by affinity chromatography using an NTA-Ni agarose column (Qiagen, Valencia, CA) were performed following the previously described procedure.37, 38 The purified SgcF was desalted using a PD-10 column (GE Healthcare, Piscataway, NJ) twice to completely remove glycerol, concentrated with a 10 K MWCO Vivaspin ultrafiltration device (Sartorius, Edgewood, NY), and stored at −80 °C in 100 μL aliquots. The purity of SgcF was assessed by SDS-PAGE on a 12% gel, and its concentration was determined from the absorbance at 280 nm using a molar absorptivity (ε= 96.9 mM−1 cm−1) calculated using the program Protparam (http://www.expasy.ch/tools/protparam.html).
Activity Assay of SgcF
HPLC-based assays were carried out in 200-μL reaction mixture containing 2 mM racemic styrene oxide and 50 mM phosphate buffer (pH 7.5). The reaction was initiated by the addition of 100 μM SgcF and incubated at 28 °C for 90 min. The reaction was quenched by extracting with ethyl acetate (3 × 200 μL). After the solvent was removed using a speed-vac, the resulting residue was dissolved in 50 μL of acetonitrile, 25 μL of which was subjected to HPLC analysis. Control reactions were carried out under the identical conditions except with SgcF that had been boiled for 5 min.
To determine the kinetic parameters of SgcF-catalyzed hydrolysis of styrene oxide, a spectrophotometric assay method was adopted to achieve continuous and accurate determination of epoxide hydrolase activity.47, 48 Prior to kinetic analysis, the optimal pH for SgcF activity was tested in three different buffers − 50 mM sodium acetate (pH 5.0, 5.5, and 6.0), 50 mM sodium phosphate buffer (pH 5.5 to pH 8.5), and 50 mM glycine-NaOH (pH 8.5, 9.0, and 10.0). The reactions were carried out in 1-mL reaction mixture containing 10 μL of 300 mM sodium periodate in DMF, 20 μL of 5 M styrene oxide in DMSO, and 50 mM buffer. The reactions were initiated by the addition of 10 μM SgcF and carried out in triplicate. The absorbance at 290 nm was monitored in a 1-mL quartz cell, and the velocity was calculated based on the rate of change of absorbance over several minutes. Steady-state kinetic parameters were obtained from reactions carried out in 50 mM phosphate buffer (pH 8.0) with varying styrene oxide concentrations [0.10 to 6.4 mM for (S)-styrene oxide and 1.0 to 24 mM for (R)-styrene oxide]. The assays were initiated by addition of SgcF [3.2 μM for (S)-styrene oxide and 9.6 μM for (R)-styrene oxide] and carried out in triplicate. The velocity was determined based on the change of the absorbance at 290 nm over several minutes. The Michaelis-Menten equation was fitted to plots of velocity of 1-phenyl-1,2-ethanediol formation versus substrate concentration to extract values for Km and kcat.
Large-scale Reaction
To prepare sufficient quantities of product for NMR characterization, the reaction was carried out in 1-mL reaction mixture containing 10 mM styrene oxide and 200 μM SgcF in 50 mM phosphate buffer (pH 8.0). During the course of the reaction, 10 mM styrene oxide and 100 μM SgcF were added every hour. The reaction was allowed to proceed at 25 °C for 8 h, after which the products were extracted from the reaction mixture with ethyl acetate (5 × 0.5 mL). The solvent was removed by evaporation under reduced pressure. Purification by a flash column chromatography finally yielded the phenyl-1, 2-ethanediol product for NMR analysis and chiral analyses. [18O]-labeled products were obtained following a similar procedure. The reactions were carried out in 0.5-mL reaction mixtures containing 80% [18O]-H2O. For (S)-styrene oxide, 5 mM substrate and 20 μM SgcF were added every 30 min. For (R)-styrene oxide, 20 mM substrate and 75 μM SgcF were added every hour.
Results
Bioinformatics Analysis
Close examination of the genes within the C-1027 biosynthetic gene cluster revealed a single gene, sgcF, whose deduced gene product showed high amino acid sequence homology to known microbial EHs.33 Most EHs belong to the α/β-hydrolase superfamily of proteins that share this common structural fold. SgcF contains several motifs that are conserved among this family of EHs: (i) the “nucleophilic elbow” motif Sm-X-Nu-X-Sm-Sm containing the Asp175 nucleophile (GGD175WGK) that functions as a member of the nucleophile-His-acid catalytic triad (D175, D336, H363); (ii) the oxyanion hole HGXP motif (HGW99P); (iii) and the G-X-Sm-X-S/T motif 18, 19, 49 (G137YGFS) that has been proposed to stabilize the interface between the lid and core domains,18 and (iv) the conserved Tyr304 in helix 5 of the lid domain. Interestingly, the second Tyr in helix 1 of the lid domain (helix α4 in Figure S2) has been changed to Trp236. Taken together, the bioinformatic analysis predicts that SgcF is an EH responsible for the hydrolysis of an enediyne intermediate bearing an oxirane ring.
Inactivation of SGCF
The ΔsgcF mutant strain SB1010 completely lost its ability to produce C-1027. Inactivation of sgcF in S. globisporus was accomplished by following the λ-red-mediated PCR-targeting method to replace the entire sgcF gene with the aac(3)IV/oriT cassette, affording the ΔsgcF mutant strain SB1010 (Figure S3A and S3B). Bioassays using M. luteus as a reference strain showed that the production of C-1027 was totally abolished in SB1010 either cultured on solid (ISP4) (Figure S3C) or in liquid (A9) media (Figure S3D). It was further confirmed by HPLC analysis that no C-1027 was produced by SB1010 when fermented in A9 media for 5 days (Figure S3E), indicating that sgcF is indispensable for C-1027 biosynthesis.
Overproduction and Purification of SgcF
The sgcF gene was amplified from the cosmid pBS100533, 43 and cloned into pCDF-2 Ek/LIC. After overproduction in E. coli BL21 (DE3), SgcF (~70 mg/L) was purified to homogeneity as an N-terminal His6-tagged fusion protein using NTA-Ni affinity column chromatography. SDS-PAGE showed a single protein band consistent with the predicted molecular weight of SgcF (44.5 KDa) (Figure S4).
Activity Assay for SgcF
Due to the unavailability of the oxirane-containing enediyne core that is predicted to be the substrate for SgcF, styrene oxide was chosen as a substrate mimic with which to test SgcF activity (Figure 3). The HPLC-based assay was carried out as described in the Experimental Section, and a single peak was eluted with a retention time identical to that of authentic 1-phenyl-1,2-ethanediol (Figure 4). The new peak was collected and analyzed by ESI-MS, yielding a [M + Na]+ ion at m/z 161.2 (calcd [M + Na]+ ion for molecular formula C8H10O2 is 161.1). The absence of a styrene oxide peak in the HPLC chromatogram was attributed to its evaporation under reduced pressure, as indicated by the control experiments performed without enzyme.
Figure 4.
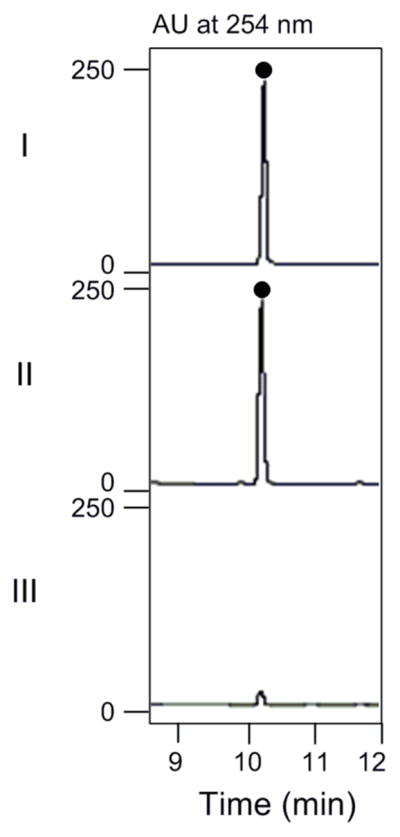
HPLC profiles for SgcF-catalyzed hydrolysis of racemic styrene oxide: (I) 1-phenyl-1,2-ethanediol standard; (II) with 100 μM SgcF for 90 min; and (III) with boiled SgcF for 90 min.
Optimization of SgcF Activity
Prior to kinetic analysis, the pH dependence of SgcF-catalyzed hydrolysis was examined. The pH profile exhibited a bell-shaped curve between pH 6.0 to 10.0 with an optimal activity at pH 8.0 (Figure S5A). As a result, all subsequent assays were performed in 50 mM phosphate buffer at pH 8.0. The reactions with different concentrations of SgcF showed that the SgcF-catalyzed hydrolysis of styrene oxide is enzyme-dependent (Figure S5B).
Enantioselectivity of SgcF
Because most EH-catalyzed reactions exhibit stereo- and/or regio-selectivity, the enantioselectivity of SgcF was investigated using enantiomerically pure (R)- and (S)-styrene oxide substrates. A spectrophotometric assay was adopted to continuously monitor the formation of 1-phenyl-1,2-ethanediol,47, 48 thereby determining the steady-state kinetic constants for each enantiomer (Figures 5A and 5B). A plot of initial velocity versus the concentration of (S)-styrene oxide displayed Michaelis-Menten kinetics yielding a Km of 0.6 ± 0.1 mM and kcat of 48 ± 1 min−1, while assays with variable (R)-styrene oxide gave a Km of 2.0 ± 0.4 mM and kcat of 4.3 ± 0.3 min−1, respectively. Thus, SgcF preferentially hydrolyzes (S)-styene oxide with a 37-fold greater specificity constant (E = 37), implicating an (S)-epoxide enediyne intermediate in C-1027 biosynthesis (Figure 3).
Figure 5.

Kinetic analysis of SgcF-catalyzed hydrolysis of (S)- and (R)-styrene oxide showing single substrate kinetic plots for (A) (S)-styrene oxide and (B) (R)-styrene oxide.
Stereochemistry of the Vicinal Diol Product
To determine the stereochemistry of the product, SgcF-catalyzed hydrolysis of racemic styrene oxide was performed on a large scale (1 mL) and the reaction was quenched by extraction of ethyl acetate when the reaction was complete as judged by thin layer chromotography. After ethyl acetate extraction and complete removal of solvent, 1H NMR analysis in CDCl3 confirmed the product as 1-phenyl-1,2-ethanediol by comparison to the authentic standard. Specifically, five protons at δ 7.3–7.4 ppm correspond to the hydrogens of the mono-substituted benzene ring, a doublet-doublet signal (δ 4.90 ppm, J = 8.1, 3.4 Hz) is associated with the proton at C-1, and two doublet-doublet signals (δ 3.83 ppm, J = 11.5, 3.4 Hz and δ 3.73 ppm, J = 11.0, 8.1 Hz) represent the two protons at C-2. Chiral HPLC analysis, however, revealed that (R)-1-phenyl-1,2-ethanediol was the dominant product (Panel III in Figure 6), a surprising finding given that no enantioselectivity would be expected if both epoxide enantiomers were hydrolyzed via the same mechanism (e.g. regioselective attack at the α-carbon). Consequently, the predominance of the (R)-vicinal diol product must arise from complementary stereo- and regioselective SgcF-catalyzed hydrolysis of each styrene oxide enantiomer. This intriguing finding prompted further investigation of this apparent enantioconvergent activity of SgcF.
Figure 6.

HPLC profiles for determination of the stereochemistry of 1-phenyl-1,2-ethanediol resulted from SgcF catalysis: (I) racemic 1-phenyl-1,2-ethanediol standard; (II) (R)-1-phenyl-1,2-ethanediol standard (◆); (III) SgcF-catalyzed hydrolysis of racemic styrene oxide; (IV) SgcF-catalyzed hydrolysis of (S)-styrene oxide; (V) (S)-1-phenyl-1,2-ethanediol standard (●); and (VI) SgcF-catalyzed hydrolysis of (R)-styrene oxide.
Investigation of Enantioconvergent Mechanisms of SgcF-Catalyzed Hydrolysis
A few EHs have been reported to produce enantiomerically-enriched diol products from racemic epoxides via complementary enantio- and regio-selectivity.30 The regioselectivity of the SgcF-catalyzed hydrolysis of (S)- and (R)-styrene oxide was investigated by performing the reaction for each individual enantiomer substrate in the presence of [18O]-H2O. 13C NMR analysis of the resultant product from SgcF-catalyzed hydrolysis of (S)-styrene oxide showed a 2.8-Hz upfield shift that was exclusively associated with the C-1 position (Figure 7C), and chiral HPLC analysis confirmed the identity of the product as pure (R)-1-phenyl-1,2-ethanediol (Panel IV in Figure 6). This indicates a mechanism involving regiospecific nucleophilic attack at C-1 of (S)-styrene oxide (Figure 7A). By contrast, the product generated from (R)-styrene oxide revealed upfield shifts at both C-1 (2.8-Hz) and C-2 (2.4-Hz) (Figure 7C), and chiral HPLC analysis showed that (R)-1-phenyl-1,2-ethanediol was produced with 20% ee (Panel VI in Figure 6). This labeling pattern is consistent with a lack of regiospecificity of the enzyme in attacking both C-1 and C-2 of (R)-styrene oxide (Figure 7B). These data establish that the enantiomerically-enriched (R)-vicinal diol product from SgcF-catalyzed hydrolysis of racemic styrene oxide originates from complementary enantio- and regioselectivities of SgcF.
Figure 7.

Regioselectivity of SgcF-catalyzed hydrolysis of (S)- and (R)-styrene oxide in [18O]-H2O: regioselective attack of the [18O]-labeled (red) nucleophile Asp175 at (A) C-1 of (S)-styrene oxide and (B) C-1 and C-2 of (R)-styrene oxide, and (C) 13C NMR spectra of C-1 and C-2 of 1-phenyl-1,2-ethanediol obtained from SgcF-catalyzed hydrolysis of (I) racemic styrene oxide in H2O, (II) (S)-styrene oxide in [18O]-H2O, and (III) (R)-styrene oxide in [18O]-H2O.
Discussion
C-1027, a chromoprotein antitumor antibiotic produced by Steptomyces globisporus, is composed of an apo-protein (CagA) and the C-1027 chromophore. The C-1027 chromophore consists of four distinct moieties – an enediyne core, a deoxy aminosugar, a benzoxazolinate, and an (S)-3-chloro-5-hydroxy-β-tyrosine moiety (Figure 1).50–52 The 9-membered enediyne core readily undergoes Bergman cycloaromatization to generate a highly reactive diradical intermediate that abstracts hydrogen atoms from DNA, leading to both double-stranded breaks (DSBs) and interstrand cross links (ICLs) that ultimately result in cell death.53–55 Although the enediyne core directly confers cytotoxicity, the three peripheral moieties appended to the core are required for activity and stability. For instance, the benzoxazolinate moiety aids in binding to CagA that protects and carries the enediyne chromophore,56, 57 while the (S)-3-chloro-5-hydroxy-β-tyrosine also contributes critical binding interactions with CagA58, 59 and modulates the reactivity of the enediyne core via π-π interactions.59 A comparison of the biosynthetic gene clusters of C-1027,33 NCS34 and maduropeptin35 suggested that these two moieties might be appended to a (R)-vicinal diol-containing C-1027 enediyne core, which in turn may originate from a (S)-enediyne core epoxide intermediate via EH-catalyzed ring-opening (Figure 3).
To test this hypothesis, we first carried out extensive bioinformatics analysis of the 56 genes within C-1027 biosynthetic gene cluster, and identified two genes, sgcF and sgcI, encoding α/β-hydrolases.33 While bioinformatics analysis alone fell short of assigning SgcI as an EH, SgcF possesses such EH characteristics as the nucleophilic elbow (Sm-X-Nu-X-Sm-Sm), the G-X-Sm-X-S/T motif, the H-G-X-P oxyanion hole, and the Asp-His-Asp catalytic triad, and hence was annotated as an EH. Interestingly, SgcF contains only one of the two Tyr residues usually conserved in the lid domain (i.e., Y304) that serve to activate the oxirane ring to nucleophilic attack, and the other is changed to Trp (i.e., W236) (Figure S2).10, 15–17, 20–22, 26 In vivo data showed that no C-1027 production was observed in the ΔsgcF mutant strain SB1010, unambiguously establishing that sgcF is indispensable for C-1027 biosynthesis. In vitro characterization of SgcF unveiled a canonical EH activity as demonstrated by the efficient hydrolysis of styrene oxide, a substrate mimic of the putative enediyne core epoxide (Figure 3). Specifically, SgcF efficiently hydrolyzes (S)-styrene oxide, displaying an apparent Km of 0.6 ± 0.1 mM and kcat of 48 ± 1 min−1, via attack at the α-position to exclusively generate the (R)-phenyl vicinal diol, consistent with the stereochemistry of the C-1027 chromophore. These findings support the role of SgcF in the proposed convergent pathway for C-1027 biosynthesis, unveiling an (R)-vicinal diol as a key intermediate (Figures 1 and 3). Interestingly, SgcF can also hydrolyze (R)-styrene oxide to afford preferentially the (R)-phenyl vicinal diol via attack at the β-position, albeit with significantly reduced efficiency (apparent Km of 2.0 ± 0.4 mM and kcat = 4.3 ± 0.3 min−1). However, the latter activity unlikely contributes to C-1027 biosynthesis in vivo since the oxygenase that catalyzes the formation of the enediyne core epoxide intermediate most probably would yield a single enediyne core epoxide, i.e., the (S)-enantiomer, as a substrate for SgcF.
EHs are believed to be involved in the biosynthesis of a variety of natural products, including polyether antibiotics like nanchangmycin60 and monensin,61 plant cutin,29 and insect pheromones.62 The large diversity of putative EHs identified from genome sequences49 implies that comparative studies may potentially reveal a great deal about the determinants of regio- and enantiospecificity of these enzymes. Indeed, comparative analyses of enediyne biosynthetic gene clusters33–35 has predicted two EHs involved in the production of NCS – NcsF1 (53% identity to SgcF) and NcsF2 (62% identity to SgcF).34 In a biosynthetic strategy that parallels SgcF, one would predict NcsF1 or NcsF2 to catalyze the hydrolysis of an (R)-enediyne core epoxide intermediate to afford an (S)-vicinal diol product for NCS biosynthesis (Figure 1). Comparative studies of SgcF and NcsF1 or F2 therefore would present a great opportunity to investigate how EHs evolve and acquire their exquisite regio- and enantioselectivity.
Most known EHs catalyze the hydrolysis of each enantiomer of a racemic mixture at a different rate to yield enantiomerically-enriched vicinal diol products. However, this method of separation, or kinetic resolution, is limited by a maximum possible yield of 50%. In contrast, SgcF can not only catalyze the hydrolysis of (S)-styrene oxide to afford (R)-1-phenyl-1, 2-ethanediol exclusively but also hydrolyze (R)-styrene oxide to yield predominately (R)-1-phenyl-1, 2-ethanediol with 20% ee. Taken together, SgcF can generate (R)-1-phenyl-1, 2-ethanediol from racemic styrene epoxide with a minimal enantiomeric excess of 60% ee. SgcF therefore represents an excellent starting point for protein engineering efforts directed towards developing a biocatalyst for preparing enantiomerically pure diols from racemic epoxides.
In conclusion, the in vitro characterization of SgcF as an EH significantly advances our understanding of C-1027 biosynthesis, supporting a (R)-vicinal diol as a key intermediate. The preference of SgcF for hydrolyzing (S)-styrene oxide to generate (R)-phenyl vicinal diol is consistent with the proposed convergent logic of C-1027 biosynthesis in which the benzoxazolinate and β-amino acid moieties are attached to an (R)-vicinal diol functionality of an (S)-enediyne core epoxide intermediate (Figure 3).39, 41 Furthermore, the complementary regioselectivity of SgcF, which leads to the enantioconvergent production of (R)-vicinal diol as the dominant product from a racemic epoxide, sets the stage to investigate the stereochemistry of EH catalysis and to engineer EHs with improved regio- and/or enantiospecificity.
Supplementary Material
Acknowledgments
We thank Dr. Y. Li, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Beijing, China for the wild-type S. globisporus strain, the Analytical Instrumentation Center of the School of Pharmacy and Dr. Q. Cui, the National Magnetic Resonance Facility at UW-Madison for support in obtaining MS and NMR data, and Prof. W. Tang, School of Phamracy, UW-Madison for the use of chiral HPLC. This work is supported in part by NIH grants CA78747 and CA113297. G.P.H. is the recipient of an NSERC postdoctoral fellowship.
Footnotes
Supporting Information Available: The structural model of AnEH (Figure S1), sequence alignment of SgcF (Figure S2), sgcF inactivation results (Figure S3), SDS-PAGE of purified recombinant SgcF (Figure S4), and pH (Figure S5A) and concentration (Figure S5B) dependence of SgcF. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shen B, Liu W, Nonaka K. Curr Med Chem. 2003;10:2317–2325. doi: 10.2174/0929867033456701. [DOI] [PubMed] [Google Scholar]
- 2.Van Lanen SG, Shen B. Curr Top Med Chem. 2008;8:448–459. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Archelas A, Furstoss R. Curr Opin Chem Biol. 2001;5:112–119. doi: 10.1016/s1367-5931(00)00179-4. [DOI] [PubMed] [Google Scholar]
- 4.Lee EY, Shuler ML. Biotechnol Bioeng. 2007;98:318–327. doi: 10.1002/bit.21444. [DOI] [PubMed] [Google Scholar]
- 5.Steinreiber A, Faber K. Curr Opin Biotechnol. 2001;12:552–558. doi: 10.1016/s0958-1669(01)00262-2. [DOI] [PubMed] [Google Scholar]
- 6.Weijers CAGM, de Bont JAM. J Mol Catal B Enzym. 1999;6:199–214. [Google Scholar]
- 7.Heikinheimo P, Goldman A, Jeffries C, Ollis DL. Structure. 1999;7:141–146. doi: 10.1016/s0969-2126(99)80079-3. [DOI] [PubMed] [Google Scholar]
- 8.Holmquist M. Curr Prot Pept Sci. 2000;1:209–235. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 9.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verscheuren KHG, Goldman A. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 10.Argiriadi MA, Morisseau C, Goodrow MH, Dowdy DL, Hammock BD, Christianson DW. J Biol Chem. 2000;275:15265–15270. doi: 10.1074/jbc.M000278200. [DOI] [PubMed] [Google Scholar]
- 11.Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Proc Natl Acad Sci U S A. 1999;96:10637–10642. doi: 10.1073/pnas.96.19.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Biochemistry. 2004;43:4716–4723. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 13.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Protein Sci. 2006;15:58–64. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mowbray SL, Elfstrom LT, Ahlgren KM, Andersson CE, Widersten M. Protein Sci. 2006;15:1628–1637. doi: 10.1110/ps.051792106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nardini M, Rick RB, Janssen DB, Dijkstra BW. J Mol Catal B Enzym. 2001;11:1035–1042. [Google Scholar]
- 16.Nardini M, Ridder IS, Rozeboom HJ, Kalk KH, Rink R, Janssen DB, Dijkstra BW. J Biol Chem. 1999;274:14579–14586. [PubMed] [Google Scholar]
- 17.Zou JY, Hallberg BM, Bergfors T, Oesch F, Arand M, Mowbray SL, Jones TA. Structure. 2000;8:111–122. doi: 10.1016/s0969-2126(00)00087-3. [DOI] [PubMed] [Google Scholar]
- 18.Barth S, Fischer M, Schmid RD, Pleiss J. Bioinformatics. 2004;20:2845–2847. doi: 10.1093/bioinformatics/bth284. [DOI] [PubMed] [Google Scholar]
- 19.Barth S, Fischer M, Schmid RD, Pleiss J. Proteins: Struct Funct Bioinform. 2004;55:846–855. doi: 10.1002/prot.20013. [DOI] [PubMed] [Google Scholar]
- 20.Rink R, Kingma J, Spelberg JHL, Janssen DB. Biochemistry. 2000;39:5600–5613. doi: 10.1021/bi9922392. [DOI] [PubMed] [Google Scholar]
- 21.Rink R, Spelberg JHL, Pieters RJ, Kingma J, Nardini M, Kellogg RM, Dijkstra BW, Janssen DB. J Am Chem Soc. 1999;121:7417–7418. [Google Scholar]
- 22.Yamada T, Morisseau C, Maxwell JE, Argiriadi MA, Christianson DW, Hammock BD. J Biol Chem. 2000;275:23082–3088. doi: 10.1074/jbc.M001464200. [DOI] [PubMed] [Google Scholar]
- 23.Hammock BD, Pinot F, Beetham JK, Grant DF, Arand ME, Oesch F. Biochem Biophys Res Commun. 1994;198:850–856. doi: 10.1006/bbrc.1994.1121. [DOI] [PubMed] [Google Scholar]
- 24.Lacourciere GM, Armstrong RN. J Am Chem Soc. 1993;115:10466–10467. [Google Scholar]
- 25.Muller F, Arand M, Frank H, Seidel A, Hinz W, Winkler L, Hanel K, Blee E, Beetham JK, Hammock BD, Oesch F. Eur J Biochem. 1997;245:490–496. doi: 10.1111/j.1432-1033.1997.00490.x. [DOI] [PubMed] [Google Scholar]
- 26.Franken SM, Rozeboom HJ, Kalk KH, Dijkstra BW. EMBO J. 1991;10:1297–1302. doi: 10.1002/j.1460-2075.1991.tb07647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu AYH, Miwa GT. Annu Rev Pharmacol Toxicol. 1980;20:513–531. doi: 10.1146/annurev.pa.20.040180.002501. [DOI] [PubMed] [Google Scholar]
- 28.Oesch F. Xenobiotica. 1972;3:305–340. doi: 10.3109/00498257309151525. [DOI] [PubMed] [Google Scholar]
- 29.Morisseau C, Hammock BD. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 30.Kim HS, Lee OK, Hwang S, Kim BJ, Lee EY. Biotechnol Lett. 2008;30:127–133. doi: 10.1007/s10529-007-9495-2. [DOI] [PubMed] [Google Scholar]
- 31.Orru RVA, Faber K. Curr Opin Chem Biol. 1999;3:16–21. doi: 10.1016/s1367-5931(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 32.Lee EY. Biotechnol Lett. 2008 doi: 10.1007/s10529-008-9727-0. [DOI] [PubMed] [Google Scholar]
- 33.Liu W, Christenson SD, Standage S, Shen B. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 34.Liu W, Nonaka K, Nie LP, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. Chem Biol. 2005;12:293–302. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 35.Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. J Am Chem Soc. 2007;129:13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christenson SD, Liu W, Toney MD, Shen B. J Am Chem Soc. 2003;125:6062–6063. doi: 10.1021/ja034609m. [DOI] [PubMed] [Google Scholar]
- 37.Lin S, Van Lanen SG, Shen B. J Am Chem Soc. 2007;129:12432–12438. doi: 10.1021/ja072311g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin SJ, Van Lanen SG, Shen B. J Am Chem Soc. 2008;130:6616–6623. doi: 10.1021/ja710601d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin SJ, Van Lanen SG, Shen B. Proc Natl Acad Sci U S A. 2009;106:4183–4188. doi: 10.1073/pnas.0808880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Lanen SG, Lin SJ, Shen B. Proc Natl Acad Sci U S A. 2008;105:494–499. doi: 10.1073/pnas.0708750105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Van Lanen SG, Ju JH, Liu W, Dorrestein PC, Li WL, Kelleher NL, Shen B. Proc Natl Acad Sci U S A. 2008;105:1460–1465. doi: 10.1073/pnas.0711625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T. Gene. 1992;111:61–68. doi: 10.1016/0378-1119(92)90603-m. [DOI] [PubMed] [Google Scholar]
- 43.Liu W, Shen B. Antimicrob Agents Chemother. 2000;44:382–392. doi: 10.1128/aac.44.2.382-392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kieser T, Bibb MJ, Buttner MM, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- 45.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Lanen SG, Dorrestein PC, Christenson SD, Liu W, Ju JH, Kelleher NL, Shen B. J Am Chem Soc. 2005;127:11594–11595. doi: 10.1021/ja052871k. [DOI] [PubMed] [Google Scholar]
- 47.Doderer K, Lutz-Wahl S, Hauer B, Schmid RD. Anal Biochem. 2003;321:131–134. doi: 10.1016/s0003-2697(03)00399-3. [DOI] [PubMed] [Google Scholar]
- 48.Mateo C, Archelas A, Furstoss R. Anal Biochem. 2003;314:135–141. doi: 10.1016/s0003-2697(02)00646-2. [DOI] [PubMed] [Google Scholar]
- 49.van Loo B, Kingma J, Arand M, Wubbolts MG, Janssen DB. Appl Environ Microb. 2006;72:2905–2917. doi: 10.1128/AEM.72.4.2905-2917.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu J, Xue YC, Xie MY, Zhang R, Otani T, Minami Y, Yamada Y, Marunaka T. J Antibiot. 1988;41:1575–1579. doi: 10.7164/antibiotics.41.1575. [DOI] [PubMed] [Google Scholar]
- 51.Otani T, Minami Y, Marunaka T, Zhang R, Xie MY. J Antibiot. 1988;41:1580–1585. doi: 10.7164/antibiotics.41.1580. [DOI] [PubMed] [Google Scholar]
- 52.Otani T, Yasuhara T, Minami Y, Shimazu T, Zhang R, Xie MY. Agric Biol Chem. 1991;55:407–417. [PubMed] [Google Scholar]
- 53.Kennedy DR, Gawron LS, Ju JH, Liu W, Shen B, Beerman TA. Cancer Research. 2007;67:773–781. doi: 10.1158/0008-5472.CAN-06-2893. [DOI] [PubMed] [Google Scholar]
- 54.Kennedy DR, Ju J, Shen B, Beerman TA. Proc Natl Acad Sci U S A. 2007;104:17632–17637. doi: 10.1073/pnas.0708274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu YJ, Zhen YS, Goldberg IH. Biochemistry. 1994;33:5947–5954. doi: 10.1021/bi00185a036. [DOI] [PubMed] [Google Scholar]
- 56.Matsumoto T, Okuno Y, Sugiura Y. Biochem Biophys Res Commun. 1993;195:659–666. doi: 10.1006/bbrc.1993.2096. [DOI] [PubMed] [Google Scholar]
- 57.Okuno Y, Otsuka M, Sugiura Y. J Med Chem. 1994;37:2266–2273. doi: 10.1021/jm00041a004. [DOI] [PubMed] [Google Scholar]
- 58.Okuno Y, Iwashita T, Sugiura Y. J Am Chem Soc. 2000;122:6848–6854. [Google Scholar]
- 59.Tanaka T, Fukuda-Ishisaka S, Hirama M, Otani T. J Mol Biol. 2001;309:267–283. doi: 10.1006/jmbi.2001.4621. [DOI] [PubMed] [Google Scholar]
- 60.Sun Y, Zhou X, Dong H, Tu G, Wang M, Wang B, ZXD Chem Biol. 2003;10:431–441. doi: 10.1016/s1074-5521(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 61.Gallimore A, Stark C, Bhatt A, Harvey B, Demydchuk Y, Bolanos-Garcia V, Fowler D, Staunton J, Leadlay P, Spencer J. Chem Biol. 2006;13:453–460. doi: 10.1016/j.chembiol.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 62.Abdel-Latief M, Garbe LA, Koch M, Ruther J. Proc Natl Acad Sci U S A. 2008;105:8914–8919. doi: 10.1073/pnas.0801559105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.