

Abstract
The secondary metabolites platensimycin (1) and platencin (2), isolated from the bacterial strain Streptomyces platensis, represent a novel class of natural products exhibiting unique and potent antibacterial activity. Platencin, though structurally similar to platensimycin, has been found to operate through a slightly different mechanism of action involving the dual inhibition of lipid elongation enzymes FabF and FabH. Both natural products exhibit strong, broad-spectrum, Gram-positive antibacterial activity to key antibiotic resistant strains, including methicillin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, and vancomycin-resistant Enterococcus faecium. Described herein are our synthetic efforts toward platencin, culminating in both the racemic and asymmetric preparation of the natural product. The syntheses demonstrate the power of the cobalt-catalyzed asymmetric Diels–Alder reaction and the one-pot reductive rearrangement of [3.2.1] bicyclic ketones to [2.2.2] bicyclic olefins.
Introduction
The initial euphoria associated with the discovery and development of penicillin was soon dampened by the realization that bacteria could evade its lethal activity through their ability to replicate and evolve rapidly into drug-resistant strains. The problems caused by bacterial drug-resistance have been exacerbated by overuse and often misuse of antibiotics. Thus, in 1941, when penicillin was first introduced, virtually all strains of Staphylococcus aureus were susceptible to the novel antibiotic, but by 1944 many strains had developed resistance by evolving enzymes (e.g. β-lactamase) capable of destroying the drug before it could reach its biological target.1 For a time, science and medicine were able to stay ahead of these menacing organisms by modifying existing antibiotics, but by the end of the twentieth century humanity was faced with alarming challenges from drug-resistant bacteria. For example, a recent study estimated 94,360 cases of invasive methicillin-resistant S. aureus (MRSA) occurred in the United States in 2005, with 18,650 of them resulting in death.2 Although the majority of such cases occur in hospital facilities there has been a significant rise in the number of reported community-acquired infections. This concerning state of affairs led to a renewed surge in research activities to discover and develop new antibiotics to combat the increasing threat of bacterial resistance.
Fatty acid biosynthesis, an essential process for the survival and propagation of bacteria, is the primary means by which bacteria form fatty acids necessary for energy storage and cellular structure building blocks. This, along with the significant differences between the human and bacterial fatty acid biosynthetic machinery, makes the bacterial fatty acid synthase (FAS) pathway an attractive strategy in the against drug-resistant bacteria.3,4 Taking advantage of modern biological techniques, a Merck team recently developed an antisense silencing RNA based assay for identifying inhibitors of the type II fatty acid biosynthesis (FAS II) pathway present in bacterial cells.3 The gene encoding the FabH and FabF condensing enzymes of FAS II was silenced, sensitizing the bacteria to inhibitors of these enzymes, and allowing for a high-throughput, whole cell, target-based assay. Screening of 250,000 natural product extracts ultimately led to the discovery of the dual FabF/FabH inhibitor platencin (1) and its congener platencin A1,5 along with the selective FabF inhibitor platensimycin (2).6
Platensimycin, isolated from Streptomyces platensis MA 7327, was discovered in a soil sample collected in Eastern Cape, South Africa, while platencin, isolated from S. platensis MA 7339, was found in a soil sample collected in Mallorca, Spain. The two antibiotics include in their structures the 3-amino-2,4-dihydroxy benzoic acid structural motif but differ in their more hydrophobic ketolide portion. Platensimycin consists of a tetracyclic framework that contains a cyclic ether, whereas platencin features an exclusively carbocyclic framework with only three rings. Like its sibling, platencin exhibits potent and broad in vitro and in vivo antibiotic activity against Gram-positive bacteria, including activity against a variety of drug-resistant bacteria such as methicillin-resistant S. aureus (MRSA, 1 μg/mL vs. 0.5 μg/mL for platensimycin) and vancomycin-resistant Enterococcus faecium (VREF, < 0.06 μg/mL vs. 0.01 μg/mL for platensimycin). Platencin also exhibited comparable in vivo efficacy in a continuous infusion mouse model against S. aureus infection with no indication of toxicity. As might be expected from their close structural relationship, platencin and platensimycin operate through a similar mechanism of action. Each was shown to inhibit lipid biosynthesis in whole cell labeling studies, but neither exhibited any activity against nucleic acid, protein biosynthesis or cell wall construction. At a molecular level, platensimycin is a potent and selective inhibitor of FabF, the elongation condensing enzyme; however, platencin inhibits this enzyme with much lower potency. Platencin derives its overall potency from a dual inhibition mechanism involving moderate inhibition of both FabF and the initiation condensing enzyme FabH.5 It should be noted that a recent report questioned the validity of the strategy to combat bacteria based on fatty acid biosynthesis since bacteria were found to sequester the needed fatty acids from their hosts.4
The structural novelty and potent activity of these antibiotics has attracted considerable attention, with a number of total syntheses of platensimycin7 and platencin8 reported to date. Several reports have also described the design and synthesis of related structures,9 some of which are also potent antibiotics.9a,b,d,f In 2008, we published our preliminary results culminating in the first total synthesis of platencin (1) through an asymmetric route.8a Herein we present the full account of our work in this area that led to considerable improvements of the originally developed route.
Results and Discussion
Our total synthesis endeavors toward platencin, first as a racemate, and then in its enantiomerically pure form, proceeded through a number of phases and evolved into a highly efficient and streamlined process. Below we describe these studies in approximately the chronological sequence they occurred.
Initial Retrosynthetic Analysis
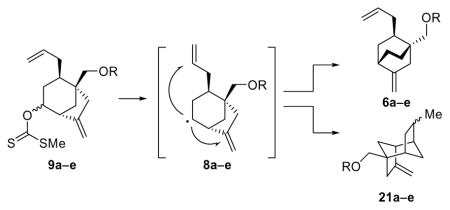
Figure 2 shows our retrosynthetic analysis of 1. Only a cursory inspection of the platencin molecule was needed to identify the first retrosynthetic disconnection, that of the amide bond, which revealed fragments 3 and 4 as potential precursors. Aniline derivative 3 was thought to be the ideal precursor for reasons of convenience and practicality given the expected sensitivity of the exocyclic olefinic bond of the final product and the neutral conditions under which the TMSE group could be removed. Applying two consecutive retrosynthetic alkylations on tricyclic fragment 4 led to enone 5 as the required key intermediate for the ketolide domain of the molecule. The latter was envisioned to arise from a ketoaldehyde, derived from 6 (R = H or protecting group) through an aldol condensation. A radical pathway starting from xanthate 9a or hydrazone 12, and proceeding through species 8 (R = H or protecting group) and 7 (R = H or protecting group) was then traced based on a homoallylic radical rearrangement.10 Our initial analysis adopted xanthate 9a as the potential precursor of the first radical species of the envisioned cascade and connected it to ketone 10. Disconnection of the indicated carbon-carbon bonds within [3.2.1] bicyclic system 10 through a retro conjugate addition of an allyl group and a retro palladium-catalyzed cyclization revealed enol ether 11 as a potential precursor.10a The latter intermediate was then traced back to hydroxymethyl enone 15 as a starting material. Our plan makes use of a hydrazone radical precursor of type 12, which can be traced through a similar retrosynthetic sequence, employing a gold(I)-catalyzed cyclization11 of an enol ether such as 14 and leading back to acetylenic hydroxymethyl enone 16.
Figure 2.

Retrosynthetic analysis of platencin [(−)-1]
First Generation Synthesis of Racemic Tricyclic Enone (±)-5
As part of our preliminary studies toward platencin (1), we explored the xanthate-based homoallylic rearrangement route to tricyclic ketone (±)-5 using racemic materials (Scheme 1).
Scheme 1.

Synthesis of Racemic Tricyclic Enone [(±)]-5]a
aReagents and conditions: a) LiHMDS (1.0 M in THF, 1.2 equiv), methyl cyanoformate (1.2 equiv), THF, −78 → 0 °C, 2 h, 92%; b) NaH (1.5 equiv), allyl bromide (1.5 equiv), THF, 0 → 25 °C, 2 h, 79%; c) LiAlH4 (1.0 M in THF, 1.2 equiv), Et2O, 0 → 25 °C, 3 h; then HCl (2 M in MeOH, 6.0 equiv), 25 °C, 16 h, 82%; d) PMBOC(NH)CCl3 (2.0 equiv), TsOH (0.1 equiv), CH2Cl2, 0 → 25 °C, 2.5 h, 85%; e) TBSOTf (2.5 equiv), Et3N (4.0 equiv), THF, 0 °C, 1 h, 91%; f) Pd(OAc)2 (0.1 equiv), O2 (balloon), DMSO, 45 °C, 12 h, 85%; g) CuBr•Me2S (2.0 equiv), allyl magnesium chloride (1.7 M in THF, 4.0 equiv), THF, −78 → −40 °C, 2 h, 86%; h) NaBH4 (2.5 equiv), MeOH, −5 → 25 °C, 30 min, 97%; i) KHMDS (0.5 M in toluene, 5.0 equiv), CS2 (10.0 equiv), MeI (10.0 equiv), THF, −78 → 25 °C, 2.5 h, 92%; j) n-Bu3SnH (2.0 equiv), AIBN (0.08 equiv), toluene, 110 °C, 8 h, 92%; k) PdCl2 (0.25 equiv), CuCl (1.5 equiv), O2 (balloon), DMF:H2O (6.6:1), 25 °C, 24 h, 81%; l) DDQ (1.2 equiv), CH2Cl2:H2O 20:1, 25 °C, 1 h, 53%; m) TPAP (0.03 equiv), NMO (6.5 equiv), CH2Cl2, 25 °C, 4 h, 54%; n) NaOH (6.0 equiv), EtOH, 25 °C, 8 h, 99%.
Thus, treatment of enone 17 with LiHMDS in THF at −78 °C followed by addition of Mander’s reagent (MeOCOCN)12 gave the corresponding carboxymethylated product which was allylated to afford compound (±)-18 in 73% overall yield. Reduction of the latter with LiAlH4 followed by acidic (aq. HCl) workup led to enone (±)-15 (82% yield), the free hydroxyl group of which was protected as a PMB ether to afford (±)-19 (85% yield). Treatment of ketone (±)-19 with TBSOTf in the presence of Et3N led to the corresponding TBS enol ether (91% yield), which underwent smooth oxidative cycloalkenylation upon exposure to a catalytic amount of Pd(OAc)2 under an atmosphere of O2 to afford bicyclic system (±)-20 in 85% yield, along with traces of the endocyclic olefinic by-product. Copper-catalyzed conjugate addition of allyl magnesium chloride to (±)-20 (CuBr•Me2S, −78 → −40 °C) gave ketone (±)-10 in 86% yield as a single diastereoisomer. Sodium borohydride reduction of (±)-10 gave a mixture of corresponding alcohols (97% yield, ca. 2:1 d.r.) that was converted to xanthates (±)-9a by the standard procedure.13 Generation of the radical species from (±)-9a by treatment with n-Bu3SnH-AIBN in toluene at 110 °C led to the desired [2.2.2] bicyclic product (±)-6a in excellent yield (92%), although the latter was contaminated with by-product 21a (ca. 4:1 ratio), arising from the alternative mode of cyclization (5-exo-trig, path b). The individual xanthate diastereomers obtained from the diasteroemerically pure alcohol precursors gave identical results, confirming that the stereochemistry of the radical precursor was inconsequential to the outcome of the reaction with regard to both yield and product distribution. The mixture of (±)-6a and 21a was inseparable by chromatography, but Wacker oxidation14 furnished ketone (±)-21 (81% yield), which was easily separated from the unreactive byproduct 21a. The product obtained by DDQ deprotection of PMB-protected intermediate (±)-21 (53% yield) was found to exist in its lactol form [(±)-22], a fact that did not deter its oxidation to keto aldehyde (±)-23 [NMO, TPAP (cat.), 54% yield].15 Furthermore, the intended aldol condensation of (±)-23 to racemic enone (±)-5 proceeded under the influence of NaOH in ethanol in excellent yield, demonstrating the viability of this radical-based strategy for the synthesis of platencin. As additional intelligence, this initial study provided useful information regarding the stability of the PMB protecting group, the performance of which, in a number of steps, particularly the Wacker oxidation, was less than stellar, prompting us to consider a more robust alternative for our first asymmetric approach to enone 5.
Asymmetric Synthesis of Enone (+)-5
In contemplating an asymmetric version of the radical rearrangement approach to enone (±)-5 we sought to prepare an enone such as 15 or 16 (Figure 2) as single enantiomers, in the knowledge that all the other centers could then be set through substrate control. One approach would be to effect an asymmetric variant of the alkylation reaction used to prepare ketoester (±)-18 (Scheme 1). Palladium-catalyzed asymmetric allylation16 reactions of similar ketoesters and their derivatives have been described in the literature;17,18 however, factors including unsatisfactory ee in our system, lack of readily available catalysts (particularly for large scale use), and the fact that any such approach would be limited to the introduction of an allyl substituent, led us to favor an asymmetric Diels–Alder reaction strategy.19 Asymmetric alkylation reactions using chiral bases or chiral phase transfer catalysts were also considered but not pursued for similar perceived drawbacks.
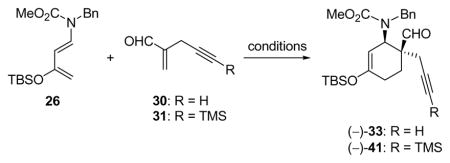
Rawal and coworkers have described the asymmetric Diels–Alder reaction of substituted 1-amino-3-silyloxybutadienes using the Jacobsen Cr(III)-salen catalyst.20 This system appeared attractive for our purposes since it was reported to offer access to highly enantiomerically enriched 4,4-disubstituted cyclohexenones. The application of this reaction to the asymmetric synthesis of 16 (Figure 2) required diene 26 and dienophile 30, the syntheses of which are summarized in Scheme 2.
Scheme 2.

Construction of Diene 26 and Dienophiles 30 and 31a
aReagents and conditions: a) 25 (1.0 equiv), 24 (2.0 equiv), TsOH (0.05 equiv), CHCl3, 65 °C, 24 h, 89%; b) TBSOTf (1.1 equiv), Et3N (3.0 equiv), Et2O, −78 → 0 °C, 1 h, 96%; c) py•SO3 (2.0 equiv), DMSO (5.0 equiv), Et3N (5.0 equiv), CH2Cl2, 25 °C, 2 h; then CH2NMe2Cl (29) (1.5 equiv), 25 °C, 12 h, R = H, 53%, R = TMS, 92%.
Diene 26 was prepared from commercially available ketone 24 according to literature procedures,21 while the rather volatile dienophile 30 was synthesized from acetylenic alcohol 27 in one pot through a two-step process involving oxidation (py•SO3, DMSO)22 and addition of Eschenmoser’s salt (29, 53% overall yield).23 Reaction of diene 26 (excess) with dienophile 30 (Scheme 3) in the presence of 0.05 mol% catalyst 32 (Figure 4) at −60 °C resulted in an impressive 92% yield of cyclohexene (−)-33, formed in 93% ee [determined by 1H NMR spectroscopic analysis of the Mosher ester derived from downstream product (−)-16]. We subsequently realized that the enantioselectivity of this reaction ranged between 85 and 93% ee, presumably due to its sensitivity to temperature and the difficulties in maintaining a steady cryogenic temperature for the extended reaction time (60 h). This deficiency, coupled to the poor reproducibility of the results on larger scales, prompted subsequent studies that resulted in considerable improvements, as discussed later. Returning to Diels–Alder product (−)-33, it was found that its intended conversion to enone (−)-16 (Scheme 3) could be achieved by reduction with LiAlH4 followed by exposure of the resulting hydroxy carbamate (34) to aqueous acid (63% overall yield).
Scheme 3.

First Generation Catalytic Asymmetric Synthesis of Tricyclic Enone [(+)-5]a
aReagents and conditions: a) 30 (1.0 equiv), 26 (1.7 equiv), 32 (0.05 equiv), 4 Å M.S., CH2Cl2, −60 °C, 60 h, 92%; b) LiAlH4 (1.0 M in THF, 1.5 equiv), Et2O, −78 → −40 °C, 2 h; then HCl (2 M in MeOH, 10.0 equiv), 25 °C, 16 h, 63%; c) SEMCl (1.2 equiv), Et3N (4.0 equiv), 4-DMAP (0.1 equiv), THF, 70 °C, 16 h, 94%; d) TIPSOTf (1.5 equiv), Et3N (3.0 equiv), −78 → 0 °C, 1 h, 97%; e) [AuCl(PPh3)] (0.02 equiv), AgBF4 (0.02 equiv), toluene/MeOH (10:1), 25 °C, 30 min, 94%; f) allyl magnesium chloride (1.7 M in THF, 4.0 equiv), CuBr•Me2S (2.0 equiv), THF, −78 °C, 1.5 h, 74%; g) NaBH4 (2.5 equiv), MeOH, −5 → 25 °C, 1 h, 97%; h) CS2 (10.0 equiv), KHMDS (0.5 M in toluene, 5.0 equiv), MeI (5.0 equiv), THF, −78 → 25 °C, 2.5 h, 100%; i) n-Bu3SnH (2.0 equiv), AIBN (0.08 equiv), toluene, 100 °C, 20 min, 92%; j) PdCl2 (0.25 equiv), CuCl (1.5 equiv), O2 (balloon), DMF/H2O (6.6:1), 25 °C, 24 h, 50% (2 steps); k) TASF (10.0 equiv), DMPU, 80 °C, 1.5 h, 63% (84% based on 25% recovered starting material); l) TPAP (0.03 equiv), NMO (6.5 equiv), CH2Cl2, 25 °C, 4 h, 54%; m) NaOH (6.0 equiv), EtOH, 25 °C, 8 h, 99%.
Figure 4.

Molecular structures of Cr(III)- and Co(III)-salen catalysts 32 and 40.
At this stage we selected the SEM group to protect the primary hydroxyl group in place of the PMB group used previously, by virtue of its robustness and linear shape, which we reasoned would not hinder any of the subsequently intended steps. Thus, treatment of (−)-16 with SEMCl in the presence of Et3N and 4-DMAP (cat.) furnished derivative (−)-35 (94% yield), which was converted to TIPS diene (−)-36 (TIPSOTf, Et3N, 97% yield) in preparation for the crucial cycloalkenylation reaction. To this end, acetylenic enol ether (−)-36 was exposed to the action of the cationic gold catalyst formed in situ from AuCl(PPh3) and AgBF4, conditions that led, within 30 min at room temperature, to the desired bicyclic enone (+)-37 in 94% yield.11 Note that our initial study toward this type of [3.2.1] bicyclic system involved a palladium-catalyzed cycloalkenylation process of a similar substrate containing a terminal olefin rather than the terminal acetylene contained within (−)-36 [i.e. (±)-19 → (±)-20, Scheme 1]. In contrasting the two processes, one can easily recognize the advantage of the latter method in terms of catalyst loading (2% gold catalyst vs. 10% palladium catalyst), reaction time (< 30 min at 25 °C vs. 12 h at 45 °C), and yield (94% vs. 85%). Conjugate addition of an allyl group to enone (+)-37 (allyl MgCl, CuBr•Me2S) proceeded smoothly to afford ketone (+)-38 as a single diastereomer in 74% yield. Ketone (+)-38 was converted, as before, to a mixture of xanthates (−)-9b (ca. 2:1 d.r.) by the standard two-step sequence (NaBH4; KHMDS, CS2, MeI) in 97% overall yield. The radical rearrangement sequence of (−)-9b proceeded as before, giving a high yield of diene 6b (92%), along with a minor quantity of byproduct 21b.24 Wacker oxidation also proceeded as expected to provide ketone (−)-39 in 50% overall yield from (−)-9b, however, the removal of the SEM group proved difficult. A systematic investigation of conditions identified nucleophilic fluoride sources as the only reagents capable of delivering the desired product, with TASF in DMPU being the optimum choice, giving up to 63% yield of lactol (−)-22 along with ca. 25% recovered starting material (−)-39. Interestingly, lactol (−)-22 was isolated as a single diastereoisomer regardless of which precursor was used. This compound crystallized from CDCl3/CH2Cl2 as colorless needles (m.p. 115–117 °C), a circumstance that allowed its X-ray crystallographic analysis (see ORTEP drawing, Figure 3) and, thus, confirmed its structure as depicted in (−)-22.25 The subsequent oxidation and aldol condensation steps proceeded well, as before, to afford enantiomerically enriched tricyclic enone (+)-5 as shown in Scheme 3.
Figure 3.

ORTEP drawing of (−)-22 derived from X-ray crystallographic analysis (non-hydrogen atoms are shown as 30% ellipsoids).
Optimization of the Catalytic Asymmetric Synthesis of Enone (+)-5
The asymmetric synthesis of enone (+)-5 described above proved effiecient enough to allow us to complete the total synthesis of platencin [(−)-1] in a highly enantioenriched form (vide infra);8a however, we identified several opportunities to improve the route at several steps while maintaining the overall strategy. These included asymmetric Diels–Alder reaction, the radical rearrangement sequence, and the protecting group choice. We were able to optimize each of these processes, which, in turn, influenced the overall efficiency of the synthesis, leading to an effective and streamlined route.
A significant practical drawback in our original asymmetric route is the volatility of dienophile 30. This enal was prepared in a highly efficient one-pot procedure that provided the product in excellent purity, but the isolated yield was only moderate, even when solvents were removed by careful distillation through a Vigreaux column. This issue was addressed by the simple inclusion of a TMS group on the alkyne. TMS-capped enal 31 was prepared from commercially available acetylene 28 using the same one-pot procedure and could be isolated by flash column chromatography or distillation in 92% yield as shown in Scheme 2. We investigated the use of both dienophiles during optimization studies to improve the practicality of the asymmetric Diels–Alder reaction with the previously discussed Cr(III)-based catalyst (32) as well as a similar Co(III)-based system (40, Figure 4).
In these investigations we were guided by the findings of the Rawal group in their development of asymmetric Diels–Alder reactions of aminosilyloxy dienes;26 our results are summarized in Table 1.
Table 1.
Optimization of the Catalytic Asymmetric Diels–Alder Reactiona
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Dienophile | Diene (equiv) | Catalyst (mol %) | Temp. (°C) | Time (h) | Yield (%) | ee (%) |
| 1 | 30 | 1.5 | 32 (5) | 25 | 60 | 85 | 75 |
| 2 | 30 | 1.5 | 32 (5) | −40 | 60 | 87 | 85 |
| 3 | 30 | 2.0 | 32 (5) | −60 | 60 | 92 | 93b |
| 4 | 30 | 1.8 | 40 (5) | 0 | 2 | 79 | 76 |
| 5 | 31 | 1.8 | 40 (5) | 0 | 1.5 | 92 | 93 |
| 6 | 31 | 1.5 | 40 (1) | 0 | 0.5 | 93 | 94 |
| 7 | 31 | 1.1 | 40 (0.1) | 0 | 0.5 | 97 | 96 |
| 8 | 31 | 1.5 | none | 25 | 0.5 | 5 | 0 |
Reactions were carried out on 0.1–1.0 mmol scale with 1.0 equiv. dienophile 30 or 31 in the presence of 4 Å M. S. in CH2Cl2.
Enantiomeric excess (ee) ranged from 86–93%.
We were able to optimize the reaction of our original dienophile (30) with diene 26 and chromium catalyst 32 (entries 1–3) to 92% yield and 93% ee, although, as mentioned above, the reaction was very slow and somewhat unreliable with respect to the ee of the product. The use of cobalt-salen catalyst 40 (entry 4) with dienophile 30 resulted in a much faster reaction at the expense of reduced ee and somewhat reduced yield; however, changing to dienophile 31 brought significant improvements. We tentatively ascribe this observation to the improved purity of the less volatile dieneophile. Further study of the cobalt-catalyzed reaction of 31 with 26 led to the optimized conditions (entry 7), which use only a slight excess of diene and give an almost quantitative yield of product (−)-41 with excellent enantioselectivity (96% ee). Noteworthy features of the Diels–Alder reaction using catalyst 40 and dienophile 31 include the much increased reaction rate (0.5 h vs. 60 h with catalyst 32) and the improved selectivity at more convenient temperatures (0 °C vs –60 °C). We also observed that the Diels–Alder reaction between dienophile 31 and diene 26 proceeded to some extent at ambient temperature in the absence of any catalyst (entry 8).
We next turned our attention to the radical rearrangement reaction. This reaction performed as expected in our original route, but the three step conversion of ketone (±)-10 or (+)-38 to (±)-6a (Scheme 1) or 6b (Scheme 3) involved the use of toxic reagents in both the radical and xanthate formation steps, and suffered from the formation of small but significant quantities of the 5-exo-trig byproducts. At this time, we were also investigating the optimum protecting group for the primary alcohol, which led to an interesting observation. When testing the radical sequence using substrates with various protecting groups, we noted that the pathway of the radical process was dependent on the nature of the protecting group used (Table 2). When smaller, linear protecting groups, such as MOM or SEM, were used, the radical sequence proceeded to give the desired [2.2.2] bicyclic products (6a–e) with good selectivity over the 5-exo-trig by-products (entries 1 and 2); the PMB ether gave similar selectivity affording an approximately 4:1 mixture (entry 3). However, both acetate and pivalate esters yielded significantly lower selectivities affording approximately 1:2 mixture of the two products (entries 4 and 5), indicating the finely balanced nature of the selectivity between the two competing reaction manifolds. Presumably, steric interactions of the ester groups position the olefinic moiety of the allyl group in such a way so as to favor the 5-exo-trig pathway leading to the undesired by-products 21a–e.
Table 2.
Effect of Protecting Group on the Radical Deoxygenation Reactiona
 | |||
|---|---|---|---|
| Entry | R (Compound) | Yield (%) | Product ratio 6:21b |
| 1 | MOM (9c) | 93 | 5.4:1 (6c) |
| 2 | SEM (9b) | 92 | 5.5:1 (6b) |
| 3 | PMB (9a) | 92 | 4:1 (6a) |
| 4 | Ac (9d) | 71 | 1:1.5 (6d) |
| 5 | Piv (9e) | 65 | 1:2 (6e) |
Reactions were carried out on 0.01 mmol scale with 2.0 equiv. n-Bu3SnH and 0.08 equiv AIBN in toluene at 100 °C.
Ratios were determined by 1H NMR (500 MHz) spectroscopic analysis.
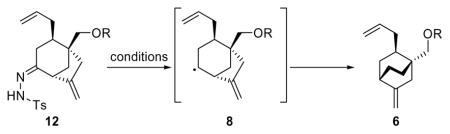
In an effort to streamline the overall conversion of the ketone intermediate to the radical rearrangement product, we explored the Wolff–Kishner type conditions for radical generation from carbonyl compounds reported by Toyota et al.27 Table 3 summarizes our optimization studies using several hydrazone intermediates prepared from the corresponding ketones [TsNHNH2, TsOH (cat.), CH2Cl2, 70 °C]. Our initial attempts to apply the reported NaBH3CN/ZnCl2 conditions27a to hydrazones 12a–c (entries 1–3) failed to furnish any of the desired products, leading only to decomposition. Reasoning that the Lewis acid conditions employed were responsible for the observed outcome, we attempted the reaction with the SEM-protected hydrazone 12d employing NaBH4 in THF (entry 4), but again observed only decomposition. Use of the milder reductant NaBH(OAc)3 was also unsuccessful leading, this time, to recovery of the starting material (entry 5). Further studies led us to return to NaBH4 as a potential reductant in a variety of solvents. Our efforts were rewarded when we employed hydrazone 12d and NaBH4 in refluxing CH2Cl2:MeOH (1:1), conditions that led to 50% yield of the desired product (−)-6b (entry 6). These conditions were then optimized (entries 8–11) to a satisfactory level (entry 11), which required portionwise addition of NaBH4 to a refluxing solution of hydrazone 12d in CH2Cl2:MeOH [20:1, 70% yield of (−)-6b]. Furthermore, by avoiding the need for a xanthate formation step, we were able to employ the hydroxy hydrazone 12 (R = H) to produce rearranged product (−)-6 (R = H) under the same optimized conditions in 74% yield (Table 3, entry 12).
Table 3.
Optimization of the Radical-Based Conversion of Hydrazones 12 to [2.2.2] Bicyclo Systems 6a
 | |||||
|---|---|---|---|---|---|
| Entry | R (compound) | Reductant (equiv) | Solvent | Temp. (°C) | Yield (%) |
| 1 | MOM (12a) | NaBH3CN (3.8)b | THF | 80 | 0c |
| 2 | PMB (12b) | NaBH3CN (3.8)b | THF | 80 | 0c |
| 3 | Piv (12c) | NaBH3CN (3.8)b | THF | 80 | 0c |
| 4 | SEM (12d) | NaBH4 (1.4) | TiHF | 90 | 0c |
| 5 | SEM (12d) | NaBH(OAc)3 (4.0) | DCE:AcOH (9:1) | 100 | 0d |
| 6 | SEM (12d) | NaBH4 (8.0) | CH2Cl2:MeOH (1:1) | 45 | 50 |
| 7 | SEM (12d) | NaBH4 (8.0) | THF:MeOH (1:1) | 70 | 47 |
| 8 | SEM (12d) | NaBH4 (8.0) | CH2Cl2:MeOH (1:1) | 25 | 0c |
| 9 | SEM (12d) | NaBH4 (8.0) | CH2Cl2:MeOH (20:1) | 45 | 55 |
| 10 | SEM (12d) | NaBH4 (8.0) | CH2Cl2:EtOH (20:1) | 45 | 60 |
| 11 | SEM (12d) | NaBH4 (2.0 × 3) | CH2Cl2:MeOH (20:1) | 45 | 70 |
| 12 | H (12) | NaBH4 (2.0 × 4) | CH2Cl2:MeOH (20:1) | 45 | 74 (6f) |
Reactions were carried out on 0.1–1.0 mmol scale.
With 3.8 equiv. ZnCl2.
Only decomposition observed.
No reaction observed.
Our optimized Diels–Alder and radical rearrangement steps were then combined to furnish our second generation asymmetric synthesis of key enone intermediate (+)-5 (Scheme 4). Thus, following the asymmetric Diels–Alder reaction of 26 with 31, product (−)-41 was converted to hydroxy enone (−)-16 in a one-pot procedure involving reduction of the aldehyde (LiAlH4, Et2O-THF), cleavage of the acetylenic TMS group by addition of MeOH and K2CO3, and, finally, elimination of the carbamate group by acidification with methanolic HCl. Hydroxy enone (−)-16 was obtained in 89% overall yield and 96% ee. A single recrystallization of the product from hexanes:EtOAc (m.p. 100–102 °C) increased the ee to > 98% ee. In preparation for the gold-catalyzed cyclization, we attemped to generate the bis-TBS derivative (−)-14 from hydroxyl enone (−)-16 by exposure of the latter to TBSOTf and Et3N. Although a smooth reaction under these conditions was observed by TLC, upon workup only starting material was recovered. Closer monitoring and NMR spectroscopic analysis of the reaction mixture in CDCl3 revealed the formation of bicyclic ketal 44 (see Scheme 5), which apparently is stable under the reaction conditions, but immediately reverts back to starting material [(−)-16] upon workup. To circumvent this troublesome circumstance we employed a one-pot protocol involving sequential treatment of hydroxyl enone (−)-16, first with TBSCl in the presence of Et3N and 4-DMAP (capping the hydroxyl group), followed by TBSOTf (forming the TBS enol ether), conditions that led to generation of the desired bis-TBS derivative (−)-14 in 97% yield. This material was then subjected to the gold-catalyzed ring closure conditions [2 mol% Au(PPh3), 2 mol% AgBF4] to produce bicyclic enone (+)-42 in 95% yield (Scheme 4). Exposure of enone (+)-42 to allylmagnesium bromide in the presence of CuBr•SMe2 at −78 → −40 °C, followed by acidic workup (aq. HCl), led to intermediate (+)-13 in 85% overall yield, thus avoiding either specific protection or deprotection steps.
Scheme 4.

Second Generation Catalytic Asymmetric Synthesis of Tricyclic Enone [(+)-5]a
aReagents and conditions: a) 31 (1.0 equiv), 26 (1.05 equiv), 40 (0.1 mol %), 4 Å M.S., CH2Cl2, 0 °C, 97%; b) LiAlH4 (1.0 M in THF, 1.5 equiv), Et2O, −78 → −40 °C, 2 h; then K2CO3 (5.0 equiv), MeOH (50 equiv), 25 °C, 5 h; then HCl (4 M in MeOH, 20.0 equiv), 25 °C, 16 h, 92%; c) TBSCl (1.5 equiv), Et3N (4.0 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 45 °C, 16 h; then TBSOTf (1.5 equiv), 0 °C, 4 h, 97%; d) [AuCl(PPh3)] (0.02 equiv), AgBF4 (0.02 equiv), toluene/MeOH (10:1), 25 °C, 30 min, 95%; e) CuBr•Me2S (2.0 equiv), allyl magnesium chloride (1.7 M in THF, 4.0 equiv), THF, −78 → −40 °C, 1.5 h; then 2 M aq. HCl (10.0 equiv), 25 °C, 4 h, 85%; f) TsNHNH2 (1.2 equiv), TsOH (0.1 mol %), CH2Cl2, 45 °C, 1 h; then NaBH4 (2.0 equiv), MeOH (7.0 equiv), 45 °C, 74%; g) DMP (2.0 equiv), pyridine (0.1 equiv), CH2Cl2, 25 °C, 1.5 h, 90%; h) PdCl2 (0.25 equiv), CuCl (1.5 equiv), O2 (balloon), DMF/H2O (6.6:1), 25 °C, 24 h, 90%; i) NaOH (6.0 equiv), EtOH, 25 °C, 8 h, 99%.
Scheme 5.

bis-Silylation of Hydroxy Enone (−)-16a
a Reagents and conditions: a) TBSOTf (2.5 equiv), Et3N (5.0 equiv), CH2Cl2, −78 → 0 °C, 1 h; b) TBSCl (1.5 equiv), Et3N (4.0 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 45 °C, 16 h; then TBSOTf (1.5 equiv), 0 °C, 4 h, 97%.
Conversion of keto alcohol (+)-13 to bicyclo [2.2.2] octane system (−)-6 was easily achieved without the need to isolate intermediate hydrazone 12 (Scheme 4). Thus, formation of the hydrazone in CH2Cl2 with TsNHNH2 and 0.1 mol% TsOH, followed by addition of MeOH and reduction as previously described, furnished (−)-6 in 74% overall yield. The alcohol was then oxidized to aldehyde (−)-43 with DMP and the latter compound was subjected to Wacker oxidation [PdCl2 (cat.), CuCl, O2] to afford keto aldehyde (−)-23 in 90% yield, thus obviating the troublesome SEM deprotection and avoiding the intermediacy of lactol (−)-22. Finally, exposure of (−)-23 to NaOH in EtOH as before furnished tricyclic enone (+)-5 in 99% yield.
Synthesis of Carboxylic Acid Coupling Partner (+)-4
Having developed a streamlined and practical catalytic asymmetric route to the platencin core (+)-5, we turned our attention to the introduction of the appendages required for the carboxylic acid coupling partner (+)-4. We initially applied our sequence originally used for the corresponding platensimycin carboxylic acid, involving sequential alkylations and metathesis as shown in Scheme 6.7a Thus, enone (+)-5 was methylated [KHMDS, MeI, THF:HMPA (4:1), −78 → 0 °C, 90% yield] to afford methyl enone (−)-45 (ca. 10:1 d.r.), which was subjected to a second alkylation with allyl iodide (KHMDS, allyl iodide, THF:HMPA (4:1), −78 → 0 °C, 86% yield, single diastereomer) to give triene (+)-46. The latter compound was converted to the desired carboxylic acid (+)-4 through a three-step procedure involving cross metathesis with boronate 4828 in the presence of Hoveyda–Grubbs catalyst 4929 and benzoquinone,30 oxidation of the resulting vinyl boronate with Me3NO to the corresponding aldehyde, and Pinnick oxidation of the latter, in 39% overall yield.31
Scheme 6.

Alkylation of Tricyclic Enone (+)-5 and Construction of Carboxylic Acid (+)-4a
aReagents and conditions: a) KHMDS (0.5 M in toluene, 1.1 equiv), MeI (8.0 equiv), THF/HMPA (4:1), −78 → 0 °C, 2 h, 90%; b) KHMDS (0.5 M in toluene, 4.0 equiv), allyl iodide (8.0 equiv), THF/HMPA (4:1), −78 → 0 °C, 3 h, 86%; c) 48 (5.0 equiv), Hoveyda–Grubbs II cat. 49 (0.1 equiv), benzoquinone (0.1 equiv), benzene, 70 °C, 1 h, 75%; d) Me3NO (5.0 equiv), THF, 70 °C, 1 h, 63%; e) NaClO2 (3.0 equiv), NaH2PO4 (5.0 equiv), 2-methyl-2-butene (10.0 equiv), t-BuOH/H2O (1:1), 25 °C, 20 min, 82%.
In a separate attempt to access the carboxylic acid (+)-4 from methylated enone (−)-45, and inspired by Corey’s platensimycin analogue work,9d we subjected this compound to enolate formation (KOt-Bu) in the presence of methyl acrylate. However, we were disappointed to observe both modest yield of the desired methyl ester product along with a number of undesired by-products ( 10 → 0 °C). We then resorted to the use of tert-butyl acrylate (less prone to polymerization than its methyl counterpart under the reaction conditions) at ambient temperature and realized a 92% yield of the targeted product [(+)-50, Scheme 7], albeit in ca. 2:1 d.r. favoring the desired stereoisomer, which was chromatographically separated and fully characterized. The required cleavage of the t-butyl ester from the latter compound demanded basic conditions [LiOH (1 N aqueous):MeOH (1:1), 50 °C, 97% yield] as the exocyclic double bond was prone to isomerization and further destructive side-reactions under acidic conditions (i.e. TFA).
Scheme 7.

Synthesis of Carboxylic Acid (+)-4 via Conjugate Additiona
aReagents and conditions: a) tert-butyl acrylate (2.0 equiv), KOt-Bu (1.0 M in t-BuOH, 2.0 equiv), THF, 25 °C, 30 min, 92% (ca. 2:1 dr); b) LiOH (1 N aqueous):MeOH (1:1), 50 °C, 6 h, 97%.
Preparation of Aniline Coupling Partner and Completion of the Total Synthesis of (−)-Platencin
The susceptibility of the core structural motif of platencin to acidic conditions dictated modification of the sequence originally employed in our total synthesis of platensimycin.7a Thus, aniline partner 3 (Scheme 8) equipped with a TMSE group was designed as a suitable coupling partner with carboxylic acid (+)-4 due to the mild conditions under which we expected to carry our the final deprotection (−TMSE).
Scheme 8.

Preparation of Aniline Derivative 3a
aReagents and conditions: a) TMSEOH (14.8 equiv), n-Bu2SnO (1.5 equiv), 70 °C, 3 h, 61%; b) H2 (balloon), 10% Pd/C (0.05 equiv), AcOH (1.0 equiv), EtOAc/MeOH (5:1), 25 °C, 15 h, 100%.
The preparation of this partner proceeded from the readily available nitro methyl ester (51) through ester exchange (n-Bu2SnO, TMSCH2CH2OH, 70 °C, 61% yield)32 and reduction (H2, 10% Pd/C, quantitative) of the resulting product, as shown in Scheme 8. The coupling of carboxylic acid (+)-4 with aniline derivative 3 proceeded under the influence of HATU and Et3N in DMF at ambient temperature to give protected platencin (−)-52, in 61% yield, from which the TMSE group was removed through the action of TASF (40 °C, 93% yield). Synthetic (−)-platencin [(−)-1] crystallized from acetone/hexanes in cubic colorless crystals (m.p. 194–197 °C dec.) upon prolonged standing. Its X-ray crystallographic analysis (see ORTEP drawing, Figure 5)33 unambiguously confirmed its structure, and, therefore, that of the natural substance, whose X-ray crystallographic analysis has not been previously reported.
Figure 5.

ORTEP drawing of (−)-1 derived from X-ray crystallographic analysis (non-hydrogen atoms are shown as 30% ellipsoids).
Conclusion
The described chemistry provides a streamlined asymmetric approach to the newly reported antibiotic platencin and its analogues. The developed synthetic strategies delivered both the racemic [(±)-1] and naturally occurring [(−)-1] forms of the natural product and allowed for the first X-ray crystallographic analysis of this antibiotic. Demonstrated by this synthesis are the power of the cobalt-catalyzed asymmetric Diels–Alder reaction26 and the one-pot reductive rearrangement of [3.2.1] bicyclic ketones to [2.2.2] bicyclic olefins through their hydrazones.27 Further applications of these synthetic technologies in chemical synthesis are foreseen.
Supplementary Material
Figure 1.

Molecular structures of platencin [(−)-1] and platensimycin [(−)-2].
Scheme 9.

Completion of the Total Synthesis of Platencin [(−)-1]a
a Reagents and conditions: a) 3 (3.2 equiv), HATU (3.2 equiv), Et3N (4.2 equiv), DMF, 25 °C, 14 h, 61%; b) TASF (2.0 equiv), DMF, 40 °C, 40 min, 93%.
Acknowledgments
We thank Drs. D.-H. Huang, R. Chadha, and G. Siuzdak for NMR spectroscopic, X-ray crystallographic and mass spectrometric assistance, respectively. We also gratefully acknowledge A. Nold and S. Ellery for assistance with HPLC, and Dr. G. V. Gondi and A. Li for helpful discussions. Financial support for this work was provided by the National Institutes of Health (USA), National Science Foundation (CHE-0603217), the Skaggs Institute for Research and Merck Sharp & Dohme (postdoctoral fellowship to D.J.E.).
Footnotes
Supporting Information Available: Complete refs. 2, 3b, 5a and 6a; experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Neu HC. Science. 1992;257:1064. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 2.Klevens RM, et al. J Am Med Assoc. 2007;298:1763. [Google Scholar]
- 3.For an excellent overview of this program, see: Singh SB, Phillips JW, Wang J. Curr Opin Drug Disc Dev. 2007;10:160.Young K, et al. Antimicrob Agents Chemother. 2006;50:519. doi: 10.1128/AAC.50.2.519-526.2006.
- 4.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Nature. 2009;458:83. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 5.For isolation of platencin, see: Wang J, et al. Proc Natl Acad Sci USA 2007104761217456595Jayasuriya H, Herath KB, Zhang C, Zink DL, Basilio A, Genilloud O, Teresa Diez M, Vicente F, Gonzalez I, Salazar O, Pelaez F, Cummings R, Ha S, Wang J, Singh SB. Angew Chem, Int Ed. 2007;46:4684. doi: 10.1002/anie.200701058.For platencin A1 isolation, see: Singh SB, Ondeyka JG, Herath KB, Zhang C, Jayasuriya H, Zink DL, Parthasarathy G, Becker JW, Wang J, Soisson SM. Bioorg Med Chem Lett. 2009;19:4756. doi: 10.1016/j.bmcl.2009.06.061.
- 6.(a) Wang J, et al. Nature. 2006;441:358. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]; (b) Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K, Wang J. J Am Chem Soc. 2006;128:11916. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]; (c) Singh SB, Herath KB, Wang J, Tsou N, Ball RG. Tetrahedron Lett. 2007;48:5429. [Google Scholar]; (d) Herath KB, Attygalle AB, Singh SB. J Am Chem Soc. 2007;129:15422. doi: 10.1021/ja0758943. [DOI] [PubMed] [Google Scholar]
- 7.For the total synthesis of platensimycin, see: Nicolaou KC, Li A, Edmonds DJ. Angew Chem, Int Ed. 2006;45:7086. doi: 10.1002/anie.200603892.For formal syntheses of platensimycin, see: Nicolaou KC, Edmonds DJ, Li A, Tria GS. Angew Chem, Int Ed. 2007;46:3942. doi: 10.1002/anie.200700586.Zou Y, Chen CH, Taylor CD, Foxman BM, Snider BB. Org Lett. 2007;9:1825. doi: 10.1021/ol070563g.Nicolaou KC, Tang Y, Wang J. Chem Commun. 2007:1922. doi: 10.1039/b704589a.Li P, Payette JN, Yamamoto H. J Am Chem Soc. 2007;129:9534. doi: 10.1021/ja073547n.Tiefenbacher K, Mulzer J. Angew Chem, Int Ed. 2007;46:8074. doi: 10.1002/anie.200702852.Lalic G, Corey EJ. Org Lett. 2007;9:4921. doi: 10.1021/ol702323s.Nicolaou KC, Pappo D, Tsang KY, Gibe R, Chen DYK. Angew Chem, Int Ed. 2008;47:944. doi: 10.1002/anie.200705080.Kim CH, Jang KP, Choi SY, Chung YK, Lee E. Angew Chem, Int Ed. 2008;47:4009. doi: 10.1002/anie.200800568.For a review on the approaches to platensimycin and related analogues, see: Tiefenbacher K, Mulzer J. Angew Chem, Int Ed. 2008;47:2548. doi: 10.1002/anie.200705303.
- 8.For total syntheses of platencin, see: Nicolaou KC, Tria GS, Edmonds DJ. Angew Chem, Int Ed. 2008;47:1780. doi: 10.1002/anie.200800066.Hayashida J, Rawal VH. Angew Chem, Int Ed. 2008;47:4373. doi: 10.1002/anie.200800756.For formal syntheses of platencin, see: Tiefenbacher K, Mulzer J. Angew Chem, Int Ed. 2008;47:6199. doi: 10.1002/anie.200801441.Yun SY, Zheng JC, Lee D. Angew Chem, Int Ed. 2008;47:6201. doi: 10.1002/anie.200801587.Waalboer DCJ, Schaapman MC, Van Delft FL, Rutjes FPJT. Angew Chem, Int Ed. 2008;47:6576. doi: 10.1002/anie.200802912.Nicolaou KC, Toh QY, Chen DYK. J Am Chem Soc. 2008;130:11292. doi: 10.1021/ja804588r.Austin KAB, Banwell MG, Willis AC. Org Lett. 2008;10:4465. doi: 10.1021/ol801647h.Tiefenbacher K, Mulzer J. J Org Chem. 2009;74:2937. doi: 10.1021/jo9001855.Varseev GN, Maier ME. Angew Chem, Int Ed. 2009;48:3685. doi: 10.1002/anie.200900447.
- 9.(a) Nicolaou KC, Lister T, Denton RM, Montero A, Edmonds DJ. Angew Chem, Int Ed. 2007;46:4712. doi: 10.1002/anie.200701548. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Tang Y, Wang J, Stepan AF, Li A, Montero A. J Am Chem Soc. 2007;129:14850. doi: 10.1021/ja076126e. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Stepan AF, Lister T, Li A, Montero A, Tria GS, Turner CI, Tang Y, Wang J, Denton RM, Edmonds DJ. J Am Chem Soc. 2008;130:13110. doi: 10.1021/ja8044376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yeung YY, Corey EJ. Org Lett. 2008;10:3877. doi: 10.1021/ol801400a. [DOI] [PubMed] [Google Scholar]; (e) Shen HC, Ding F, Singh SB, Parthasarathy G, Soisson SM, Ha SN, Chen X, Kodali S, Wang J, Dorso K, Tata JR, Hammond ML, MacCoss M, Colletti SL. Bioorg Med Chem Lett. 2009;19:1623. doi: 10.1016/j.bmcl.2009.02.006. [DOI] [PubMed] [Google Scholar]; (f) Wang J, Lee V, Sintim HO. Chem Eur J. 2009;15:2747. doi: 10.1002/chem.200802568. [DOI] [PubMed] [Google Scholar]; (g) Jang KP, Kim CH, Na SW, Kim H, Kang H, Lee E. Bioorg Med Chem Lett. 2009;19:4601. doi: 10.1016/j.bmcl.2009.06.092. [DOI] [PubMed] [Google Scholar]
- 10.(a) Toyota M, Wada T, Fukumoto K, Ihara M. J Am Chem Soc. 1998;120:4916. [Google Scholar]; (b) Toyota M, Asano T, Ihara M. Org Lett. 2005;7:3929. doi: 10.1021/ol051411t. [DOI] [PubMed] [Google Scholar]; (c) Nonhebel DC. Chem Soc Rev. 1993:347. [Google Scholar]; (d) Dowd P, Zhang W. Chem Rev. 1993;93:2091. [Google Scholar]
- 11.Staben ST, Kennedy-Smith JJ, Huang D, Corkey BK, LaLonde RL, Toste FD. Angew Chem, Int Ed. 2006;45:5991. doi: 10.1002/anie.200602035. [DOI] [PubMed] [Google Scholar]
- 12.Mander LN, Sethi SP. Tetrahedron Lett. 1983;24:5425. [Google Scholar]
- 13.(a) Barton DHR, McCombie SW. J Chem Soc Perkin Trans 1. 1975:1574. [Google Scholar]; (b) Trost BM, Cramer N, Bernsmann H. J Am Chem Soc. 2007;129:3086. doi: 10.1021/ja070142u. [DOI] [PubMed] [Google Scholar]
- 14.(a) Park PK, O’Malley SJ, Schmidt DR, Leighton JL. J Am Chem Soc. 2006;128:2796. doi: 10.1021/ja058692k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smidt J, Sieber R. Angew Chem. 1959;71:261. [Google Scholar]; (c) Takacs JM, Jiang X-T. Curr Org Chem. 2003;7:369. [Google Scholar]; (d) Tsuji J. Synthesis. 1984:369. [Google Scholar]
- 15.Griffith WP, Ley SV, Whitcombe GP, White AD. J Chem Soc Chem Commun. 1987:1625. [Google Scholar]
- 16.(a) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (a) Sawamura M, Ito Y. Chem Rev. 1992;92:857. [Google Scholar]; (b) Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Fandrick DR. Aldrichimica Acta. 2007;40:59. [Google Scholar]
- 17.(a) Trost BM, Radinov R, Grenzer EM. J Am Chem Soc. 1997;119:7879. [Google Scholar]; (b) Trost BM, Bream RN, Xu J. Angew Chem, Int Ed. 2006;45:3109. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]
- 18.Varseev and Maier have recently described the use of an allyl enol carbonate to prepare an intermediate similar to 18 in up to 87 % ee, see ref. 8i.
- 19.Asymmetric alkylation reactions using chiral bases or chiral phase transfer catalysts were also considered but not pursued for similar reasons.
- 20.(a) Kozmin SA, Rawal VH. J Am Chem Soc. 1997;119:7165. [Google Scholar]; (b) Janey JM, Iwama T, Kozmin SA, Rawal VH. J Org Chem. 2000;65:9059. doi: 10.1021/jo005619y. [DOI] [PubMed] [Google Scholar]; (c) Huang Y, Iwama T, Rawal VH. Org Lett. 2002;4:1163. doi: 10.1021/ol0255716. [DOI] [PubMed] [Google Scholar]
- 21.(a) Huang Y, Iwama T, Rawal VH. J Am Chem Soc. 2000;122:7843. [Google Scholar]; (b) Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628. doi: 10.1021/ja017863s. [DOI] [PubMed] [Google Scholar]
- 22.Parikh JR, Doering WvE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 23.Kinast G, Tietze LF. Angew Chem, Int Ed. 1976;15:239.This reagent is easily prepared on a large scale using a procedure previously reported; see: Danishefsky S, Chackalamannil S, Harrison P, Silvestri M. J Am Chem Soc. 1985;107:2474.
- 24.Tricyclic by-product 21b was characterized by NMR spectroscopic analysis; structures of the other corresponding by-products, such as 21a, 21c–e, were assigned by analogy.
- 25.CCDC-671740 contains the supplementary crystallographic data for compound (−)-22. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 26.Huang Y, Iwama T, Rawal VH. J Am Chem Soc. 2002;124:5950. doi: 10.1021/ja026088t. [DOI] [PubMed] [Google Scholar]; (b) McGilvra JD, Rawal VH. Synlett. 2004;13:2440. [Google Scholar]
- 27.(a) Toyota M, Asano T, Ihara M. Org Lett. 2005;7:3929. doi: 10.1021/ol051411t. [DOI] [PubMed] [Google Scholar]; (b) Taber DF, Wang Y, Stachel SJ. Tetrahedron Lett. 1993;34:6209. [Google Scholar]; (c) Hutchins RO, Milewski CA, Maryanoff BE. J Am Chem Soc. 1973;95:3662. [Google Scholar]; (d) Zard SZ, Miranda LD. Chem Commun. 2001:1068. [Google Scholar]; (e) Taber DE, Wang Y, Pahutski TF., Jr J Org Chem. 2000;65:3861. doi: 10.1021/jo991866u. [DOI] [PubMed] [Google Scholar]
- 28.(a) Njardarson JT, Biswas K, Danishefsky SJ. Chem Commun. 2002:2759. doi: 10.1039/b209941a. [DOI] [PubMed] [Google Scholar]; (b) Morrill C, Grubbs RH. J Org Chem. 2003;68:6031. doi: 10.1021/jo0345345. [DOI] [PubMed] [Google Scholar]
- 29.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
- 30.Hong SH, Sanders DP, Lee CW, Grubbs RH. J Am Chem Soc. 2005;127:17160. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- 31.(a) Lindgren BO, Nilsson T. Acta Chem Scand. 1973;27:888. [Google Scholar]; (b) Bal BS, Childers WE, Jr, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- 32.Baumhof P, Mazitschek R, Giannis A. Angew Chem, Int Ed. 2001;40:3672. doi: 10.1002/1521-3773(20011001)40:19<3672::aid-anie3672>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 33.CCDC-740150 contains the supplementary crystallographic data for compound (−)-1. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.