

Abstract
Malignant transformation was demonstrated in UROtsa cells following 52 wk exposure to 50 nM monomethylarsonous acid (MMAIII); the result was the malignantly transformed cell line, URO-MSC. URO-MSC cells were used to study the induction of DNA damage and the alteration of DNA repair enzymes in both the presence of MMAIII [URO-MSC(+)] and after subsequent removal of MMAIII [URO-MSC(-)] following chronic, low-level exposure. In the presence of MMAIII, URO-MSC(+) cells demonstrated a sustained increase in DNA damage following 12 wk exposure; in particular, a significant increase in DNA single strand breaks at 12 wk exposure consistently elevated through 52 wk. The persistence of DNA damage in URO-MSC cells was assessed after a 2 wk removal of MMAIII. URO-MSC(-) cells demonstrated a decrease in DNA damage compared to URO-MSC(+); however, DNA damage in URO-MSC(-) remained significantly elevated when compared to untreated UROtsa and increased in a time-dependent manner. Reactive oxygen species (ROS) were demonstrated to be a critical component in the generation of DNA damage determined through the incubation of ROS scavengers with URO-MSC cells. Poly (ADP-ribose) polymerase (PARP) is a key repair enzyme in DNA single strand break repair. URO-MSC(+) resulted in a slight increase in PARP activity after 36 wk MMAIII exposure, suggesting the presence of MMAIII is inhibiting the increase in PARP activity. In support, PARP activity in URO-MSC(-) increased significantly, coinciding with a subsequent decrease in DNA damage demonstrated in URO-MSC(-) compared to URO-MSC(+). These data demonstrate that chronic, low-level exposure of UROtsa cells to 50 nM MMAIII results in: the induction of DNA damage that remains elevated upon removal of MMAIII; increased levels of ROS that play a role in MMAIII induced-DNA damage; and decreased PARP activity in the presence of MMAIII.
Keywords: Arsenic, Monomethylarsonous acid, Bladder cancer, UROtsa, DNA damage
1. Introduction
Numerous research efforts are focused on determining the critical role of the arsenic metabolite, monomethylarsonous acid (MMAIII), in the induction of bladder carcinogenesis. Multiple mechanisms have been implicated in the MMAIII-induced transformation of the human bladder urothelial cell model, UROtsa, following chronic, low-level exposure. Acute exposure to MMAIII have been shown to generate reactive oxygen species (ROS) resulting in oxidative stress and perturbations of cell signaling pathways involved in tumorigenesis (Eblin et al., 2006, 2007). However, the ability of chronic, low-level MMAIII to induce DNA damage and alter DNA repair activity has not been extensively studied. The research herein was carried out to determine the ability of chronic, low-level MMAIII to induce DNA damage, generate elevated levels of ROS implicated in damaging DNA, evaluate the specific type of DNA damage occurring, and investigate the effect of MMAIII in the alteration of DNA repair activity. Of particular importance was the assessment of these parameters in the presence and absence of MMAIII following chronic, low-level exposures. The ability of MMAIII exposure to permanently alter normal cell function after the subsequent removal of an environmentally relevant exposure, exemplifies the carcinogenic potential of the biomethylated metabolite, MMAIII.
Inorganic arsenic is a known human carcinogen associated with pleiotropic toxicities including cancers of the skin, lung, liver, and bladder, in addition to non-carcinogenic health effects (Chen et al., 1988; Brown and Ross, 2002; Marshall et al., 2007). Sources of inorganic arsenic exposure include air, water, and food from both natural and anthropogenic sources (Ramirez-Solis et al., 2004). Following exposure, inorganic arsenic undergoes a series of biotransformation processes alternating between reduction of pentavalent arsenic to trivalent arsenic and sequential methylation steps resulting in monomethylarsenic acid (MMAIII,V) and dimethylarsenic acid (DMAIII,V) metabolites (Aposhian, 1997; Healy et al., 1998; Vahter, 2002). Of particular interest in these studies, the toxicity of the trivalent metabolite, MMAIII, is 20 times greater than that of arsenite (AsIII) (Styblo et al., 2002; Bredfeldt et al., 2006). The level of MMAIII used in this study [50 nM MMAIII or approximately 3.75 ppb] represents an environmentally relevant exposure level (the current EPA inorganic arsenic standard for drinking water is set at 10 ppb), and a concentration which falls within the range of MMAIII detected in the urine of humans exposed to inorganic arsenic in their water supply (Aposhian et al., 2000; Mandal et al., 2001, 2004). Despite the classification of arsenic as a human carcinogen, the exact mechanisms of arsenic carcinogenicity are not well understood due, in part, to the multitude of complex factors attributed to playing a role in the induction of carcinogenesis.
Numerous mechanisms have been implicated in arsenical-induced malignant transformation. Exposure to MMAIII has been linked to the alteration of signaling events including processes of the MAPK, NFkB, JAK-STAT, AP-1, and WNT signaling pathways (Ludwig et al., 1998; Simeonova et al., 2001; Ahlborn et al., 2008; Tun-Kyi et al., 2008; Jensen et al., 2009). Other mechanisms include sulfhydryl depletion, altered DNA repair, the formation of DNA adducts and/or damage as a result of oxidative stress, and the alteration of DNA methylation and histone modifications in promoter regions (National Research Council, 2001; Bredfeldt et al., 2004; Jensen et al., 2008, 2009). In particular, chronic exposure to low-level MMAIII in UROtsa cells has been shown to produce oxidative stress as a result of elevated ROS. ROS have been demonstrated to play a role in altered signaling pathways involved in proliferation and anchorage-independent growth; all of which contribute to the MMAIII-induced malignant transformation of UROtsa cells (Eblin et al., 2007, 2008, and 2009).
Several lines of evidence have shown that inorganic and organic arsenicals can produce ROS and free radicals such as superoxide (O2-), hydrogen peroxide (H2O2), hydroxyl radical (·OH), singlet oxygen (1O2), and lipid peroxyl radical, LOO. (Shi et al., 2004a, 2004b; Eblin et al., 2006; 2008). The generation of ROS can cause structural alterations in DNA, affect cytoplasmic and nuclear signal transduction pathways, and modulate activity of proteins involved in cell proliferation, differentiation, and apoptosis (Wiseman and Halliwell, 1996; Leonard et al., 2004; Beyersmann and Hartwig, 2008). With respect to genotoxicity, the application of radical scavengers has revealed the involvement of arsenical-induced ROS in the induction of DNA damage in arsenite exposed rats (Ramanathan et al., 2003). ROS may have a significant role in the incidence of DNA damage following arsenical exposure, providing evidence against a direct genotoxic mechanism of action involved in arsenical-induced cancer. Previous research has demonstrated that ROS can act as intermediates in the DNA damaging activity in supercoiled phiX174 DNA and DNA in peripheral human lymphocytes following acute, low-level MMAIII exposure (Nesnow et al., 2002; Schoen et al., 2004). However, the ability of the biomethylated metabolite, MMAIII, to induce a genotoxic insult following chronic, low-level exposure in a human bladder model has not been adequately examined.
The ability of trivalent arsenicals to attach to zinc binding structures prevalent in DNA repair proteins may provide a target for arsenical exposure, resulting in the alteration/inhibition of critical DNA repair enzymes involved in maintaining genomic integrity (Kitchin et al., 2008). DNA damage in the form of single strand breaks is repaired through the base excision repair process. A critical repair enzyme involved in base excision repair is poly (ADP-ribose) polymerase (PARP) (Caldecott, 2007). The capacity of chronic, low-level MMAIII exposure to decrease activity of such repair enzymes in the presence of a genotoxic insult, such as ROS, could be a potential mechanism in arsenical-induced carcinogenesis.
The primary route of arsenic elimination is urinary excretion; therefore, the bladder is a key target organ of toxicity as a result of exposure to both inorganic arsenic and multiple biomethylated metabolites (Tapio and Grosche, 2006). The immortalized, non-tumorigenic human urothelial cell line, UROtsa, and the MMAIII-induced malignant transformed variant, URO-MSC, were used as in vitro models to study the molecular mechanisms leading to the development of MMAIII-induced malignant transformation (Petzoldt et al., 1995; Sens et al., 2004; Bredfeldt et al., 2006). URO-MSC cells in the presence of MMAIII [URO-MSC(+)] were used to investigate the generation of ROS, the induction of DNA damage, and the alteration of the DNA repair enzyme, PARP, following exposure to chronic, biologically relevant concentrations of MMAIII. The persistence of DNA damage and the alteration of DNA repair activity was also studied in URO-MSC cells after the 2 wk withdrawal of MMAIII [URO-MSC(-)] to examine the biological alterations in URO-MSC(-) cells following previous chronic MMAIII exposure. Analysis in the absence of MMAIII was necessary to ensure that the experimentation focused on the persistent arsenical-induced alteration of biological processes within the UROtsa cell line, and not the chemical effect caused by the presence of MMAIII.
2. Materials and Methods
2.1 Reagents
Dulbecco's Modified Eagle Medium, fetal bovine serum, antibiotic-antimycotic, and 1X trypsin-EDTA (0.25%) were acquired from Gibco Invitrogen Corporation (Carlsbad, CA). Diiodomethylarsine (MMAIII iodide, CH3AsI2) was prepared by the Synthetic Chemistry Facility Core (Southwest Environmental Health Sciences Center, Tucson, AZ) using the method of Millar et al., 1960. Etoposide was obtained from Trevigen, Inc. (Gaithersburg, MD). Water used in studies was distilled and de-ionized.
2.2 Dosing solutions
Preparation of dosing solution and procedures were derived from Bredfeldt et al., 2006. Pure MMAIII was stored in ampules at 4 °C. Fresh stock solutions of 25 mM MMAIII were made and diluted to a final concentration of 5 μM prior to dosing (1 to 100 dilution) to obtain a final concentration of 50 nM MMAIII. All dosing solutions were sterile filtered with a 0.2 μm acrodisc and stored in sealed, sterile tubes at 4 °C that were opened only for dosing in a sterile cell culture hood. As previously reported by Gong et al., 2001, MMAIII solutions in distilled, de-ionized water were stable for approximately 4 months at 4 °C with no degradation observed when monitored using HPLC-ICP MS.
2.3 Cell culture
UROtsa cells were a generous gift from Drs. Donald and Maryann Sens (University of North Dakota). URO-MSC cells were created in our laboratory as reported by Bredfeldt et al., 2006. Cell culture conditions were derived from those previously described by Rossi et al., 2001 and Bredfeldt et al., 2004. UROtsa cells were cultured in a growth medium of DMEM containing 5% v/v FBS and 1% antibiotic–antimycotic. Culture medium was changed every 2 days. Cultured cells were incubated in an atmosphere that was 5% CO2:95% air at 37 °C. Confluent cells were removed from plates with trypsin–EDTA (0.25%) and subcultured at a ratio of 1:3. All UROtsa and URO-MSC cells used in this study tested negative for the presence of mycoplasma contamination. MMAIII-treated cells were continuously cultured in a medium enriched with 50 nM MMAIII and dosed every other day to ensure continuous presence of MMAIII. Parallel cultures of UROtsa cells were maintained in MMAIII-free medium and served as passage-matched controls. Experimentation was carried out with MMAIII in the culture medium to investigate cellular alterations in the presence of MMAIII; and after withdrawal of MMAIII from culture medium to examine biological changes in UROtsa cells altered after removal of MMAIII. UROtsa cells chronically exposed to low-level MMAIII were evaluated in the presence of MMAIII at the time of experimentation, and after a 2 wk removal of MMAIII following previous chronic, low-level exposure (Fig. 1). Persistence of MMAIII in cell culture conditions was determined by exposing UROtsa cells to 50 nM MMAIII and then measuring the presence of the various arsenic species in both cell lysates and media 1-4 days after initial MMAIII exposure using HPLC/ICP-MS (Bredfeldt et al., 2004). Results demonstrated MMAIII was below the limit of detection (0.5 ppb) after 3 days. The 2 wk removal of MMAIII and cell culture splitting upon cell confluency prior to experimentation was to ensure that MMAIII was no longer present within URO-MSC(-) cells. Previous studies by Bredfeldt et al., 2004 evaluated the ability of UROtsa to metabolize AsIII to MMAIII, MMAV, and DMAV. The most abundant metabolite produced in those studies was MMAIII. UROtsa cells treated for 24 h with 50 nM MMAIII did not metabolize MMAIII to more toxic products, as detected by HPLC/ICP-MS. Demonstrating that MMAIII is the chemical responsible for the observed effects as >90% of the dose remained in the form of MMAIII. DMAIII was not detected in these same UROtsa cells treated with 50 nM MMAIII.
Figure 1.
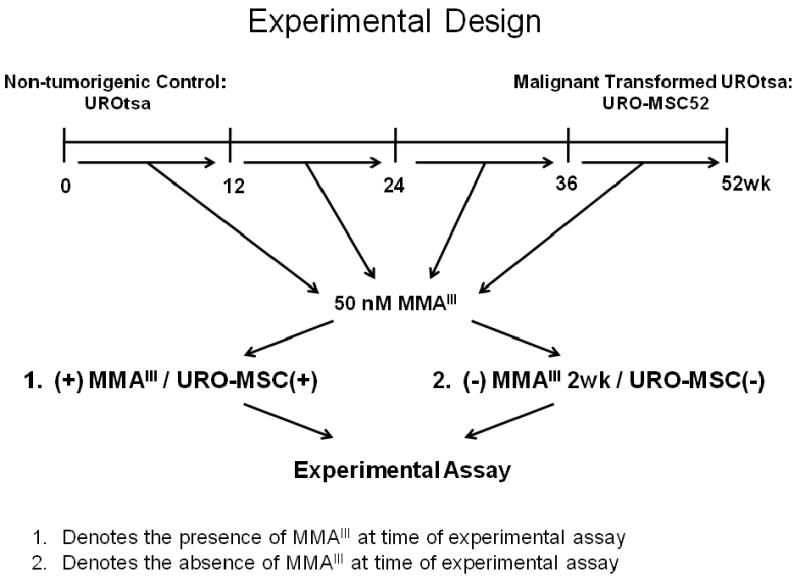
Summary of experimental design. Non-tumorigenic UROtsa cells were chronically exposed to 50 nM MMAIII for 52 wk resulting in the malignantly transformed URO-MSC cell line. Cells were exposed to MMAIII for 12, 24, 36 and 52 wk to examine the progressive changes following MMAIII exposure. For experimentation purposes, cells were analyzed in the presence of MMAIII [URO-MSC12(+), URO-MSC24(+), URO-MSC36(+), URO-MSC52(+)], or URO-MSC cells were grown in the absence of MMAIII for 2 wk following previous chronic exposure [URO-MSC12(-), URO-MSC24(-), URO-MSC36(-), URO-MSC52(-)] to analyze the biological alterations in UROtsa cells in the presence and absence of the MMAIII.
2.4 Alkaline comet assay
The comet assay was performed using a modified protocol of Trevigen Inc. (Gaithersburg, MD) to measure DNA strand breaks. URO-MSC cells at 12, 24, 36, and 52 wk in the presence of MMAIII [URO-MSC12(+), URO-MSC24(+), URO-MSC36(+), and URO-MSC52(+)] and after a 2 wk removal of MMAIII [URO-MSC12(-), URO-MSC24(-), URO-MSC36(-), and URO-MSC52(-)] were utilized and cultured in 100 × 20 mm tissue culture dishes (BD Falcon, Franklin Lakes, NJ) at 5 × 105 cells per plate and allowed to reach 90% confluency. Adherent cells were removed via trypsin-EDTA (0.25%), quenched with 5% FBS/Media, and centrifuged at 400 rpm for 5 min. Cells were resuspended in 1X PBS (Ca2+ and Mg2+ free) and counted via trypan blue exclusion assay (Bredfeldt et al., 2006). Cells were washed in ice cold 1X PBS (Ca2+ and Mg2+ free) and resuspended at 1 × 105 cell/ml in ice cold 1X PBS (Ca2+ and Mg2+ free). Cells (1 × 105) were combined with a low melting point agarose at a ratio of 1 to 10, allowed to solidify onto a comet slide, and placed in prechilled lysis solution overnight. Comet slide was placed in alkali solution (1 h) followed by horizontal electrophoresis. After electrophoresis, slides were rinsed in distilled H2O and immersed in 70% ethanol for 5 min and allowed to dry. Prior to fluorescence microscopy, samples were stained with SYBR green from Trevigen Inc. (Gaithersburg, MD).
2.5 Image analysis
Comet images were viewed via epifluorescence microscopy at excitation and emissions of 494/521 nm. The images were captured using an Olympus IMT-2 inverted fluorescence microscope, using a 20× objective. The camera used was a Hamamatsu ORCA-100 CCD camera (chip resolution 1280 × 1024) in bin2 mode, with the actual image resolution set at 640 × 512 using the SimplePCI version 6.5 (Hamamatsu) instrument control software. At least 75 randomly selected cells were analyzed per n-value. An image analysis system based on the Matlab computer program (Mathworks, Natick, MA) developed by Dr. Joceline Lega (Department of Mathematics, University of Arizona) was used to analyze individual cells to determine mean comet profiles, assess mean comet tail length, and average comet moment.
2.6 Spectrofluorometric analysis of endogenous ROS
URO-MSC cells exposed to 50 nM MMAIII for 12, 24, and 52 wk in the presence of MMAIII or following removal of MMAIII (2 wk) were grown to confluency, trypsinized, and counted. Cells (200 cells/μl) were added to Eppendorf tubes (1.5 ml), washed with PBS, and loaded with 25 μM CM-H2DCFDA (Molecular Probes/Invitrogen, Carlsbad, CA) for 30 min at 37 °C. Cells were washed and re-suspended in PBS. Cell suspensions were added to a 96-well plate and fluorescence emission measured at 530 nm on a SpectraMax Gemini XS multi-well plate reader (Molecular Devices Co., Sunnyvale, CA) with excitation filter set at 485 nm. Temperature was maintained at 37 °C and readings were performed every minute for a total of 1 h (Wang and Joseph, 1999).
2.7 Effect of ROS scavengers on DNA damage in URO-MSC cells
UROtsa cells exposed 50 nM MMAIII for 12 or 52 wk followed by removal of MMAIII for 2 wk [URO-MSC12(-)/URO-MSC52(-)] were treated with antioxidants as previously described (Eblin et al., 2009): potassium iodide (KI; 5 mM), superoxide dismutase (SOD; 100 units/ml), or catalase (CAT; 200 units/ml) every 2 days. After a 14 day incubation of URO-MSC12(-)/URO-MSC52(-) cells with the various antioxidants, cells were removed from culture flasks via trypsinization and counted via trypan blue exclusion assay. DNA damage was detected using the alkaline comet assay. A minimum of 75 randomly selected cells were analyzed per n-value, n=4, for each condition with protocol performed similar to methods section 2.3 and section 2.4 as previously described.
2.8 Analysis of Single Stranded DNA damage via flow cytometry
A monoclonal antibody, F7-26, from Cell Technology Inc. (Mountain View, CA) was used to detect single stranded DNA in UROtsa cells chronically exposed to MMAIII for 12, 24, 36, and 52 wk in the presence of MMAIII and after the 2 wk removal of MMAIII exposure. Cell samples were cultured in 75 cm2 tissue culture flasks from Sarstedt (Newton, NC) until confluence was reached. Cells were removed via trypsin-EDTA (0.25%), and resuspended in serum enriched media. Cell count was performed using the trypan blue exclusion assay. URO-MSC(+) and URO-MSC(-) cells: 12, 24, 36, 52 wk MMAIII exposure and UROtsa control (n=4) cells (1 × 106) were transferred into individual 5 ml polystyrene round bottom tubes (BD Falcon, Franklin Lakes, NJ) and fixed using 1 ml fixative solution (6:1 methanol/PBS solution) for 48 h. Samples were denatured using 1× denaturing buffer, formamide (Sigma Co., St. Louis, MO), for 10 min at 80 °C. Samples were then blocked and stained using a mouse anti-ssDNA (primary) antibody and anti-mouse IgM FITC labeled (secondary) antibody (Cell Technology, Mountain View, CA). After subsequent staining and washes, cells (1 × 106) were placed in sterile PBS. Samples were analyzed at the Arizona Cancer Center Cytometry Core Facility using a FACScan cytometer (BD Biosciences, San Jose CA) equipped with a 15 mW Argon laser. Cells were analyzed via excitation/emission wavelengths at 488/530 nm. The protocol for the detection of ssDNA using the mouse anti-ssDNA monoclonal antibody was adapted from Frankfurt et al., 1996 and Cell Technology Inc. (Mountain View, CA).
2.9 Poly (ADP-ribose) polymerase (PARP) Activity Assay
PARP activity in chronic MMAIII exposed UROtsa cells was determined using a semi-quantitative ELISA in a 96 well format. Relative PARP activity was determined through detection of poly (ADP-ribose) polymer, PAR, which was immobilized onto histone proteins on the bottom of the 96-well plates. URO-MSC(+) and URO-MSC(-) cells at 12, 24, 36, and 52 wk MMAIII exposure were seeded at 5 × 103 cells/200 μl media and allowed to grow overnight in a 96-well flat-bottom plate. Adherent cells were lysed with cell extraction buffer (100 μl/well) and protein levels quantified via the bicinchoninic acid assay. Lysates were incubated with PARP substrate cocktail containing NAD+, which allows for the generation of PAR based on relative PARP activity within the samples. An anti-PAR monoclonal antibody (50 μl), a goat anti-mouse IgG-HRP conjugate (50 μl), and horseradish peroxidase substrate were used to generate a colorimetric signal. 50 μl of TACS-Sapphire was added to the samples and allowed to incubate for 10 min; reaction was stopped by adding 50 μl of phosphoric acid. Calculated absorbance was correlated with relative PARP activity. Samples were read using a VERSAmax plate reader (Molecular Devices Co., Sunnyvale, CA) at an absorbance of 450 nm. The protocol for the detection of PARP activity was adapted from Trevigen Inc. (Gaithersburg, MD).
2.10 Statistics
Graphs were generated in Microsoft Office Excel (Microsoft Corp., Redmond, WA). Comet assay data analyzed included an n=4 for each experimental value with a minimum of 75 cells assessed per each n-value. Detection of ROS via spectrofluorometric analysis included an n=3 for each experimental value. Detection of ssDNA damage via flow cytometry was expressed as the average of four experiments. PARP activity was expressed as the average of three experiments. Error bars within each column represent the standard error of the mean (± SEM). Statistical analysis utilized and p-value cutoff for each experiment is noted in corresponding figure legends. Statistical significance was determined using the Student's t-test for comparison of samples, ANOVA followed by Bonferroni's multiple comparison test, or Cruzick's non-parametric trend analysis which is an extension of the Wilcoxon rank-sum test using Stata10 (College Station, TX).
3. Results
3.1 DNA damage assessed through the alkaline comet assay
The majority of studies examining DNA damage as a result of exposure to inorganic or trivalent methylated arsenic species have focused on the ability of these arsenicals to induce DNA strand breaks following acute exposure (Liu et al., 2000; Schwerdtle et al., 2003; Chai et al., 2007); however, our model focuses on the biological changes taking place within the non-tumorigenic, urothelial cell line, UROtsa, during chronic, low-level exposure to the biomethylated metabolite, MMAIII. The URO-MSC cell lines were used to examine the extent of DNA damage in the presence of MMAIII through utilization of the alkaline comet assay. The DNA damaging agent, etoposide, was utilized as a positive control. Etoposide, a topoisomerase II inhibitor, was utilized as a positive control due to the ability to induce DNA single- and double- strand breaks (Sinha et al., 1988; Van Maanen et al., 1988). The average comet tail moment is defined as the product of the tail length and the fraction of total DNA in the tail. The mean comet moment of URO-MSC cells in the presence of MMAIII is first elevated at 12 wk exposure [URO-MSC12(+)] and remains consistently elevated through 52 wk exposure [URO-MSC52(+)] (Fig. 2).
Figure 2.
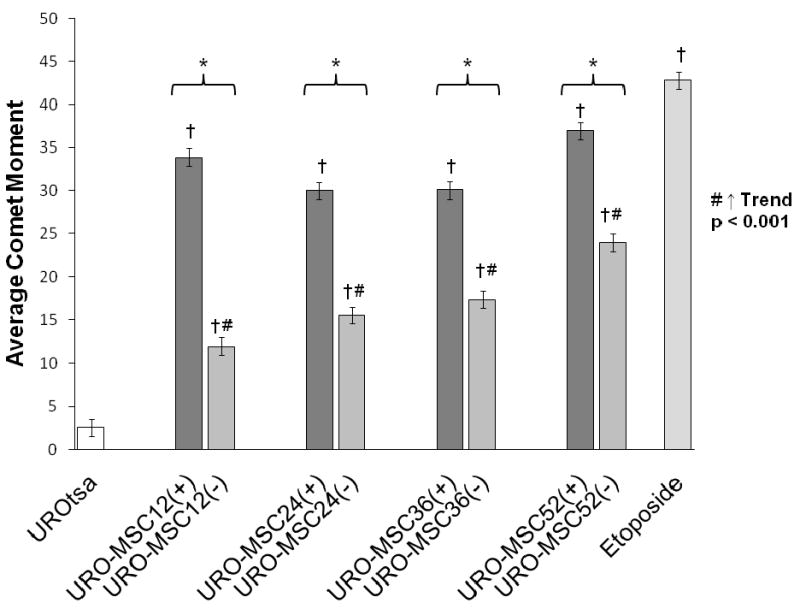
Comparison of the average comet moment in URO-MSC cells in the presence/absence of MMAIII following chronic exposure. Graph depicts changes in comet moment between MMAIII exposed URO-MSC cells and untreated UROtsa control (n=4), each n value ≥ 75 cells. (*) Marks statistically significant difference (p ≤ 0.05) between URO-MSC(+) and URO-MSC(-) determined using Student's t-test. (†) Marks statistically significant difference in DNA comet moment of URO-MSC(+)/URO-MSC(-) cells with UROtsa control identified with ANOVA followed by Bonferroni's multiple comparison test with p ≤ 0.05 considered statistically significant. Non-parametric test for trend demonstrates a time-dependent increase in comet moment of URO-MSC(-) cells, (#) marks statistically significant (p ≤ 0.001) upward trend. Error bars within each column represent the standard error of the mean (± SEM).
To determine if the changes in DNA damage were related to the presence of MMAIII, or whether the changes were associated with the transformation of UROtsa cells, the alkaline comet assay was performed on URO-MSC cells after MMAIII was removed from culture conditions for 2 wk. The average comet moment of URO-MSC(-) cells decreased in contrast to URO-MSC(+) cells in the presence of MMAIII with the same length of exposure; however, the DNA damage in URO-MSC cells removed from MMAIII increased in a time-dependent manner following previous exposure to MMAIII (Fig. 2). These data demonstrate that in the presence of MMAIII DNA damage levels in URO-MSC(+) cells remain elevated and sustained throughout chronic exposure. In addition, although DNA damage levels in URO-MSC(-) cells decrease compared to URO-MSC(+) cells, the disparity in DNA damage levels between the two decrease as the length of exposure to MMAIII increases.
3.2 Role of ROS in DNA damage following chronic, low-level exposure to MMAIII
At environmentally relevant concentrations arsenite and MMAIII can generate DNA damaging ROS (Eblin et al., 2006). Previous studies by Eblin et al., 2006, 2008 have focused on the generation of ROS species following acute exposures in the presence of MMAIII; however, the elevation of ROS in URO-MSC cells following chronic, low-level MMAIII exposure has not been analyzed. To establish ROS as a key factor involved in generating DNA damage seen in URO-MSC cells it was necessary to determine whether URO-MSC(+)/URO-MSC(-) cells have elevated levels of ROS. In the presence of MMAIII, URO-MSC(+) cells have significantly elevated levels of ROS at 12 wk exposure [URO-MSC12(+)] which remain elevated through 52 wk in the presence of MMAIII (Fig. 3). URO-MSC(-) cells exposed to MMAIII for 12, 24, and 52 wk followed by cell culture growth for a period of 2 wk in the absence of MMAIII were assessed for the production of endogenous ROS. URO-MSC12(-), URO-MSC24(-), and URO-MSC52(-) cells have elevated levels of endogenous ROS in a time-dependent manner when compared to untreated UROtsa control, despite removal of MMAIII following previous chronic exposure (Fig. 3).
Figure 3.
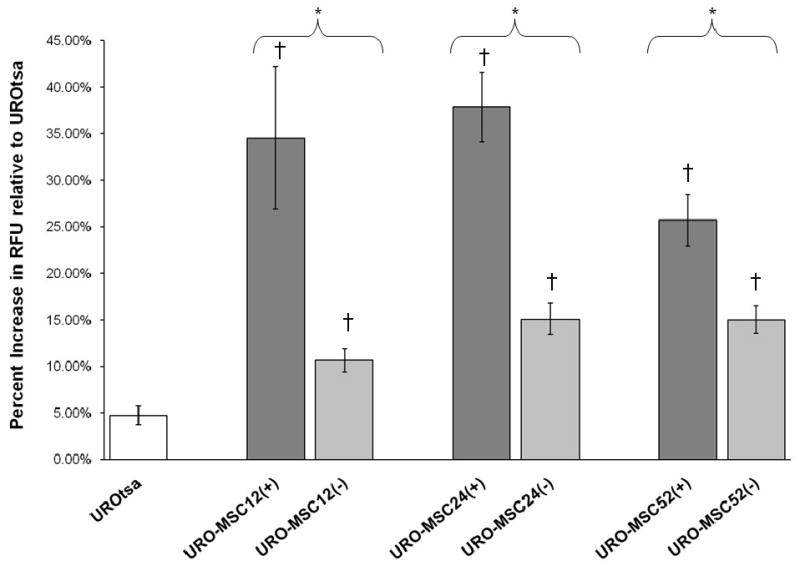
Detection of ROS generation following chronic MMAIII exposure in URO-MSC(+) and URO-MSC(-) cells. Spectrofluorometric analysis of ROS in URO-MSC12(+), URO-MSC24(+), and URO-MSC52(+) demonstrate a constitutive increase in ROS levels beginning at 12 wk exposure through 52 wk. URO-MSC12(-), URO-MSC24(-), and URO-MSC52(-) cells demonstrate a general time-dependent increase in ROS following previous chronic exposure to MMAIII. (*) Marks statistically significant difference (p ≤ 0.05) between URO-MSC(+) and URO-MSC(-) determined using Student's t-test. (†) Marks statistically significant changes in ROS levels (n=3) of treated URO-MSC(+)/URO-MSC(-) samples compared to untreated UROtsa control identified with ANOVA followed by Bonferroni's multiple comparison test; p ≤ 0.05 was considered statistically significant. Error bars within each column represent the standard error of the mean (± SEM).
To demonstrate that ROS may be responsible for the increase in DNA damage seen in URO-MSC cells chronically exposed to MMAIII, it was necessary to determine whether ROS scavengers would lead to a decrease in DNA damage in URO-MSC cells cultured in the absence of MMAIII (2 wk). Because URO-MSC12(-) cells demonstrated the initial increase in DNA damage and elevation in ROS levels following chronic MMAIII exposure, these cells were chosen to demonstrate the role of ROS in generating DNA damage. To further exemplify the involvement of ROS in damaging DNA, the malignantly transformed URO-MSC52(-) cells demonstrating the greatest levels of DNA damage were also incubated with ROS scavengers. URO-MSC12(-)/URO-MSC52(-) cells were incubated with the ROS scavengers: KI (5 mM), SOD (100 units/ml), and CAT (200 units/ml) every 2 days for a 14 day period. URO-MSC12(-)/URO-MSC52(-) cells showed a significant decrease in DNA damage as measured using the alkaline comet assay following incubation with SOD, CAT, or KI (Fig. 4a, 4b). These data show that DNA damage following MMAIII exposure is partially dependent on the generation of ROS. Interestingly, previous studies have indicated that superoxide, O2-, is the primary ROS species produced by arsenite exposure. Superoxide formation in biological systems has the potential to result in the production of H2O2, 1O2, and ˙OH, all of which have significant roles in the generation of oxidative stress, perturbations in signaling pathways, and DNA damage (Wiseman and Halliwell, 1996; Buetler et al., 2004; Leonard et al., 2004; Shi et al., 2004a, 2004b; Beyersmann and Hartwig, 2008). Incubation of URO-MSC cells with SOD, KI, or CAT significantly reduced the levels of DNA damage, supporting the role of arsenical-induced O2-, ·OH, and H2O2 in producing a genotoxic insult.
Figure 4.
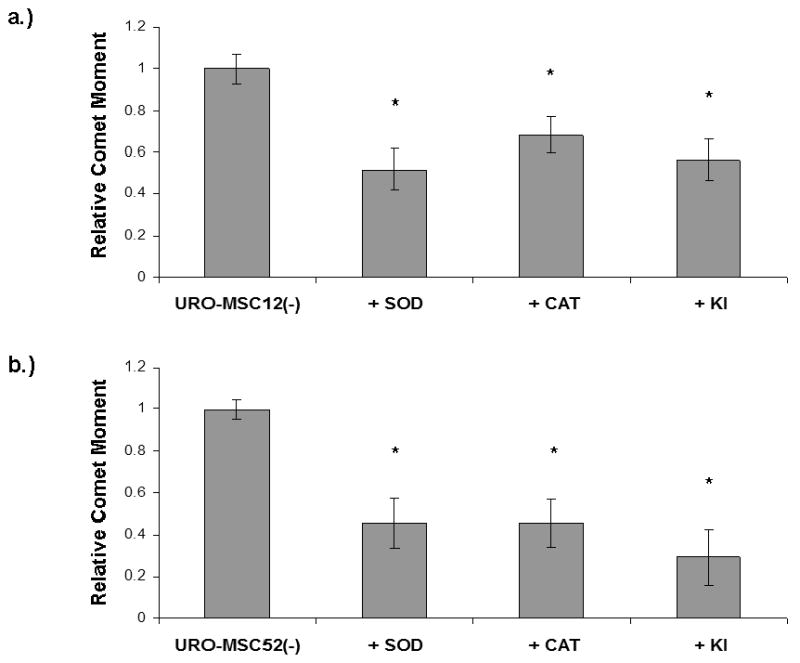
a.) Decrease in DNA damage in URO-MSC12(-) cells incubated with ROS scavengers following chronic, low-level exposure to MMAIII. b.) Decrease in DNA damage in URO-MSC52(-) cells incubated with ROS scavengers following chronic, low-level exposure to MMAIII. Graphs depict average comet moment of URO-MSC12(-)/URO-MSC52(-) cells after treatment with ROS scavengers: SOD, CAT, and KI. Average comet moment is decreased in URO-MSC12(-)/URO-MSC52(-) cells incubated with SOD (100 units/ml), CAT (200 units/ml), or KI (5 mM) for 2 wk. Statistically significant differences in DNA comet moment were identified with ANOVA followed by Bonferroni's multiple comparison test; p ≤ 0.01 was considered statistically significant and marked by asterisk (*). Error bars within each column represent the standard error of the mean (± SEM).
3.3 Analysis of DNA single strand breaks
To verify the extent of single strand DNA (ssDNA) breaks, flow cytometry was conducted using a mouse primary antibody directed against ssDNA breaks. Results demonstrate a statistically significant increase in single stranded DNA damage in URO-MSC12(+), URO-MSC24(+), URO-MSC36(+), and URO-MSC52(+) cells compared to UROtsa control, consistent with the level of damage seen after treatment with the positive control, etoposide (Fig. 5). By 12 wk of exposure to MMAIII, a 3-fold increase in ssDNA was present, and the increase was maintained throughout continuous exposure to MMAIII. URO-MSC12(-), URO-MSC24(-), URO-MSC36(-), and URO-MSC52(-) cells out of MMAIII demonstrate a time-dependent increase in ssDNA damage (Fig. 5). The ssDNA damage closely resembles the results seen when MMAIII-induced DNA damage was measured using the alkaline comet assay (Fig. 2). Taken together, these data demonstrate that not only does significant damage to DNA occur in the presence of low-level MMAIII, but even upon removal of chronic, low-level MMAIII exposure URO-MSC(-) cells maintain an increase in DNA damage. The DNA damage levels in URO-MSC(-) cells remain elevated relative to untreated UROtsa levels after the removal of the MMAIII exposure and increase in a time-dependent manner. As the length of exposure to MMAIII increases in URO-MSC cells, the difference in the levels of ssDNA damage between URO-MSC(+) cells and URO-MSC(-) cells decreases.
Figure 5.
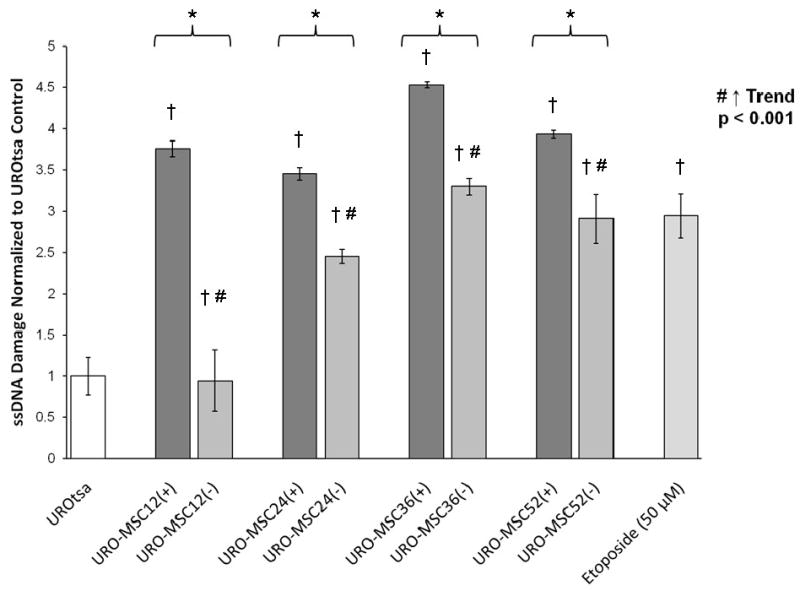
Comparison of ssDNA damage in the presence/absence of MMAIII following chronic, low-level exposure to MMAIII in URO-MSC cells. (*) Marks statistically significant difference (p ≤ 0.05) between URO-MSC(+) and URO-MSC(-) determined using Student's t-test. (†) Marks statistically significant difference in DNA single strand breaks of URO-MSC(+)/URO-MSC(-) cells compared to UROtsa control identified with ANOVA followed by Bonferroni's multiple comparison test with p ≤ 0.05 considered statistically significant. Non-parametric test for trend demonstrates a time-dependent increase in ssDNA breaks of URO-MSC(-) cells, (#) marks statistically significant (p ≤ 0.001) upward trend. Error bars within each column represent the standard error of the mean (± SEM).
3.4 PARP repair activity following chronic MMAIII exposure
Inactivation of DNA repair enzymes via trivalent arsenic species is believed to occur through an interaction with potential molecular targets containing zinc finger domains (Hartwig et al., 2003a; 2003b). One particular DNA repair enzyme of interest involved in the repair of single stranded DNA damage is poly (ADP-ribose) polymerase, PARP. In the presence of MMAIII, PARP activity in URO-MSC(+) cells increased with exposure; however, levels are only significantly increased above UROtsa control levels in URO-MSC36(+), and URO-MSC52(+) cells (Fig. 6). PARP activity levels in URO-MSC(+) cells are comparable to levels of PARP in UROtsa cells incubated with etoposide, which was utilized as a positive control. In regards to DNA repair activity in the absence of MMAIII, relative PARP activity significantly increases in a time-dependent manner in cultured URO-MSC24(-), URO-MSC36(-), and URO-MSC52(-) cells. The results demonstrate that in the presence of MMAIII, URO-MSC24(+), URO-MSC36(+), and URO-MSC52(+) cells have a general decrease in PARP activity compared to URO-MSC(-) cells devoid of MMAIII; yet, significant difference is only reached in URO-MSC52(+) cells compared to URO-MSC52(-) cells.
Figure 6.
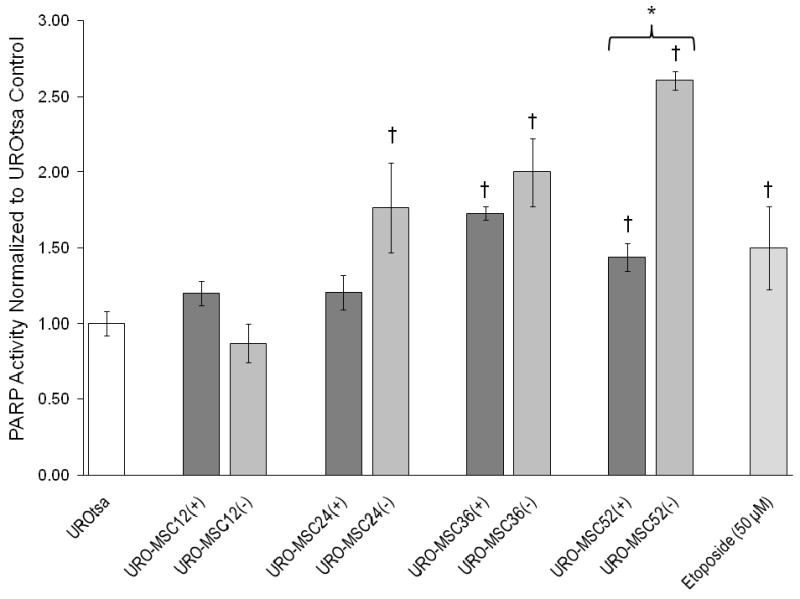
Comparison of relative PARP activity in the presence/absence of MMAIII following chronic, low-level exposure to MMAIII in URO-MSC cells. Graph depicts changes in PARP activity between MMAIII exposed URO-MSC cells and untreated UROtsa control. (*) Marks statistically significant difference (p ≤ 0.05) between URO-MSC(+) and URO-MSC(-) determined using Student's t-test. (†) Marks statistically significant difference in PARP activity of URO-MSC(+)/URO-MSC(-) cells compared to UROtsa control identified with ANOVA followed by Bonferroni's multiple comparison test with p ≤ 0.05 considered statistically significant. Error bars within each column represent the standard error of the mean (± SEM).
4. Discussion
Inorganic arsenicals do not react with DNA directly to cause DNA damage, nor are they significantly mutagenic in most classical mutagenesis assays at low concentrations (Hei et al., 1998; Klein et al., 2007). However, the arsenic species, AsIII and MMAIII, have been noted to play an indirect role in DNA damage via the generation of ROS (Hei et al., 1998; Schwerdtle et al., 2003; Shi et al., 2004a, 2004b; Eblin et al., 2006; Klein et al., 2007). The induction of chromosomal aberrations, DNA strand breaks, and oxidative DNA base damage have been noted following low-level arsenic exposure in cultured mammalian cell lines (Schoen et al., 2004).
To determine whether chronic exposure to 50 nM MMAIII results in an increase in DNA damage, the extent of DNA damage in UROtsa cells in the presence of MMAIII after 12, 24, 36, and 52 wk exposure was analyzed via alkaline comet assay. DNA damage was significantly increased in UROtsa cells after 12 wk exposure [URO-MSC12(+)]. This increase in DNA damage in the presence of MMAIII remained statistically elevated and at comparable levels through 52 wk exposure [URO-MSC24(+), URO-MSC36(+), and URO-MSC52(+)] (Fig. 2). To examine the biological implications of chronic, low-level MMAIII on the ability to generate DNA damage in the absence of MMAIII, the alkaline comet assay was carried out in URO-MSC cells after a 2 wk removal of MMAIII. The DNA damage levels were significantly decreased compared to URO-MSC(+) cells at the time of experimentation; however, these levels remain significantly elevated above control and increased with prolonged exposure to MMAIII. The linear increase in DNA damage demonstrated in URO-MSC(-) cells coincide with growing evidence of malignant transformation within UROtsa cells following chronic, low-level exposure to MMAIII. Bredfeldt et al., 2006 demonstrated that URO-MSC cells obtain characteristics of anchorage independent growth after 24 wk MMAIII exposure and tumor growth in immunocompromised mice after 52 wk MMAIII exposure. The combination of the DNA damage data in the presence and absence of MMAIII exposure strengthens the possibility of an indirect genotoxic insult as a result of arsenical-induced ROS.
Chronic MMAIII exposure can alter cell signaling events, which can lead to an upregulation of endogenous ROS as a result of secondary ROS generation in URO-MSC cells no longer exposed to MMAIII. Although experimentation has been shown to produce elevated levels of ROS in the presence of MMAIII, the secondary ROS generation in the absence of MMAIII has the potential to regenerate elevated levels of ROS resulting in subsequent DNA damage (Eblin et al., 2006). Previous studies, have demonstrated the inactivation of ROS with antioxidants in URO-MSC cells results in a decrease in colony formation in soft agar, suggesting that activated Ras and NADPH oxidase could play a role in producing secondary ROS. Eblin et al. 2009, has also established the importance of constitutive activation of the MAPK pathway and subsequent ROS generation leading to the upregulation of Ras in URO-MSC cells following chronic MMAIII exposure. Activation of Ras through ROS can act through a positive feedback mechanism, resulting in increased ROS production (Serù et al., 2004). This is important as it establishes a possible mechanism for maintaining elevated endogenous ROS following chronic MMAIII exposure, which may contribute to the induction of DNA damage, even after the removal of MMAIII.
Chronic, low-level exposure to MMAIII results in elevated levels of ROS. Results indicate that in the presence of MMAIII, URO-MSC(+) cells have significantly elevated levels of ROS at 12 wk exposure [URO-MSC12(+)] which remain elevated through 52 wk in the presence of MMAIII [URO-MSC52(+)]. URO-MSC(-) cells have elevated levels of endogenous ROS, which increase in a time-dependent manner when compared to untreated UROtsa cells despite removal of MMAIII following previous chronic exposure (Fig. 3). Following acute exposure Eblin et al., 2006 demonstrated that 50 nM MMAIII in UROtsa cells results in the generation of ROS; furthermore, pretreatment of these cells with superoxide dismutase or catalase ameliorate the increase in ROS. The production of ROS in UROtsa cells following chronic MMAIII exposure has the potential to induce malignant transformation. Previous investigations have demonstrated that acute exposure to MMAIII results in increased ROS and numerous alterations of growth signaling pathways including the activation of signaling cascades involving stress-related cellular proliferation (Eblin et al., 2006, 2007; Wang et al., 2007). Furthermore, evidence of the involvement of ROS as a potential player involved in cellular hyperproliferation and anchorage independent growth, two common characteristics of malignant transformation, have been established (Eblin et al., 2009).
Support for the importance of ROS in the induction of DNA damage following chronic, low-level MMAIII exposure comes from the partial decrease in DNA damage in URO-MSC12(-) and URO-MSC52(-) cells incubated with ROS scavengers. Incubation of URO-MSC12(-)/URO-MSC52(-) cells with the ROS scavengers SOD, CAT, and KI each resulted in a significant decrease in the level of DNA damage produced (Fig. 4a, 4b). Previous studies have indicated that superoxide, O2-, is the primary ROS species produced by AsIII exposure (Buetler et al., 2004; Shi et al., 2004). Due to the ability of O2- to form other free radicals (H2O2 and ·OH) involved in damaging DNA, blocking O2- with SOD could potentially decrease the ability of downstream free radicals to generate further DNA damage. These results establish the significance of ROS in the induction of DNA damage demonstrated in URO-MSC cells.
As the generation of DNA damage in URO-MSC cells was shown to be dependent on the MMAIII-induced ROS, it was important to determine the type of DNA damage being generated in order to investigate the importance of DNA repair enzymes particular to the type of DNA damage present. Utilizing a monoclonal antibody generated against ssDNA, URO-MSC(+) cells had a similar trend in DNA damage as URO-MSC12(+), URO-MSC24(+), URO-MSC36(+), and URO-MSC52(+) that were previously demonstrated using the comet assay (Fig. 5). Single stranded DNA damage was first elevated at 12 wk MMAIII exposure and consistently elevated through 52 wk. In the absence of MMAIII there was a decrease in ssDNA damage, but with prolonged previous, chronic exposure to MMAIII the difference between ssDNA damage levels between URO-MSC(+) and URO-MSC(-) cells decreased. In the absence of MMAIII, URO-MSC12(-), URO-MSC24(-), URO-MSC36(-), and URO-MSC52(-) resulted in a time-dependent increase in ssDNA damage (Fig. 5) similar to the results demonstrated using the comet assay (Fig. 2).
Arsenical exposure not only contributes to the generation of ROS involved in DNA damage, but has also been shown to inhibit DNA repair mechanisms specific to ROS-induced damage (Snow et al., 1999). As the role of MMAIII-induced generation of ROS and subsequent involvement in DNA damage has been investigated, it was important to determine if the presence/absence of MMAIII resulted in altered repair activity of PARP, a key repair enzyme in DNA single strand break repair. In URO-MSC(+) cells, PARP activity only slightly increased following chronic exposure to 50 nM MMAIII. PARP activity levels were significantly elevated above control UROtsa levels after 36 wk exposure; however, these levels remained decreased when compared to URO-MSC(-) cells removed from MMAIII exposure. URO-MSC cells following a 2 wk removal of MMAIII had a time-dependent increase in PARP activity. The time-dependent increase in PARP activity in URO-MSC(-) cells may be elevated as a result of the elevated levels of DNA damage. PARP activity levels in URO-MSC(-) cells may be elevated to account for the increase in DNA damage. These data demonstrate that in the presence of MMAIII, PARP activity levels are reduced, which could explain the vast elevation of DNA damage; however, after removal of MMAIII exposure, PARP activity levels elevate allowing for subsequent initiation of DNA repair activity. The increase in PARP activity may explain the subsequent decrease in DNA damage levels seen in URO-MSC(-) cells devoid of MMAIII when compared to URO-MSC(+) cells.
In summary, exposure to 50 nM MMAIII results in an increase in ROS following chronic exposure. Of great significance, the concentrations of MMAIII used in this study represent an environmentally relevant level of exposure. In addition, ROS upregulation was determined to play a role in the induction of DNA damage; in particular, the ability to generate ssDNA damage. The DNA damage demonstrated in URO-MSC12(-)/URO-MSC52(-) can be decreased through treatment with the ROS scavengers SOD, KI, and CAT which block O2-, ·OH, and break down H2O2, respectively. These data demonstrate that chronic, low-level MMAIII-induced ROS have the capacity to cause genotoxic insult. Following chronic, low-level MMAIII exposure in URO-MSC cells, DNA damage in the presence of MMAIII is significantly elevated after 12 wk and remains elevated through 52 wk. Upon removal of MMAIII for 2 wk, DNA damage levels in URO-MSC cells remain elevated and increase in a time-dependent manner when compared to control UROtsa despite the absence of MMAIII. In 2004, Yamauchi et al. demonstrated that the length of inorganic arsenic exposure dictates the ability of individuals to recover from oxidative DNA damage. In the URO-MSC(-) cells following previous chronic, low-level exposure DNA damage levels remain elevated, possibly due to an increase in the length of time necessary to recover. The decreased levels of PARP activity in the presence of MMAIII may also explain the significantly elevated levels of DNA damage first occurring at 12 wk in the presence of 50 nM MMAIII. When MMAIII is removed for 2 wk, a time-dependent increase in PARP activity may account for the decrease in DNA damage levels in URO-MSC(-) cells out of MMAIII exposure. Taken together, the increased DNA damage as a result of MMAIII-induced ROS in concert with an overall decrease in repair capacity in the presence of MMAIII may play a key role in generating an indirect genotoxic insult and an overall contribution in the progression of arsenic-induced bladder carcinogenesis.
Acknowledgments
The authors would like to thank Drs. Donald and Mary Ann Sens and Dr. Scott Garret for the UROtsa cells and assistance with culturing conditions, Dr. Joceline Lega (Department of Mathematics, University of Arizona) for the comet analysis software, Doug Cromey (SWEHS Imaging Core Facility) for his help with image analysis, and the Synthetic Chemistry Facility Core (Southwest Environmental Health Sciences Center, Tucson, AZ) for the Diiodomethylarsine (MMAIII). The analyses for the biotransformation and persistence of MMAIII were performed by the Analytical Section of the Hazard Identification Core from our NIEHS-supported Superfund Basic Research Program Grant (ES 04940). We wish to acknowledge Michael Kopplin for performing the metal analyses. The research herein was made possible by the NIEHS Superfund Basic Research Program (ES 04940) and the Southwest Environmental Health Sciences Center (ES 06694). SMW is funded by an NIEHS Training Grant (ES 07091).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors maybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlborn GJ, Nelson GM, Ward WO, Knapp G, Allen JW, Ouyang M, Roop BC, Chen Y, O'Brien T, Kitchin KT, Delker DA. Dose response evaluation of gene expression profiles in the skin of K6/ODC mice exposed to sodium arsenite. Toxicol Appl Pharmacol. 2008;227:400–416. doi: 10.1016/j.taap.2007.10.029. [DOI] [PubMed] [Google Scholar]
- Aposhian HV. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu Rev Pharmacol Toxicol. 1997;37:397–419. doi: 10.1146/annurev.pharmtox.37.1.397. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Gurzau ES, Le XC, Gurzau A, Healy SM, Lu X, Ma M, Yip Li, Zakharyan RA, Maiorina RM, Dart RC, Tircus MG, Gonzalez-Ramirez D, Morgan DL, Avram D, Aposhian MM. Occurrence of monomethylarsonous acid in urine of humans exposed to inorganic arsenic. Chem Res Toxicol. 2000;13:693–697. doi: 10.1021/tx000114o. [DOI] [PubMed] [Google Scholar]
- Beyersmann D, Hartwig A. Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch Toxicol. 2008;82(8):493–512. doi: 10.1007/s00204-008-0313-y. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Kopplin MJ, Gandolfi AJ. Effects of arsenite on UROtsa cells: low-level arsenite causes accumulation of ubiquitinated proteins that is enhanced by reduction in cellular glutathione levels. Toxicol Appl Pharmacol. 2004;198(3):412–418. doi: 10.1016/j.taap.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Jagadish B, Eblin KE, Mash EA, Gandolfi AJ. Monomethylarsonous acid induced transformation of human bladder cells. Toxicol Appl Pharmacol. 2006;216:69–79. doi: 10.1016/j.taap.2006.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetler TM, Krauskopf A, Ruegg UT. Role of superoxide as a signaling molecule. News Physiol Sci. 2004;19(3):120–123. doi: 10.1152/nips.01514.2003. [DOI] [PubMed] [Google Scholar]
- Brown KG, Ross GL. Arsenic, Drinking Water, and Health: A Position Paper of the American Council on Science and Health. Reg Tox Pharm. 2002;36:162–174. doi: 10.1006/rtph.2002.1573. [DOI] [PubMed] [Google Scholar]
- Caldecott KW. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Rep. 2007;6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Chai CY, Huang YC, Hung WC, Kang WY, Chen WT. Arsenic salt-induced DNA damage and expression of mutant p53 and COX-2 proteins in SV-40 immortalized human uroepithelial cells. Mutagenesis. 2007;22(6):403–408. doi: 10.1093/mutage/gem035. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Kuo TL, Wu MM. Arsenic and cancers. Lancet. 1988;1:414–415. doi: 10.1016/s0140-6736(88)91207-x. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bowen ME, Cromey DW, Bredfeldt TG, Mash EA, Lau SS, Gandolfi AJ. Arsenite and monomethylarsonous acid generate oxidative stress response in human bladder cell culture. Toxicol Appl Pharmacol. 2006;217:7–14. doi: 10.1016/j.taap.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Buffington S, Gandolfi AJ. Mitogenic Signal Transduction Caused by Monomethylarsonous Acid in Human Bladder Cells: Role in Arsenic-Induced Carcinogenesis. Toxicol Sci. 2007;95(2):321–330. doi: 10.1093/toxsci/kfl160. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Hau AM, Jensen TJ, Futscher BW, Gandolfi AJ. The role of reactive oxygen species in arsenite and monomethylarsonous acid-induced signal transduction in human bladder cells: Acute studies. Toxicol. 2008;255:107–114. doi: 10.1016/j.tox.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblin KE, Jensen TJ, Wnek SM, Buffington SE, Futscher BW, Gandolfi AJ. Reactive Oxygen Species regulate properties of transformation in UROtsa cells exposed to monomethylarsonous acid by modulating MAPK signaling. Toxicol. 2009;255:107–114. doi: 10.1016/j.tox.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankfurt OS, Robb JA, Sugarbaker EV, Villa L. Monoclonal Antibody to Single-Stranded DNA Is a Specific and Sensitive Cellular Marker of Apoptosis. Experimental Cell Res. 1996;226:387–397. doi: 10.1006/excr.1996.0240. [DOI] [PubMed] [Google Scholar]
- Gong Z, Lu X, Cullen WR, Le C. Unstable Trivalent Arsenic Metabolites, Monomethylarsonous Acid and Dimethylarsinous Acid. J Anal At Spectrom. 2001;16:1409–1413. [Google Scholar]
- Hartwig A, Blessing H, Schwerdtle T, Walter I. Modulation of DNA repair processes by arsenic and selenium compounds. Toxicol. 2003a;193:161–169. doi: 10.1016/j.tox.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Hartwig A, Pelzer A, Asmuss M, Burkle A. Very Low Concentrations of Arsenite Suppress Poly (ADP-ribosyl)ation In Mammalian Cells. Int J Cancer. 2003b;104:1–6. doi: 10.1002/ijc.10911. [DOI] [PubMed] [Google Scholar]
- Healy SM, Casarez EA, Ayala-Fierro F, Aposhian HV. Enzymatic Methylation of Arsenic Compounds. Toxicol Appl Pharmacol. 1998;148:65–70. doi: 10.1006/taap.1997.8306. [DOI] [PubMed] [Google Scholar]
- Hei TK, Liu SX, Waldren C. Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc Natl Acad Sci USA. 1998;95:8103–8107. doi: 10.1073/pnas.95.14.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Novak P, Eblin KE, Gandolfi AJ, Futscher BW. Epigenetic remodeling during arsenical-induced malignant transformation. Carcinogenesis. 2008;29(8):1500–8. doi: 10.1093/carcin/bgn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Wozniak RJ, Eblin KE, Wnek SM, Gandolfi AJ, Futscher BW. Epigenetic mediated transcriptional activation of WNT5A participates in arsenical-associated malignant transformation. Toxicol Appl Pharmacol. 2009;255(12):107–14. doi: 10.1016/j.taap.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchin KT, Wallace K. The role of protein binding of trivalent arsenicals in arsenic carcinogenesis and toxicity. J Inorg Biochem. 2008;102:532–539. doi: 10.1016/j.jinorgbio.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Klein CB, Leszczynska J, Hickey C, Rossman TG. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol Appl Pharmacol. 2007;222:289–297. doi: 10.1016/j.taap.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard SS, Harris GK, Shi X. Metal-induced oxidative stress and signal transduction. Free Radic Biol Med. 2004;37:1921–1942. doi: 10.1016/j.freeradbiomed.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Ludwig S, Hoffmeyer A, Goebeler M, Kilian K, Hafner H, Neufeld B, Han J, Rapp UR. The stress inducer arsenite activates mitogen-activated protein kinases extracellular signal-regulated kinases 1 and 2 via a MAPK kinase 6/p38-dependent pathway. J Biol Chem. 1998;273:1917–1922. doi: 10.1074/jbc.273.4.1917. [DOI] [PubMed] [Google Scholar]
- Lui F, Jan KY. DNA damage in arsenite- and cadmium-treated bovine aortic endothelial cells. Free Radic Biol Med. 2000;28(1):55–63. doi: 10.1016/s0891-5849(99)00196-3. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Ogra Y, Suzuki KT. Identification of dimethylarsonous and monomethylarsonous acids in human urine of the arsenic-affected areas in West Bengal, India. Chem Res Toxicol. 2001;14:371–378. doi: 10.1021/tx000246h. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Ogra Y, Anzai K, Suzuki KT. Speciation of arsenic in biological samples. Toxicol Appl Pharmacol. 2004 Aug 1;198:307–318. doi: 10.1016/j.taap.2003.10.030. [DOI] [PubMed] [Google Scholar]
- Marshall G, Ferreccio C, Yuan Y, Bates MN, Steinmaus C, Selvin S, Liaw J, Smith AH. Fifty-year study of lung and bladder cancer mortality in Chile related to arsenic in drinking water. J Natl Cancer Inst. 2007;99:920–928. doi: 10.1093/jnci/djm004. [DOI] [PubMed] [Google Scholar]
- Millar IT, Heany H, Heinehey DM, Fernelius WC. Methyliiodoarsine Inorg Synth. 1960;6:113–115. [Google Scholar]
- National Research Council. Arsenic in Drinking Water (2001 update) National Academy Press; Washington, DC: 2001. [Google Scholar]
- Nesnow S, Roop BC, Lambert G, Kadiiska M, Mason RP, Cullen WR, Mass MJ. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. Chem Res Toxicol. 2002;15(12):1627–1634. doi: 10.1021/tx025598y. [DOI] [PubMed] [Google Scholar]
- Petzoldt JL, Leigh IM, Duffy PG, Sexton C, Masters JR. Immortalisation of human urothelial cells. Urol Res. 1995;23:377–380. doi: 10.1007/BF00698738. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis A, Mukapadhyay R, Rosen BP, Stemmler TL. Experimental and Theoretical Characterization of Arsenite in Water: Insights into the Coordination Environment of As-O. Inorg Chem. 2004;43:2954–2959. doi: 10.1021/ic0351592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan K, Shila S, Kumaran S, Panneerselvam C. Ascorbic acid and α-tocopherol as potent modulators on arsenic induced toxicity in mitochondria. J of Nutritional Biochemistry. 2003;14:416–420. doi: 10.1016/s0955-2863(03)00076-7. [DOI] [PubMed] [Google Scholar]
- Rossi MR, Masters JR, Park S, Todd JH, Garrett SH, Sens MA, Somji S, Nath J, Sens DA. The immortalized UROtsa cell line as a potential cell culture model of human urothelium. Environ Health Perspect. 2001;109:801–808. doi: 10.1289/ehp.01109801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen A, Beck B, Sharma R, Dube E. Arsenic toxicity at low doses: epidemiological and mode of action considerations. Toxicol Appl Pharmacol. 2004;198:253–267. doi: 10.1016/j.taap.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Schwerdtle T, Walter I, Mackiw I, Hartwig A. Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA. Carcinogenesis. 2003;24(5):967–974. doi: 10.1093/carcin/bgg018. [DOI] [PubMed] [Google Scholar]
- Sens DA, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic Cadmium- and Arsenite-Induced Malignant Transformation of Human Bladder Urothelial Cells. Toxicol Sci. 2004;79:56–63. doi: 10.1093/toxsci/kfh086. [DOI] [PubMed] [Google Scholar]
- Serù R, Mondola P, Damiano S, Svegliati S, Agnese S, Avvedimento EV, Santillo M. HaRas activates the NADPH oxidase complex in human neuroblastoma cells via extracellular signal-related kinase ½ pathway. J Neurochem. 2004;91:613–622. doi: 10.1111/j.1471-4159.2004.02754.x. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Kashon ML, Kommineni C, Crecelius E, Luster MI. Quantitative relationship between arsenic exposure and AP-1 activity in mouse urinary bladder epithelium. Toxicol Sci. 2001;60:279–284. doi: 10.1093/toxsci/60.2.279. [DOI] [PubMed] [Google Scholar]
- Sinha BK, Haim N, Dusre L, Kerrigan D, Pommier Y. DNA Strand Breaks Produced by Etoposide (VP-16,213) in Sensitive and Resistant Human Breast Tumor Cells: Implications for the Mechanism of Action. Cancer Res. 1988;48:5096–5100. [PubMed] [Google Scholar]
- Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004a;255:67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- Shi H, Hudson LG, Liu KJ. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radical Biol Med. 2004b Sep 1;37:582–593. doi: 10.1016/j.freeradbiomed.2004.03.012. Review. [DOI] [PubMed] [Google Scholar]
- Snow E, Hu Y, Yan C, Chouchane S. Modulation of DNA repair and glutathione levels in human keratinocytes by micromolar arsenite. In: Chappell WR, Abernathy CO, Calderon R, editors. Proceedings of 4th International Conference on Arsenic Exposure and Health Effects; San Diego, CA. 1999. [Google Scholar]
- Styblo M, Drobna Z, Jaspers I, Lin S, Thomas DJ. The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ Health Perspect. 2002;110(Suppl 5):767–771. doi: 10.1289/ehp.110-1241242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapio S, Grosche B. Arsenic in the etiology of cancer. Mutat Res. 2006;612(3):215–246. doi: 10.1016/j.mrrev.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Tun-Kyi A, Qin JZ, Oberholzer PA, Navarini AA, Hassel JC, Dummer R, Döbbeling U. Arsenic trioxide down-regulates antiapoptotic genes and induces cell death in mycosis fungoides tumors in a mouse model. Annals of Oncology. 2008;19(8):1488–1494. doi: 10.1093/annonc/mdn056. [DOI] [PubMed] [Google Scholar]
- Vahter M. Mechanisms of arsenic biotransformation. Toxicol. 2002:181–182. 211–217. doi: 10.1016/s0300-483x(02)00285-8. [DOI] [PubMed] [Google Scholar]
- Van Maanen JMS, Retel J, De Vries J, Pinedo HM. Mechanism of action of antitumour drug etoposide: a review. J Natl Cancer Inst. 1988;80:1526–1533. doi: 10.1093/jnci/80.19.1526. [DOI] [PubMed] [Google Scholar]
- Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical Biology & Medicine. 1999;27(56):612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi AJ, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol Appl Pharmacol. 2007;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313:17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi H, Aminaka Y, Yoshida K, Sun G, Pi J, Waalkes MP. Evaluation of DNA damage in patients with arsenic poisoning: urinary 8-hydroxydeoxyguanine. Toxicol Appl Pharmacol. 2004;198:291–296. doi: 10.1016/j.taap.2003.10.021. [DOI] [PubMed] [Google Scholar]