

Abstract
The association of neurofibromatosis 1 (NF1), juvenile xanthogranulomas (JXG), and juvenile myelomonocytic leukemia (JMML) has been previously reported. We describe herein this triad in a Caucasian male infant with a pathogenic mutation in the NF1 gene (neurofibromin). The clinical course from initial presentation to final diagnosis is detailed; the physical features and hematological characteristics are discussed. The patient underwent bone marrow transplantation and is currently in remission. Children with concurrent cutaneous café-au-lait and JXG lesions should be evaluated and monitored closely for the possible development of JMML.
Keywords: neurofibromatosis 1, juvenile xanthogranuloma, juvenile myelomonocytic leukemia, hepatomegaly, café-au-lait spots
INTRODUCTION
The association of neurofibromatosis 1 (NF1), juvenile xanthogranulomas (JXG), and juvenile myelomonocytic leukemia (JMML) was first described in 1958 by Royer et al [1]. A subsequent comprehensive review of the literature in 1995 concluded that this triad is more common than previously suspected and that children with both NF1 and JXG have been estimated to have a 20–32 times higher risk of developing JMML than do those with NF1 alone [2] . In the general population, the incidence of JMML is 1.2 cases per million children per year. The incidence of JMML in patients with NF1 is approximately 14% [3,4]. To date, there have been a limited number of cases of NF1 patients reported with JXG and JMML [5].
JXGs present as yellowish-red nodules and usually arise during the first 6 months of life with predilection for the head and neck. The incidence of JXG in the general population of normal children is quite high and it approximates 1–2%; JXG is rarely associated with systemic manifestations and, for the most part, represents a self-limited dermatologic disorder [6].
NF1 is an autosomal dominant disorder characterized by the presence of café-au-lait spot macules (CALS), macrocephaly, neurofibromas, cognitive deficits and a predisposition to certain neoplasias. The overall incidence of this disorder is 1/3000 live births; approximately 50% of cases are the result of spontaneous mutations [7]. The gene responsible for this disorder is neurofibromin 1, which is located on the long arm of chromosome 17.
JMML is a myelodysplastic and myeloproliferative disorder that encompasses the diagnoses formerly referred to as “juvenile chronic myelogenous leukemia (JCML)”, “chronic myelomonocytic leukemia of infancy”, and “infantile monosomy 7”. JMML accounts for 1–2% of childhood leukemias and in the United States there are about 3 cases per million children diagnosed annually [8,10].
We describe a patient with an initial presentation of CALS and JXG who was found to have JMML. The course of the diagnostic work-up is discussed along with his family history, DNA analysis, and outcome.
CASE REPORT
A 9 month old male was referred with a history of a scalp “mole” that was eventually diagnosed as a JXG based on appearance, clinical examination and subsequent dermatologic consultation. On examination he was found to also have 7 CALS and within 1 month developed 2 more JXG lesions. Additional physical findings included hepatosplenomegaly and undescended testicles. A complete blood cell count was notable for anemia (hemoglobin 10.8 gm/dl) and leukocytosis (26.7 K/ul) with 5% neutrophils, 62% lymphocytes, 28% monocytes, 4% eosinophils, and 1% basophils. The platelet count was normal. Flow cytometric immunophenotyping of the nucleated cells in the peripheral blood showed 17% were granulocytic, 62% lymphoid, and 1% erythroid precursors. There were also 2% blasts, 4% immature monocytic cells and 13% atypical monocytic cells, for a total of 20% myelomonocytic-type leukemic cells. The leukocyte alkaline phosphatase score was 4, which is below the cut-off for a reactive process. Interphase fluorescence in situ hybridization (FISH) demonstrated monosomy 7 in 44% of the peripheral blood mononuclear cells. The cerebrospinal fluid had no evidence of tumor. A chest x-ray showed diffuse interstitial lung disease and an ultrasound of the abdomen demonstrated an enlarged liver and spleen. A surveillance MRI revealed a spinal cyst that has not changed over the past 2 years. The clinico-histopathologic features met the World Health Organization criteria for the diagnosis of JMML[9].
The family history on the paternal side was significant for a distant cousin who died of “chronic myelogenous leukemia” at 10 years of age and had CALS. The patient’s father and two paternal uncles underwent a diagnostic work-up for NF1 that included MRI, ophthalmology and dermatology evaluations, and blood tests. The father’s imaging studies revealed the presence of two paraspinal, non-vertebral, tumors within the muscle at C3–C4 described as fat containing ganglioneuromas. One of the paternal uncles had a single eye lesion consistent with a Lisch nodule. Although the family history on the paternal side was suggestive of NF1, none of the relatives met criteria for the clinical diagnosis of this condition. The distant cousin died of presumed JCML and had CALS, which is highly suspicious for the diagnosis of NF1.
In order to definitively confirm for the family the etiology of the patient’s clinical presentation, we tested his DNA for mutations in the PTPN11 and NF1 genes. The patient was found to have a pathogenic mutation in the NF1 gene (c.4270-2A>G), while the PTPN11 did not reveal mutations. The parents were tested for the NF1 mutation and were found to be non-mutated (blood samples). Given the positive family history on the paternal side (suggestive of NF1 because of the distant cousin; although father and uncle did not meet criteria), we performed a buccal swab on the father (whose blood was negative for NF1 mutations) but no mutations were found. The buccal swab was performed as a non-invasive way to test for mosaicim.
The patient underwent allogeneic bone marrow transplantation with only mild graft vs. host disease in his skin. Although he developed increased CALS post-transplant, his JMML has remained in remission since that time (currently 22 months post transplant).
DISCUSSION
We describe an infant who was referred to us for evaluation of a “scalp mole”. He was shown to have an NF1 mutation and JMML. The suspected mole was determined to be a JXG. Notably, children with NF1 and JXG have been estimated to have a 20–30 fold higher risk for JMML than patients with NF1 without JXG [2]. JMML is a potentially fatal disorder that affects children usually less than 2 years of age [8,9] . Early diagnosis and treatment with stem cell transplantation is important for a successful outcome [10]. In this patient the association of JXG, NF1 and JMML was established early due to the rapid progression of the JXGs, the characteristic peripheral blood findings, clinical features of hepatosplenomegaly and interstitial infiltrates and the suspicious family history on the paternal side.
We were unable to establish a diagnosis of NF1 in the father and the paternal uncles due to lack of criteria, and negative DNA tests. Thus, the most likely explanation at this time is that our patient’s NF1 mutation is the result of a sporadic event. However, the possibility of germline mosaicism must be considered when counseling this couple for future pregnancies.
The patient is presently asymptomatic and continues to be under the care of specialists in hematology-oncology, genetics, and dermatology. No other member of his family on the paternal side has been similarly affected to date, but they also continue to be closely monitored. The occurrence of the triad of NF1, JXG, and JMML that we describe has been reported previously. Since the first reported case in 1958 [1] there have been approximately 20 additional cases in the literature. The specific diagnosis of JXG has been made clinically in most cases with only some reports describing histopathological analysis. Children with NF1 and JXG have a 20–30 fold higher risk for JMML than patients with NF1 without JXG [2]. The importance of evaluating this group of patients for JMML is highlighted once more by our case report and the review of the literature. Children with concurrent cutaneous café-au-lait and JXG lesions should be evaluated and monitored closely for the possible development of JMML.
Figure 1.
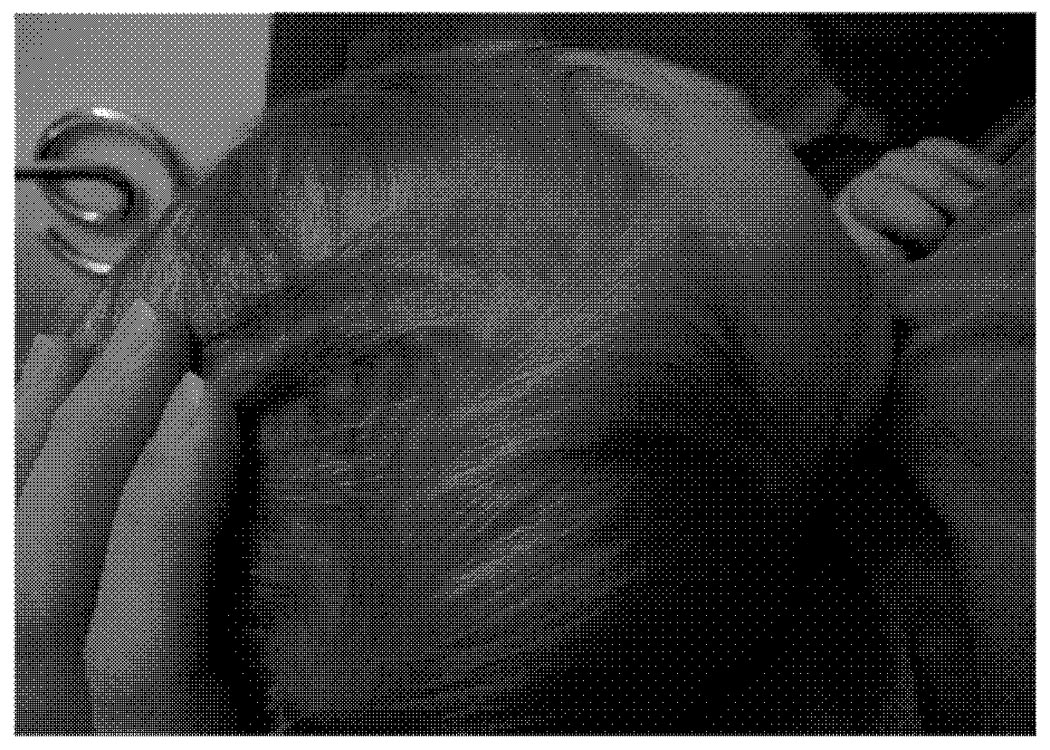
The patient was referred for the scalp lesion shown in the picture; lesion was clinically determined to be a JXG.
Figure 2.
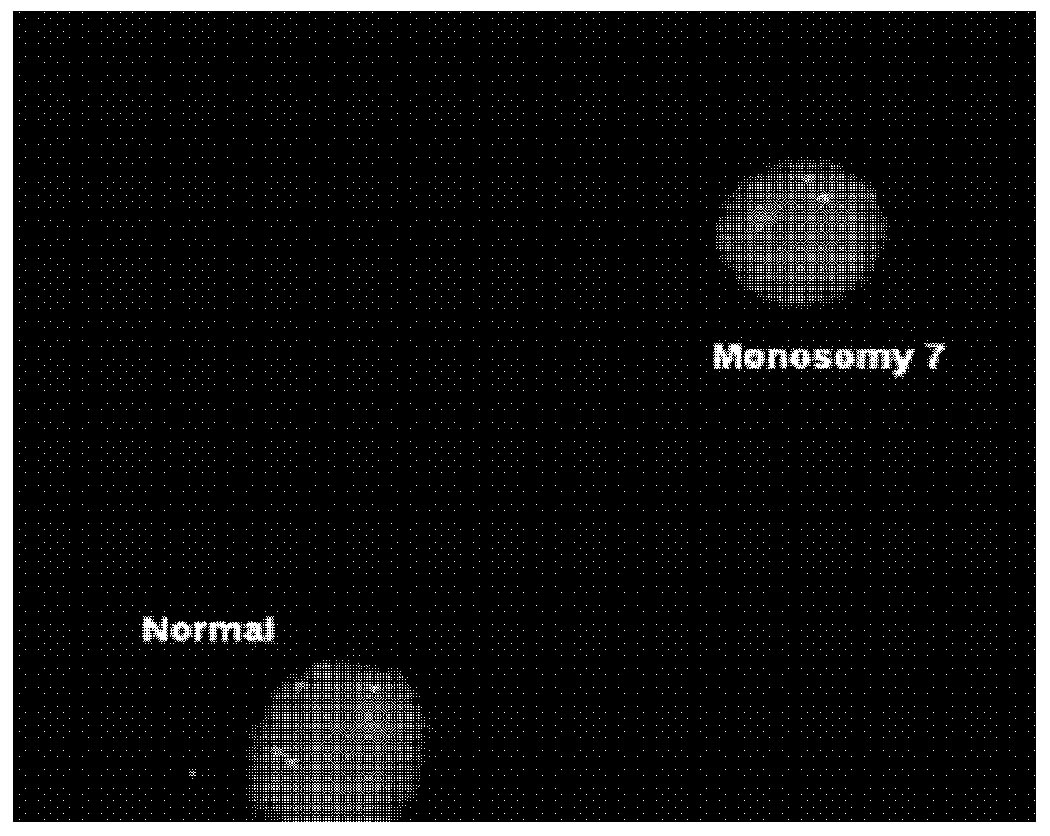
Interphase FISH with the LSI D7S522 (7q31) SpectrumOrange/CEP 7 SpectrumGreen Probe (Abbott Molecular, Inc.) revealed monosomy 7 in 44% of the peripheral blood mononuclear cells.
Acknowledgement
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and National Institute of Child Health and Human Development.
Footnotes
Conflict of Interest
The authors have none to declare.
REFERENCES
- 1.Royer P, Blondet C, Guihard J. Xantholeucemie du nourrisson et neurofibromatose de Recklinghausen. Ann Pediatr. 1958;24:1504–1513. [PubMed] [Google Scholar]
- 2.Zvulunov A, Barak Y, Metzker A. Juvenile Xanthogranuloma, neurofibromatosis, and juvenile chronic myeloid leukemia: world statistical analysis. Ach Dermatol. 1995;131:904–908. [PubMed] [Google Scholar]
- 3.Burgdorf WHC, Zelger B. JXG, NF1, and JMML: alphabet soup or a clinical issue. Peditr Dermatol. 2004;21:174–176. doi: 10.1111/j.0736-8046.2004.21219.x. [DOI] [PubMed] [Google Scholar]
- 4.Stiller CA, Chessels JM, Fitchett M. Neurofibromatosis and childhood leukemia/lymphoma ; a population-based UKCCSG study. Br J Cancer. 1994;70:969–973. doi: 10.1038/bjc.1994.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated with neurofibromatosis 1: 14 patients without evidence of hematologic malignancies. Pediatr Dermatol. 2004;21:97–101. doi: 10.1111/j.0736-8046.2004.21201.x. [DOI] [PubMed] [Google Scholar]
- 6.Stover DG, Alapati S, Regueira O, Turner C, Whitlock JA. Treatment of Xanthogranulomas. Pediatr Blood Cancer. 2008;51(1):130–133. doi: 10.1002/pbc.21523. Review. [DOI] [PubMed] [Google Scholar]
- 7.Gerber PA, Antal AS, Neumann NJ, Homey B, Matuschek C, Peiper M, Budach W, Bölke E. Neurofibromatosis. Eur J Med Res. 2009 17;14(3):102–105. doi: 10.1186/2047-783X-14-3-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urs L, Qualman SJ, Kahwash SB. Juvenile myelomonocytic leukemia: report of seven cases and review of literature. Pediatr Dev Pathol. 2009;12(2):136–142. doi: 10.2350/08-04-0456.1. 2008. [DOI] [PubMed] [Google Scholar]
- 9.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Fourth Edition. Lyon, France: IARC Press; 2008. [Google Scholar]
- 10.Niemeyer CM, Arico M, Basso G Members of the European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Chronic Meylomonocytic Leukemia in Childhood: A Retrospective Analysis of 110 Cases. Blood. 1997;89:3534–3543. [PubMed] [Google Scholar]