

Abstract
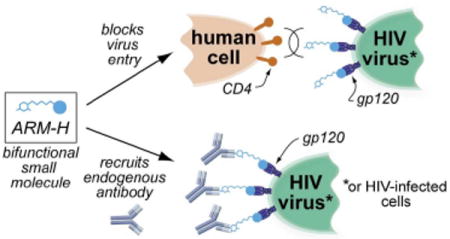
HIV/AIDS is a global pandemic for which new treatment strategies are desperately needed. We have designed a novel small molecule with the potential to interfere with HIV survival through two mechanisms: (1) by recruiting antibodies to gp120-expressing virus particles and infected human cells, thus enhancing their uptake and destruction by the human immune, and (2) by binding the viral glycoprotein gp120, inhibiting its interaction with the human protein CD4, and preventing virus entry. Here we demonstrate that ARM-H is capable of simultaneously binding gp120, a component of the Env surface viral glycoprotein (found on the surface of both HIV and virus-infected cells) and anti-2,4-dinitrophenyl antibodies (already present in the human bloodstream). The ternary complex formed between antibody, ARM-H, and gp120 is immunologically active and leads to the complement-mediated destruction of Env-expressing cells. Furthermore, ARM-H prevents virus entry into human T-cells, and should therefore be capable of inhibiting virus replication through two mutually reinforcing mechanisms (inhibition of virus entry and antibody-mediated killing). These studies demonstrate the viable anti-HIV activity of antibody-recruiting small molecules, and have the potential to initiate novel paradigms in HIV treatment.
In recent years, antibody-based therapeutics have become important instruments in treating human disease.1 These approaches suffer from certain limitations including severe side effects, lack of oral bioavailability, and high cost.2 Thus, alternative small-molecule-based methods that exploit the powerful, cytolytic potential of antibodies already present in the human bloodstream could address many of these disadvantages.
Here we report that a rationally designed bifunctional small molecule,3 called “antibody-recruiting molecule targeting HIV” (ARM-H), is capable of redirecting anti-dinitrophenyl (anti-DNP) antibodies, a population of antibodies present in high concentrations in the human bloodstream,4 to the HIV gp120 env-gene product (Fig. 1). The Env glycoprotein, a complex between gp120 and membrane-bound gp41, is expressed on both the surface of the HIV virus and on virus-infected cells.5 The gp120 component of Env mediates the first step in viral entry into human cells by binding the protein CD4. We demonstrate here that a ternary complex formed between anti-DNP antibodies, ARM-H and Env-expressing cells can mediate the complement-dependent destruction of these cells. Further, since ARM-H binds gp120 competitively with CD4, it also inhibits the entry of live HIV virus into human T-cells. Thus, ARM-H has the potential to interfere with the survival of HIV through multiple complementary mechanisms.
Figure 1.
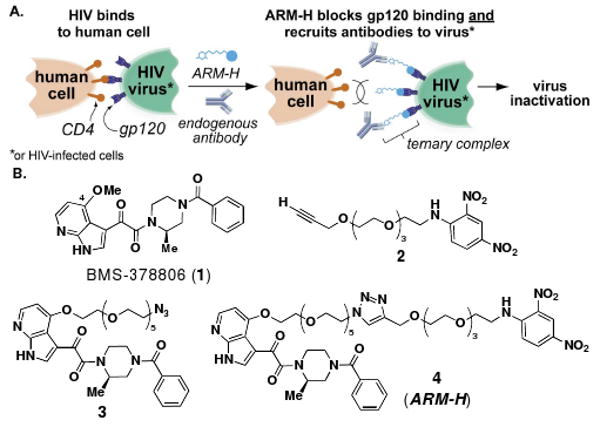
(A) Schematic depiction of the reported approach to HIV targeting. The synthetic bifunctional small-molecule ARM-H is designed to function through two autonomous mechanisms: (1) by binding the viral glycoprotein gp120, inhibiting its interaction with CD4, and (2) by recruiting antibodies to the HIV virus and to infected cells for destruction by the human immune system. (B) Chemical structures of small molecules employed in these studies.
Our design of ARM-H began with the small molecule BMS-378806 (1, Scheme 1), a known inhibitor of the CD4-gp120 interaction.6 On the basis of previously reported structure-activity relationships in which the carbon atom of the C4 methoxy group could be replaced with various bulky substituents,7 we hypothesized that it might be possible to derivatize 1 at this site with a linker attached to DNP without sacrificing the compound's ability to inhibit viral entry. This hypothesis was supported by analysis of a published computational docking model, which suggests that the C4 methoxy group in 1 points toward solvent in the complex.8 Thus we reasoned that BMS-378806 could be re-engineered to recruit anti-DNP antibodies to gp120-expressing particles (infected cells or viruses), increasing their “visibility” to the human immune system. Based upon this strategy, we prepared compound 4 in high yield (38% overall) via azide-alkyne cycloaddition9,10 of 2 and 3, which were derived in turn from known intermediates (Schemes S2 and S3).6
The ability of ARM-H to inhibit CD4 binding to HIV-1 gp120 was assessed first in an ELISA assay (Fig. 2A).11 Here ARM-H was able to out-compete the CD4-gp120 interaction with an IC50 of approximately 8.7 μM, and this is only slightly less potent than the parent compound, BMS-378806 (IC50 = 1.3 μM). On the basis of this observation, we then investigated ARM-H's ability to inhibit the entry of live HIV-1 virus into the human MT-2 T-cell line. Although in this assay ARM-H once again proved less potent than BMS-378806 (ARM-H IC50 = 6.4 μM versus 0.32 μM for BMS-378806, Fig. S1),12 it demonstrated equivalent potency to d4T, which is currently a mainstay in HIV pharmacotherapy (IC50 = 4.2 μM, Fig. S1). Notably, ARM-H demonstrated no observable cytotoxicity in control MT-2 cultures lacking HIV virus (Fig. 2B, white circles).
Figure 2.
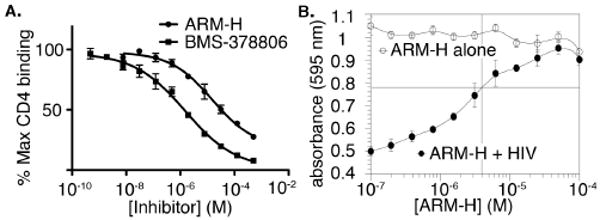
ARM-H outcompetes the gp120-CD4 interaction in vitro and with live HIV-1 virus assay. (A) Competition ELISA monitoring the binding of sCD4 to immobilized gp120. (B) HIV-1 viral replication assay. UV absorption at 595 nm, increased by the metabolic action of live MT-2 cells on an assay reagent,13 is monitored as a surrogate for cell viability in the presence of increasing concentrations of ARM-H alone (white circles), or ARM-H plus live HIV-1 virus (black circles).
We next investigated the ability of ARM-H to recruit antibodies to gp120 both in vitro, and in tissue culture. To this end, initial ELISA experiments (Fig. S2) demonstrated a concentration-dependent increase in anti-DNP antibody binding to the ARM-H–gp120 complex, but not to gp120 alone. Thus, ARM-H is capable of templating a ternary complex between gp120 and anti-DNP antibody.
To demonstrate that this ternary association could form in a complex cellular milieu, we evaluated the ability of ARM-H to recruit AlexaFluor488-labeled anti-DNP antibody to HIV-Env-expressing Chinese hamster ovary cells (CHO-gp120)14 by immunofluorescence microscopy. As depicted in Fig. 3, a strong fluorescent signal was observed when CHO-gp120 cells were incubated with ARM-H and labeled anti-DNP antibodies (panels a-b).15 This fluorescence was absent from both CHO-gp120 cells not treated with ARM-H (panels c-d), and CHO cells not coding for HIV-env gene expression (CHO-WT cells, panels e-f). Furthermore, the intense fluorescence observed in panel b was considerably diminished in the presence of the competing ligands soluble CD4 (sCD4, panels g-h), BMS-378806 (panels i-j), and a DNP-containing alkyne that lacks the gp120-binding motif (2, panels k-l). Taken together, these results provide strong evidence that ARM-H is capable of recruiting anti-DNP antibodies to cells expressing the Env glycoprotein in a fashion that depends upon its simultaneous binding both to gp120 and to anti-DNP antibodies.
Figure 3.
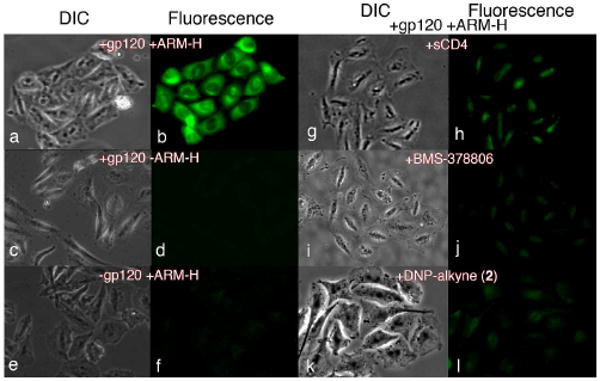
Immunofluorescence Microscopy. Chinese Hamster Ovary (CHO) cells expressing HIV-1 Envelope (Env) glycoprotein (+gp120) and control cells lacking Env (-gp120) were stained with AlexaFluor488-conjugated anti-DNP antibody (15 μg/mL). Microscopy was then performed after treatment with the following reagents, as indicated: ARM-H (10 μM), sCD4 (4 μg/mL), BMS-378806 (BMS, 500 μM), or DNP-alkyne 2 (50 μM). Fluorescence signal indicates ARM-H-mediated recruitment of antibody to gp120-expressing cell.
Finally, we explored whether the ternary complex formed between anti-DNP antibody, ARM-H (4), and live Env-expressing cells could activate complement proteins and mediate cellular death. Complement proteins are known to lyse cells by forming pores in lipid membranes, and have been shown to play a critical role in inactivating HIV in humans.16,17 Thus, rabbit complement proteins were added to gp120-positive CHO cells in the presence of ARM-H and a fixed concentration anti-DNP antibody (Fig. 4). Substantial cell killing was observed which exhibited a significant dependence on ARM-H concentration (data in red).18,19 Notably, in the absence of anti-DNP antibody and complement-preserved serum (data in green), in cells lacking the Env glycoprotein (CHO-WT, data in black), or in the presence of 3, which lacks the DNP group (data in blue), no cell death was observed suggesting that termolecular complex formation is necessary for complement-dependent cytotoxicity, and that ARM-H itself is not toxic to cells. Positive control experiments, in which cells were directly labeled with 2,4-dinitrobenzenesulfonic acid (Fig. S4), were found to yield comparable levels of CDC to those observed for ARM-H, providing a benchmark for assay results depicted in Fig. 4. Thus, ARM-H is capable of recruiting a functional complement-dependent cytotoxic response against Env-expressing cells.
Figure 4.
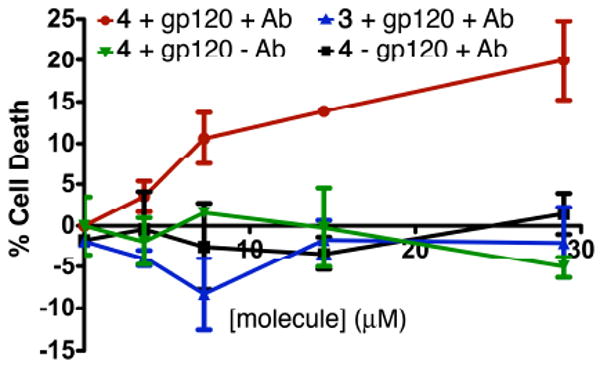
ARM-H-mediated killing of live HIV Env-expressing CHO cells. Env-expressing CHO cells (+gp120) or control cells lacking Env (-gp120) were treated with ARM-H (4) or control compound (3) at the indicated concentrations in the presence (+Ab) or absence (-Ab) of 10% complement-preserved rabbit serum and rat anti-DNP IgG (50 μg/mL). Values on the Y-axis correspond to the percent of cell death (versus background). Data depicted represents the mean ± standard deviation of triplicate data from a representative experiment. The depicted trends were reproduced on at least three separate occasions.20
Thus, we have shown that the bifunctional small molecule ARM-H can both recruit anti-DNP antibodies to gp120-expressing cells and inhibit the gp120–CD4 interaction. Data supporting these conclusions include: (1) ARM-H binds to gp120 competitively with CD4 and decreases viral infectivity in an MT-2 cell assay, (2) the small molecule can guide the formation of a ternary complex between anti-DNP antibodies and Env-expressing cells, and (3) antibodies present in this ternary complex can promote the complement-mediated killing of Env-expressing cells. Critically, no non-specific cytotoxicity was observed in either MT-2 or CHO cell lines in response to ARM-H. Also, ARM-H-mediated inhibition of HIV entry and CDC activity are both operative at concentrations ranging from 6-30 μM, confirming that ARM-H could function simultaneously through dual mechanisms under therapeutic conditions.
While a few methods for recruiting naturally occurring antibodies to cancer cells,21-27 bacteria,28-31 and viruses,32-34 have appeared in the literature, this area remains underexplored. In the HIV realm, all such approaches have relied upon protein- or peptide-based antibody targeting constructs. For example, Schultz, et al. first demonstrated that anti-DNP antibodies could be redirected to immobilized protein targets (gp120 and streptavidin) as a therapeutic strategy toward HIV.32 More recent work in this vein has employed peptide–α-Gal conjugates to target human anti-Gal antibodies to HIV-infected cells.33,34 These peptide conjugates were shown to be effective in killing Env-expressing cells, but were also found to exhibit some non-specific cytotoxicity. 28 ARM-H is unique in that it represents a small-molecule-based anti-HIV strategy, and targets the virus lifecycle through mutually reinforcing molecular mechanisms – it both prevents virus entry and targets Env-expressing cells for immune recognition and clearance. In general, small molecules have certain advantages over proteins from a therapeutic standpoint due to their low propensity for immunogenicity, high metabolic stability, ready large-scale production, and relatively low cost. Small molecule antibody-recruiting therapeutics such as ARM-H would have additional benefits over available treatment approaches to HIV. For example, directing HIV-infected cells and virus particles to Fcγ receptors on antigen presenting cells could enhance the presentation of viral antigens on MHC proteins, and contribute to long-lasting anti-HIV immunity.26,35 Further, because anti-DNP antibodies are already present in the human bloodstream, no pre-vaccination would be necessary for ARM-H activity. Also, the binding of bifunctional small-molecule targeting agents to antibodies should prolong their plasma half-life, thus increasing their effectiveness.27 Elucidation of the molecular details governing the interactions between ARM-H, gp120, and anti-DNP antibodies will assist in optimization efforts as well as in the evaluation of this strategy in more complex biological models for HIV infection. These and other investigations are currently ongoing in our laboratories.
Supplementary Material
Acknowledgments
The authors wish to thank Jacob Appelbaum and Phillip Lichtor for helpful suggestions, Caitlin Sengelaub for technical assistance, and Professor Guido Ferrari (Duke University) for helpful discussions. This work was funded by the National Institutes of Health through the NIH Director's New Innovator Award Program, grant number DP22OD002913 (D.A.S.). K.S.A. acknowledges NIH grant GM49551 for funding support. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CHO-WT (here called CHO-gp120) from Dr. Carol Weiss and Dr. Judith White and MT-2 cells, catalog no. 237, from Douglas Richman.
Footnotes
Supporting Information Available: Detailed experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brekke OH, Sandlie I. Nat Rev Drug Discov. 2003;2:52–62. doi: 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- 2.Allen TM. Nat Rev Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 3.Corson TW, Aberle N, Crews CM. ACS Chem Biol. 2008;3:677–692. doi: 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antibodies recognizing the 2,4-dinitrophenyl (DNP) epitope have been estimated to constitute 1% of circulating IgM and 0.8% of circulating IgG (see Karjalainen K, Makela O. Eur J Immunol. 1976;6:88–93. doi: 10.1002/eji.1830060204. and Farah FS. Immunology. 1973;25:217–226.). The prevalence of anti-DNP antibodies has been estimated at between 18-90% of humans (see Ortega E, Kostovetzky M, Larralde C. Mol Immunol. 1984;21:883–888. doi: 10.1016/0161-5890(84)90143-3. and Jormalainen S, Makela O. Eur J Immunol. 1971;1:471–478. doi: 10.1002/eji.1830010613.).
- 5.Miranda LR, Schaefer BC, Kupfer A, Hu ZX, Franzusoff A. Proc Natl Acad Sci U S A. 2002;99:8031–8036. doi: 10.1073/pnas.122696599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Zhang Z, Wallace O, Deshpande M, Fang H, Yang Z, Zadjura L, Tweedie D, Huang S, Zhao F, Ranadive S, Robinson B, Gong Y, Ricarrdi K, Spicer T, Deminie C, Rose R, Wang HH, Blair W, Shi P, Lin P, Colonno RJ, Meanwell NA. J Med Chem. 2003;46:4236–4239. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 7.Wang JS, Le N, Heredia A, Song HJ, Redfield R, Wang LX. Org Biomol Chem. 2005;3:1781–1786. doi: 10.1039/b415159c. [DOI] [PubMed] [Google Scholar]
- 8.Kong R, Tan J, Ma X, Chen W, Wang C. Biochim Biophys Acta. 2006;1764:766–772. doi: 10.1016/j.bbapap.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 9.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 10.Tornøe C, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 11.Ho H, Fan L, Nowicka-Sans B, McAuliffe B, Li C, Yamanaka G, Zhou N, Fang H, Dicker I, Dalterio R, Gong Y, Wang T, Yin Z, Ueda Y, Matiskella J, Kadow J, Clapham P, Robinson J, Colonno R, Lin P. J Virol. 2006;80:4017–4025. doi: 10.1128/JVI.80.8.4017-4025.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The increase in potency of BMS-378806 in MT-2 cells versus ELISA assays may be the result of a cooperative enhancement in binding to viral envelope gp120, which exists as a trimer (ELISA studies were performed using monomeric gp120). The steric bulk of ARM-H due to the C4 tether may impede binding of more than one ARM-H molecule per gp120 trimer.
- 13.More detail regarding assay conditions and troubleshooting can be found in the Supporting Information.
- 14.Weiss C, White J. J Virol. 1993;67:7060–7066. doi: 10.1128/jvi.67.12.7060-7066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Because it was necessary to permeabilize cells prior to labeling, intracellular gp120 is also observed in these micrographs.
- 16.Aasa-Chapman MMI, Holuigue S, Aubin K, Wong M, Jones NA, Cornforth D, Pellegrino P, Newton P, Williams I, Borrow P, Mcknight A. J Virol. 2005;79:2823–2830. doi: 10.1128/JVI.79.5.2823-2830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerencer M, Burek V, Crowe BA, Barrett NP, Dorner F. Microb Pathog. 1998;25:253–266. doi: 10.1006/mpat.1998.0233. [DOI] [PubMed] [Google Scholar]
- 18.The modest levels of complement-dependent cytotoxicity observed for ARM-H in Figure 4 are similar to values reported in other systems (see Ref. 34) and may result from the low levels of Env expression on the CHO-gp120 cells (see Ref. 14). Also, due to assay incompatibilities, CDC data corresponding to ARM-H concentrations greater than 30 μM could not be obtained. More details can be found in the Supporting Information.
- 19.Characteristic auto-inhibition of ternary complex formation at high levels of bifunctional molecule, arising from excess free bifunctional material driving equilibria toward formation of binary complexes, was not reliably observed in these assays (see Supporting Information for more detail). For more information on auto-inibitory behavior in ternary complexes, see Mack ET, Perez-Castillejos R, Suo Z, Whitesides GM. Anal Chem. 2008;80:5550–5555. doi: 10.1021/ac800578w. and references contained therein.
- 20.More detail regarding assay conditions and troubleshooting can be found in the Supporting Information.
- 21.Carlson C, Mowery P, Owen R, Dykhuizen EC, Kiessling L. ACS Chem Biol. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 22.Owen R, Carlson C, Xu J, Mowery P, Fasella E, Kiessling L. Chembiochem. 2007;8:68–82. doi: 10.1002/cbic.200600339. [DOI] [PubMed] [Google Scholar]
- 23.Popkov M, Gonzalez B, Sinha S, Barbas C. Proc Natl Acad Sci U S A. 2009;106:4378–4383. doi: 10.1073/pnas.0900147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popkov M, Rader C, Gonzalez B, Sinha S, Barbas C. Int J Cancer. 2006;119:1194–1207. doi: 10.1002/ijc.21924. [DOI] [PubMed] [Google Scholar]
- 25.Low P, Henne W, Doorneweerd D. Accounts Chem Res. 2008;41:120–129. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, You F, Vlahov I, Westrick E, Fan M, Low PS, Leamon CP. Mol Pharm. 2007;4:695–706. doi: 10.1021/mp070050b. [DOI] [PubMed] [Google Scholar]
- 27.Rader C, Sinha SC, Popkov M, Lerner RA, Barbas CF. Proc Natl Acad Sci U S A. 2003;100:5396–5400. doi: 10.1073/pnas.0931308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertozzi CR, Bednarski MD. J Am Chem Soc. 1992;114:5543–5546. [Google Scholar]
- 29.Bertozzi CR, Bednarski MD. J Am Chem Soc. 1992;114:2242–2245. [Google Scholar]
- 30.Li J, Zacharek S, Chen X, Wang JQ, Zhang W, Janczuk A, Wang PG. Bioorg Med Chem. 1999;7:1549–1558. doi: 10.1016/s0968-0896(99)00099-1. [DOI] [PubMed] [Google Scholar]
- 31.Krishnamurthy VM, Quinton LJ, Estroff LA, Metallo SJ, Isaacs JM, Mizgerd JP, Whitesides GM. Biomaterials. 2006;27:3663–3674. doi: 10.1016/j.biomaterials.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Shokat KM, Schultz PG. J Am Chem Soc. 1991;113:1861–1862. [Google Scholar]
- 33.Naicker KP, Li H, Heredia A, Song H, Wang L. Org Biomol Chem. 2004;2:660–664. doi: 10.1039/b313844e. [DOI] [PubMed] [Google Scholar]
- 34.Perdomo MF, Levi M, llberg MS, Vahlne A. Proc Natl Acad Sci U S A. 2008:6. [Google Scholar]
- 35.Rawool DB, Bitsaktsis C, Li Y, Gosselin DR, Lin Y, Kurkure NV, Metzger DW, Gosselin EJ. J Immunol. 2008;180:5548–5557. doi: 10.4049/jimmunol.180.8.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.