

Abstract
Growth suppressive effect of diallyl trisulfide (DATS), a promising cancer chemopreventive constituent of garlic, against cultured human cancer cells correlates with checkpoint kinase 1 (Chk1)-mediated mitotic arrest, but the fate of the cells arrested in mitosis remains elusive. Using LNCaP and HCT-116 human cancer cells as a model, we now demonstrate that the Chk1-mediated mitotic arrest resulting from DATS exposure leads to apoptosis. The DATS exposure resulted in G2 phase and mitotic arrest in both LNCaP and HCT-116 cell lines. The G2 arrest was accompanied by down-regulation of cyclin-dependent kinase 1 (Cdk1), cell division cycle (Cdc) 25B, and Cdc25C leading to Tyr15 phosphorylation of Cdk1 (inactivation). The DATS-mediated mitotic arrest correlated with inactivation of anaphase-promoting complex/cyclosome as evidenced by accumulation of its substrates cyclinB1 and securin. The DATS treatment increased activating phosphorylation of Chk1 (Ser317) and transient transfection with Chk1-targeted siRNA conferred significant protection against DATS-induced mitotic arrest in both cell lines. The Chk1 protein knockdown also afforded partial yet statistically significant protection against apoptotic DNA fragmentation and caspase-3 activation resulting from DATS exposure in both LNCaP and HCT-116 cells. Even though DATS treatment resulted in stabilization and Ser15 phosphorylation of p53, the knockdown of p53 protein failed to rescue DATS-induced mitotic arrest. In conclusion, the results of the present study indicate that Chk1-dependence of DATS-induced mitotic arrest in human cancer cells is not influenced by the p53 status and cells arrested in mitosis upon DATS exposure are driven to apoptotic DNA fragmentation.
Keywords: diallyl Trisulfide, mitotic Arrest, apoptosis
INTRODUCTION
Epidemiological observations support the premise that dietary intake of Allium vegetables (e.g., garlic) may lower the risk of different types of malignancies including stomach, esophageal, and prostate cancer [1–3]. For example, the risk of prostate cancer was found to be significantly lower in men consuming >10 g/day of total Allium vegetables than in men with total Allium vegetable intake of <2.2g/day in a population-based, case-control study [3]. Anticarcinogenic effect of Allium vegetables is attributed to organosulfur compounds (OSCs), which are released upon processing (e.g., cutting or chewing) of these vegetables [4]. Allium vegetable-derived OSCs, including diallyl sulfide (DAS), diallyl disulfide (DADS) and diallyl trisulfide (DATS), have been shown to inhibit cancer in animal models induced by a variety of chemical carcinogens [5–8]. For example, oral gavage of 200 mg DAS/kg body weight for 3 days prior to challenge with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone significantly decreased pulmonary tumor incidence and multiplicity in female A/J mice [7]. Dietary feeding of DADS suppressed the incidence and multiplicity of azoxymethane-induced invasive colon cancer in rats [8]. Naturally occurring OSCs are also effective in affording protection against benzo[a]pyrene-induced forestomach and pulmonary carcinogenesis in mice [5]. The mechanisms by which DAS, DADS, and DATS inhibit chemically-induced cancer are well-explained and involve an increase in expression of phase 2 carcinogen detoxifying enzymes (e.g., glutathione transferases and quinone reductase) and/or inhibition of cytochrome P450-dependent monooxygenases [9–13].
More recent studies, including those from our laboratory, have indicated that OSCs inhibit growth of human cancer cells in culture and in xenograft model in vivo [14–24]. The mechanism by which OSCs suppress growth of human cancer cells is not fully understood but known cellular responses to OSCs include G2 and mitotic arrest, disruption of microtubule network, apoptosis induction, and suppression of angiogenesis [12–28]. The G2/M phase cell cycle arrest and apoptosis induction was first documented by Milner and colleagues in DADS-treated human colon cancer cells [14,15]. Subsequent studies from our laboratory using human prostate and lung cancer cells revealed that DATS was a much more potent suppressor of cancer cell proliferation compared with either DAS or DADS [17,28]. We also found that the DATS-mediated apoptosis in cancer cells is dependent on generation of reactive oxygen species and regulated by activation of c-Jun N-terminal kinases and induction of Bax/Bak [17,24,28]. The generation of reactive oxygen species in DATS-treated cells is accomplished by an increase in labile iron pool due to proteasome-mediated degradation of iron storage protein ferritin [22]. Apoptosis resulting from DATS exposure is significantly attenuated by pharmacological inhibition of c-Jun N-terminal kinase, overexpression of anti-oxidative enzyme catalase, and siRNA knockdown of Bax and Bak [17,24,28]. Consistent with these results, the DATS-mediated inhibition of PC-3 human prostate cancer xenograft growth in vivo is accompanied by induction of Bax and Bak [27]. In addition, the DATS-induced apoptosis correlates with proteolytic cleavage of caspase-9 and caspase-3 and blunted in the presence of pharmacological inhibitors of these caspases [21]. More recent studies from our laboratory have demonstrated that oral gavage of 1 and 2 mg DATS/day, three times per week for 13 weeks, significantly inhibits incidence and burden of poorly differentiated carcinoma and pulmonary metastasis multiplicity in a transgenic mouse model of prostate cancer without causing any harmful side effects [29].
We showed previously that DATS treatment activates mitotic arrest in human prostate cancer cells lacking functional p53 (PC-3 and DU145) in association with activation of checkpoint kinase 1 (Chk1) [20,23]. However, it is unclear if the DATS-mediated and Chk1-dependent mitotic arrest is unique to prostate cancer cells or influenced by the p53 status, which is a known downstream target of Chk1 [30]. In addition, the ultimate fate of the cells arrested in mitosis following DATS exposure remains elusive. In the present study, we have addressed these mechanistically intriguing questions relevant to the mechanism of anticancer effect of DATS using LNCaP (a human prostate cancer cell line expressing wild-type p53), HCT-116 (a human colon carcinoma cell line expressing wild-type p53), and p53−/− variant of HCT-116 cells.
MATERIALS AND METHODS
Reagents
DATS (purity ~99%) was purchased from LKT Laboratories (St. Paul, MN). Oligofectamine was from Invitrogen (Carlsbad, CA); penicillin/streptomycin antibiotic mixture and fetal bovine serum (FBS) were from GIBCO (Grand Island, NY); propidium iodide, 4′,6-diamidino-2-phenylindole (DAPI), and phosphatase inhibitors were from Sigma (St. Louis, MO); RNaseA was from Promega (Madison, WI); and protease inhibitor cocktail was from BD Pharmingen (San Diego, CA). The antibodies against cyclinB1, cyclin-dependent kinase 1 (Cdk1), phospho-(Tyr15)-Cdk1, cell division cycle (Cdc) 25B, Cdc25C, Cdc20 and Cdc27 were from Santa Cruz Biotechnology (Santa Cruz, CA); the antibodies against Chk1 and phospho-(Ser317)-Chk1 were from Cell Signaling (Beverly, MA); antibody against securin was from MBL (Woburn, MA); anti-Cdh1 antibody was from NeoMarkers (Fremont, CA); an antibody specific for detection of phospho-(Ser10)-histone H3 was from Upstate Biotechnology (Lake Placid, NY); and anti-actin antibody was from Sigma.
Cell Culture
Monolayer culture of LNCaP cells was maintained in RPMI 1640 medium supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 0.2% glucose and antibiotics. The wild-type HCT-116 cells and p53−/− variant of HCT-116 cells were maintained in McCoy’s 5A medium supplemented with 10% FBS and antibiotics. Stock solution of DATS was prepared in dimethyl sulfoxide (DMSO) and an equal volume of DMSO (final concentration 0.1%) was added to controls. Cells were maintained at 37°C in an atmosphere of 5% CO2 and 95% air.
Analysis of Cell Cycle Distribution
Cell cycle distribution in control (DMSO-treated) and DATS-treated cells was determined by flow cytometry following staining with propidium iodide as described by us previously [18,20]. Briefly, cells (3×105) were plated, allowed to attach by overnight incubation, and exposed to DMSO (control) or desired concentrations of DATS for specified time periods. Both floating and adherent cells were collected, washed with phosphate-buffered saline (PBS), fixed with 70% ethanol, and incubated for 30 min at room temperature with propidium iodide and RNaseA. Cell cycle distribution was determined using a Coulter Epics XL Flow Cytometer (Miami, FL). Cells in different phases of the cell cycle were computed for DMSO-treated control and DATS-treated cultures.
Microscopic Analysis of Mitotic Cells
The mitotic figures were visualized by fluorescence microscopy after staining DMSO-treated control and DATS-treated cells with anti-α-tubulin antibody and DAPI. Briefly, (2×105) were grown on coverslips, and allowed to attach by overnight incubation at 37°C. The cells were then exposed to DMSO or 40 μM DATS for 4, 8, or 16 h at 37°C, washed with PBS, and fixed at 4°C overnight in 2% paraformaldehyde. Subsequently, the cells were permeabilized with 0.1% Triton-X100 for 15 min at room temperature, washed with PBS, and blocked with PBS containing 0.5 % (w/v) bovine serum albumin and 0.15 % (w/v) glycine (BSA buffer) for 1 h at room temperature. Cells were then treated with anti-α-tubulin antibody (1:4000 dilution in BSA buffer) for 1 h at room temperature, washed with BSA buffer, and incubated with 1 μg/mL Alexa Fluor 568-conjugated secondary antibody (Molecular Probes, Eugene, OR) for 1 h at room temperature followed by counter staining with DAPI. Slides were mounted and mitotic figures were examined under a Leica DM IRB fluorescence microscope at 40× objective lens magnification. A total of 300 cells were scored for mitotic figures.
Immunoblotting
Cells were treated with DATS and lysed as descried by us previously [31]. Lysate was cleared by centrifugation at 14,000 rpm for 15 min. Lysate proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto membrane. The membrane was treated with a solution containing 10 mM Tris (pH 7.4), 150 mM NaCl, 0.05% Tween-20 and 5% non-fat dry milk, and incubated with the desired primary antibody for 1–2 h at room temperature or overnight at 4°C. The membrane was treated with appropriate secondary antibody for 1 h at room temperature. The immunoreactive bands were visualized by enhanced chemiluminescence method. The blots were stripped and re-probed with anti-actin antibody to normalize for differences in protein loading. To quantify changes in protein levels, the intensity of the immunoreactive bands was determined by densitometric scanning and corrected for actin control.
RNA Interference
RNA interference of Chk1 was performed using Chk1-specific siRNA duplexes (GCG UGC CGU AGA CUG UCC AdTdT) from Dharmacon (Lafayette, CO). The p53-specific siRNA was purchased from Santa Cruz Biotechnology (cat # sc-29435). For transfection, cells were seeded in 6-well plates and transfected at 30–50% confluency with 200 nM siRNA using Oligofectamine according to the manufacturer’s recommendations. Cells transfected with a non-specific siRNA (UUC UCC GAA CGU GUC ACG UdTdT; Qiagen) were used as controls. Twenty-four hours after transfection, the cells were treated with DMSO or desired concentration of DATS for specific time periods. Both floating and adherent cells were collected, washed with PBS, and processed for immunoblotting of desired protein or analysis of cytoplasmic histone-associated DNA fragmentation and caspase-3 activation.
Determination of Apoptosis
Apoptosis induction by DATS was assessed by analysis of cytoplasmic histone-associated DNA fragmentation using a kit from Roche Diagnostics according to the manufacturer’s instructions. Activation of caspase-3 was determined by flow cytometry using a kit from Cell Signaling as described by us previously [32]. Briefly, the cells (3×105) were plated in T25 flasks and allowed to attach by overnight incubation. The cells were then treated with DMSO (control) or desired concentration of DATS for specified time periods. Subsequently, the cells were collected by trypsinization and processed for flow cytometric analysis of caspase-3 activation according to the manufacturer’s instructions.
Flow Cytometric Analysis of Ser10 Phosphorylated Histone H3
The effect of DATS treatment on Ser10 phosphorylation of histone H3, a sensitive marker for mitotic cells, was determined by flow cytometry as described by Widrow and Laird [33] with some modifications. Briefly, the cells were treated with DATS as described above, fixed in 70% ethanol at 4°C, suspended in 1 mL of 0.25% Triton X-100 in PBS, and incubated on ice for 15 min. The cells were collected, suspended in 100 μL of PBS containing 1% BSA and 0.75 μg of polyclonal anti-phospho-(Ser10)-histone H3 antibody, and incubated at 4°C overnight with gentle shaking. The cells were then rinsed with PBS containing 1% BSA and incubated in dark with secondary antibody for 45 min at room temperature. The cells were washed with PBS containing 1% BSA, and treated with propidium iodide and RNaseA for 30 min. Cellular fluorescence was measured using a Coulter Epics XL Flow Cytometer.
RESULTS
DATS Treatment Caused G2 Phase and Mitotic Arrest in Cancer Cells
We have shown previously that DATS treatment causes G2/M phase cell cycle arrest in PC-3 and DU145 human prostate cancer cells [18], which lack functional p53. Because p53 tumor suppressor is involved in regulation of G2/M transition in response to different stimuli [34], we reasoned that the DATS-induced G2/M phase cell cycle arrest might be influenced by the p53 status. We explored this possibility using LNCaP cells line, which expresses wild-type p53. Figure 1A depicts time course kinetic effect of DATS treatment on cell cycle distribution in LNCaP cells. Exposure of LNCaP cultures to 40 μM DATS resulted in statistically significant enrichment of G2/M fraction that was accompanied by a decrease in G0/G1 cells. The G2/M phase cell cycle arrest was evident as early as 4 h after DATS treatment and persisted for up to 16 h post-exposure (Figure 1A). For example, the percentage of G2/M fraction upon treatment of LNCaP cultures with 40 μM DATS for 4, 8, and 16 h was higher by about 1.6- to 1.8-fold compared with corresponding DMSO-treated control. The G2/M phase cell cycle arrest in LNCaP cell line was also observed at 20 μM DATS concentration (results not shown). DATS treatment also caused significant increase in fraction of sub-diploid apoptotic cells at 8 and 16 h time points (Figure 1A). The DATS-mediated enrichment of G2/M and sub-diploid fractions was not unique to prostate cancer cells because similar effects were observed in wild-type p53 expressing human colon cancer cell line HCT-116 (Figure 1B).
Figure 1.

Diallyl trisulfide (DATS)-treated cancer cells were arrested in G2/M phase of the cell cycle. Cell cycle distribution in (A) LNCaP and (B) HCT-116 cultures following treatment with DMSO (control) or DATS (20 or 40 μM) for 4, 8 or 16 h (LNCaP) or 16 h (HCT-116). Results are mean ± SE (n=3). *, P < 0.05, significantly different compared with corresponding DMSO-treated control by t-test (LNCaP) or one-way ANOVA followed by Dunnett’s test (HCT-116). The experiment was performed twice with triplicate measurements. The results were consistent and representative data from a single experiment are shown.
Next, we raised the question of whether DATS-treated LNCaP cells were arrested in mitosis, which would not be revealed by the flow cytometric analysis of cell cycle distribution. We addressed this question by immunofluorescence microscopy following staining of control and DATS-treated LNCaP cells with anti-α-tubulin antibody and DAPI. The DMSO-treated control LNCaP cells exhibited intact microtubule network and lack of chromatin condensation (Figure 2A). On the other hand, the cells treated with 40 μM DATS exhibited chromatin condensation and disruption of the α-tubulin network. The α-tubulin staining in DATS-treated LNCaP cells was restricted to the periphery of the DAPI-stained nuclei (Figure 2A). The mitotic figures from control and DATS-treated LNCaP cultures were scored and the results are summarized in Figure 2B. The fraction of mitotic cells was increased by about 1.8-, 2.4-, and 8.0-fold upon treatment of LNCaP cells with 40 μM DATS for 4, 8, and 16 h, respectively, compared with corresponding DMSO-treated control (Figure 2B). The Ser10 phosphorylation of histone H3 is a sensitive marker for mitotic cells and correlates with mitotic chromosome condensation [35]. As can be seen in Figure 2C, DATS treatment (20 and 40 μM) resulted in a marked increase in Ser10 phosphorylation of histone H3 in both LNCaP and HCT-116 cell lines especially at the 16 h time point. Collectively, these results indicated that the DATS-induced G2 and mitotic arrest was neither specific for prostate cancer cells nor influenced by the p53 status.
Figure 2.
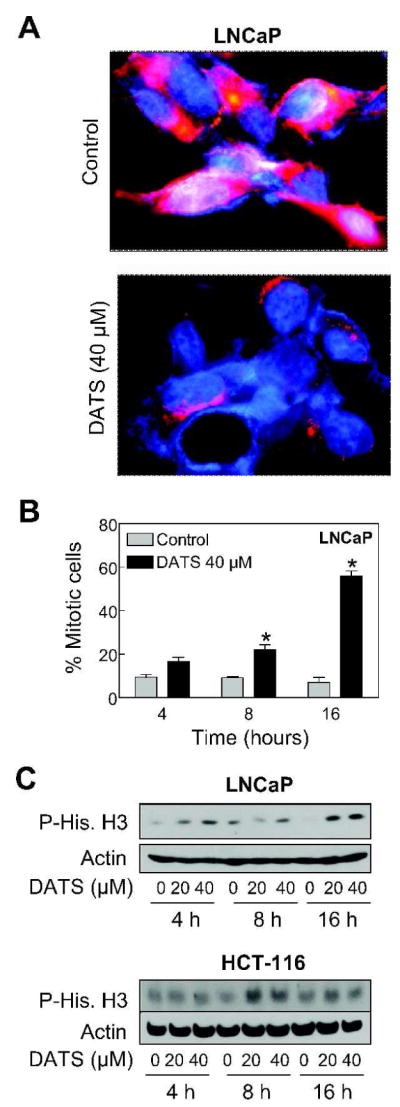
Diallyl trisulfide (DATS) treatment caused mitotic arrest in LNCaP and HCT-116 cells. (A) Representative fluorescence microscopic images for α-tubulin (red fluorescence) and DAPI (blue fluorescence) staining in LNCaP cultures treated for 16 h with DMSO (control) or 40 μM DATS. (B) Quantitation of mitotic figures with condensed chromatin and disrupted α-tubulin network in LNCaP cultures treated for 4, 8, and 16 h with DMSO (control) or 40 μM DATS. A total of 300 cells from four different slides were scored for mitotic figures. Results are mean ± SE (n=4). *, P < 0.05, significantly different compared with corresponding DMSO-treated control by t-test. (C) Immunoblotting for phospho-(Ser10)-histone H3 using lysates from LNCaP and HCT-116 cells treated with DMSO (control) or the indicated concentrations of DATS for 4, 8, or 16 h. The blots were stripped and re-probed with anti-actin antibody to ensure equal protein loading. Similar results were observed in two experiments.
DATS Treatment Caused Accumulation of Tyr15 phosphorylated Cdk1 in LNCaP Cells
Eukaryotic cell cycle progression involves sequential activation of Cdks, whose activity is dependent upon their association with regulatory cyclins [36,37]. A complex between Cdk1 and cyclinB1 is important for entry into mitosis in most organisms [36,37]. The activity of Cdk1/cyclinB1 kinase is negatively regulated by reversible phosphorylations at Thr14 and Tyr15 of Cdk1 [36,37]. Dephosphorylation of Thr14 and Tyr15 of Cdk1, and hence activation of the Cdk1/cyclinB1 kinase complex, is catalyzed by Cdc25 family of dual specificity phosphatases [38]. We proceeded to determine the effect of DATS treatment on levels of G2/M regulatory proteins using LNCaP cells to gain insights into the mechanism of cell cycle arrest. The DATS exposure resulted in an increase in the protein level of cyclinB1, which was transient and peaked between 8 and 16 h of treatment at both 20 and 40 μM concentrations (Figure 3A). On the other hand, DATS treatment caused a marked decrease in the protein levels of Cdk1, Cdc25B, and Cdc25C (Figure 3A). The DATS-mediated Tyr15 phosphorylation of Cdk1 (inactive) was evident as early as 4 h after DATS treatment (Figure 3B) and peaked between 8 and 16 h (Figure 3A). Because Tyr15 phosphorylation of Cdk1 is indicative of inactivation of Cdk1/cyclinB1 complex, it is reasonable to conclude that DATS treatment causes transient inactivation of this kinase leading to G2 arrest.
Figure 3.

Diallyl trisulfide (DATS) treatment caused inactivation of anaphase-promoting complex/cyclosome in LNCaP and HCT-116 cells. Immunoblotting for cyclinB1, cdk1, Cdc25B, Cdc25C, and/or Tyr15 phosphorylated Cdk1 (inactive) using lysates from (A) LNCaP cells and (C) HCT-116 cells treated with DMSO (control) or the indicated concentrations of DATS for 4, 8, and 16 h. Immunoblotting for components of APC/C (Cdc27, Cdc20, cdk1) and APC/C substrate securin using lysates from (B) LNCaP and (D) HCT-116 cells treated with DMSO (control) or the indicated concentrations of DATS for 4, 8, and 16 h. The blots were stripped and re-probed with anti-actin antibody to ensure equal protein loading. Immunoblotting for each protein was performed at least twice using independently prepared lysates and the results were consistent. Representative data from a single experiment are shown. Numbers on top of bands, densitometric quantitation relative to corresponding DMSO-treated control.
DATS Treatment Inhibited Anaphase Promoting Complex/Cyclosome (APC/C) in LNCaP Cells
The mitotic exit depends on active APC/C, which is a multi-subunit ubiquitin-protein ligase complex [39,40]. Cdc27 is one of the core subunits of APC/C which becomes phosphorylated in mitosis [41]. To test whether DATS-mediated mitotic arrest was due to inactivation of APC/C, we determined the effect of DATS treatment on protein levels of APC/C components (Cdc27, Cdc20, and Cdh1) and APC/C substrate securin by immunoblotting. As can be seen in Figure 3B, immunoblotting for Cdc27 protein revealed increased level as well as appearance of bands with reduced electrophoretic mobility suggesting phosphorylation in DATS-treated LNCaP cells and confirming mitotic arrest in our model. While DATS treatment did not have an appreciable effect on Cdc20 expression, an increase in phosphorylation of Cdh1 was observed in LNCaP cells exposed to DATS as evidenced by the appearance of a band with reduced electrophoretic mobility (Figure 3B). The increase in Cdh1 phosphorylation was evident as early as 4 h after treatment with 40 μM DATS (Figure 3B). The protein levels of APC/C substrate securin was higher in DATS-treated LNCaP cells at 4 and 8 h time points compared with corresponding controls (Figure 3B). These results indicated inactivation of APC/C in DATS-treated LNCaP cells.
DATS Treatment Caused Phosphorylation of Cdk1 and Inactivation of APC/C in HCT-116 Cells
We also determined the effect of DATS treatment on levels of proteins involved in regulation of G2/M progression and APC/C components/substrate using HCT-116 cells. Similar to LNCaP cells, DATS treatment resulted in increase in cyclinB1 level, down-regulation of Cdk1, and transient increase in Tyr15 phosphorylation of Cdk1 (Figure 3C). Likewise, the DATS-mediated Tyr15 hyperphosphorylation of Cdk1 in HCT-116 cells was observed at both 20 and 40 μM concentrations and peaked between 8 and 16 h (Figure 3C). Even though DATS treatment resulted in phosphorylation of Cdh1 in HCT-116 cells, the kinetics of this effect was different between LNCaP and HCT-116 cells. In HCT-116 cells, the hyperphosphorylation of Cdh1 was not evident until 16 h (Figure 3D) whereas DATS-mediated increase in the level of P-Cdh1 in the LNCaP cell line was observed at 4 h especially at the 40 μM concentration (Figure 3B). Consistent with data in LNCaP, DATS treatment resulted in accumulation of APC/C substrate securin in the HCT-116 cell line. These observations indicated that the DATS-mediated mitotic arrest was caused by inactivation of APC/C in both LNCaP and HCT-116 cells.
Chk1 Protein Knockdown Conferred Protection against DATS-induced Mitotic Arrest in LNCaP and HCT-116 Cells
To test whether DATS-induced mitotic arrest in LNCaP and HCT-116 cells was regulated by activation of Chk1, we first determined the effect of DATS treatment on activating phosphorylation (Ser317) of Chk1. As can be seen in Figure 4A, DATS treatment resulted in rapid and sustained increase in Ser317 phosphorylation of Chk1 at both 20 and 40 μM concentrations. The basal phosphorylation of Chk1 was very weak in DMSO-treated control LNCaP cells (Figure 4A). Likewise, DATS treatment increased Ser317 phosphorylation of Chk1 in HCT-116 cells, which peaked between 8 and 16 h (Figure 4B). The DATS-mediated increase in Ser317 phosphorylation of Chk1 was not due to an increase in its protein level in either cell line (Figure 4A,B). Next, we utilized siRNA technology to gain further insight into the role of Chk1 activation in DATS-mediated mitotic arrest. Transient transfection of LNCaP cells with a Chk1-specific siRNA resulted in about 70% depletion of Chk1 protein level compared with cells transfected with a non-specific siRNA (Figure 4C). Similar to un-transfected cells, the DATS treatment (40 μM, 8 h) resulted in accumulation of APC/C substrates cyclinB1 and securin and hyperphosphorylation of mitotic marker histone H3 over DMSO-treated control in non-specific siRNA transfected LNCaP cells (Figure 4C). The DATS-mediated accumulation of cyclinB1 and securin protein levels and Ser10 phosphorylation of histone H3 were markedly suppressed in Chk1 depleted LNCaP cells (Figure 4C). Knockdown of Chk1 protein level also conferred a marked protection against DATS-mediated accumulation of cyclinB1 and securin and Ser10 phosphorylation of histone H3 in HCT-116 cells (Figure 4D). These results indicated that the DATS-mediated mitotic arrest was Chk1-dependent in both LNCaP and HCT-116 cells.
Figure 4.

Diallyl trisulfide (DATS)-induced mitotic arrest was partially but markedly reversed by knockdown of checkpoint kinase 1 (Chk1) protein. Immunoblotting for total Chk1 and Ser317 phosphorylated Chk1 using lysates from (A) LNCaP and (B) HCT-116 cells treated with DMSO (control) or the indicated concentrations of DATS for 4, 8, and 16 h. Immunoblotting for total Chk1, APC/C substrates (cyclinB1 and securin), and Ser10 phosphorylated histone H3 (a marker of mitotic cells) using lysates from (C) LNCaP and (D) HCT-116 cells transiently transfected with a non-specific siRNA or Chk1-targeted siRNA and treated for 8 h with either DMSO (control) or 40 μM DATS. The blots were stripped and re-probed with anti-actin antibody to ensure equal protein loading. Numbers on top of the band, change in protein level relative to corresponding DMSO-treated control (panels A and B) and DMSO-treated non-specific siRNA transfected cells (panels C and D). Similar results were observed in replicate experiments.
Cells Arrested in Mitosis by DATS Treatment Were Driven to Apoptosis
The relationship between DATS-induced cell cycle arrest and apoptosis has not been addressed previously either by us or other investigators. To address this question, we determined the effect of Chk1 protein knockdown on DATS-mediated apoptosis using LNCaP and HCT-116 cells. As can be seen in Figure 5A, twenty-four hour exposure of non-specific siRNA transfected LNCaP cells to 20 and 40 μM DATS resulted in about 5 to 6-fold increase in release of histone-associated DNA fragments into the cytosol (a measure of apoptosis) compared with DMSO-treated control. The extent of cytoplasmic histone-associated DNA fragmentation resulting from a similar DATS treatment was significantly lower in LNCaP cells transfected with the Chk1-specific siRNA in comparison with non-specific siRNA transfected cells (Figure 5A). Consistent with these results, Chk1 protein knockdown conferred significant protection against DATS-mediated activation of caspase-3 in LNCaP cells (Figure 5B). The DATS-induced cytoplasmic histone-associated DNA fragmentation (Figure 5C) as well as activation of caspse-3 (Figure 5D) was also significantly suppressed by Chk1 protein knockdown in HCT-116 cells. However, the protection against DATS-mediated DNA fragmentation by Chk1 knockdown was relatively more pronounced in the HCT-116 cells than in the LNCaP cell line. While the precise mechanism underlying the difference in response between LNCaP and HCT-116 cells remains elusive, several possibilities exist to explain this discrepancy. One possible explanation relates to the genetic differences between LNCaP and HCT-116 cells. Second, the LNCaP cell line is relatively more sensitive to DNA fragmentation and caspase-3 activation by DATS exposure compared with HCT-116 cells (Fig. 5A,C). It is also plausible that the signal transduction leading to apoptosis is different between LNCaP and HCT-116 cells. Nonetheless, these results indicated that cells arrested in mitosis upon DATS exposure were driven to apoptosis.
Figure 5.
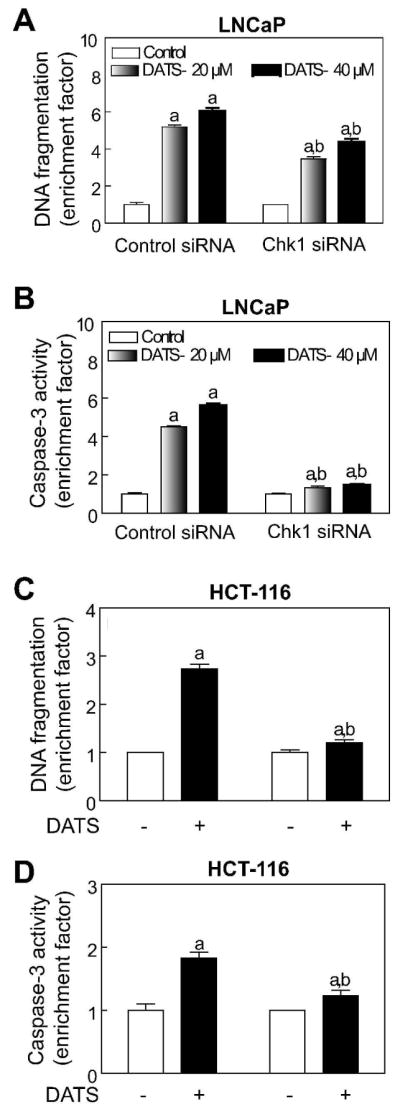
Checkpoint kinase 1 (Chk1) protein knockdown protected against diallyl trisulfide (DATS)-induced apoptosis. Cytoplasmic histone-associated DNA fragmentation in (A) LNCaP and (C) HCT-116 cells transiently transfected with a non-specific siRNA or Chk1-specific siRNA and treated for 24 h with either DMSO (control) or DATS (20 and 40 μM for LNCaP and 40 μM for HCT-116). Activation of caspase-3 in (B) LNCaP and (D) HCT-116 cells transiently transfected with a non-specific siRNA or Chk1-specific siRNA and treated for 24 h with either DMSO (control) or DATS (20 and 40 μM for LNCaP and 40 μM for HCT-116). Results are expressed as enrichment factor relative to DMSO-treated control in each cell line. Data are mean ± SE (n=3). Significantly different, P < 0.05, compared with acorresponding DMSO-treated control and bbetween DATS-treated non-specific siRNA transfected and DATS-treated Chk1-specific siRNA transfected cells by one-way ANOVA followed by Bonferroni’s test. Each experiment was performed twice with comparable results.
p53 Protein Was Dispensable for DATS-induced Mitotic Arrest
The p53 tumor suppressor is involved in regulation of G2/M transition in response to a variety of stresses [34]. The transcriptional activity and stability of p53 is regulated by phosphorylation at multiple sites [30]. For example, Ser15 phosphorylation of p53 leads to its stabilization due to reduced interaction with Mdm2 [30]. Because p53 is a downstream target of Chk1 [30], it was of interest to determine whether the presence of wild-type p53 affects Chk1-dependence of DATS-induced mitotic arrest. As can be seen in Figure 6A, DATS treatment (20 and 40 μM) caused an increase in protein level as well as Ser15 phosphorylation of p53 in the LNCaP cell line. We used siRNA technology to determine possible involvement of p53 in DATS-induced mitotic arrest. The level of p53 protein was decreased by about 70% in LNCaP cells transiently transfected with a p53-specific siRNA (Figure 6B). However, the DATS-mediated stabilization of cyclinB1 and Ser10 phosphorylation of histone H3 was maintained in cells with p53 protein knockdown (Figure 6B). Consistent with these results, the DATS treatment (40 μM, 8 h) resulted in increased Ser10 phosphorylation of histone H3 not only in wild-type HCT116 cells but also in its p53−/− variant (Figure 6C). Based on these results, we conclude that p53 protein is dispensable for DATS-induced mitotic arrest.
Figure 6.
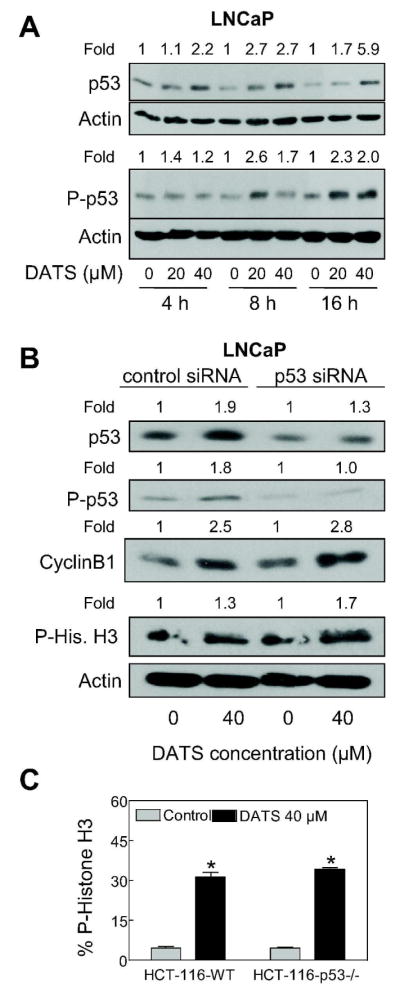
p53 was dispensable for diallyl trisulfide (DATS)-induced mitotic arrest. (A) Immunoblotting for total p53 and Ser15 phosphorylated p53 using lysates from LNCaP cells treated with DMSO (control) or the indicated concentrations of DATS for 4, 8, and 16 h. (B) Immunoblotting for total p53, Ser15 phosphorylated p53, cyclinB1, and Ser10 phosphorylated histone H3 using lysates from LNCaP cells transiently transfected with a non-specific siRNA or p53-specific siRNA and treated for 8 h with either DMSO (control) or 40 μM DATS. The blots were stripped and re-probed with anti-actin antibody to ensure equal protein loading. Numbers on top of band, changes in the levels of total or phosphorylated proteins relative to corresponding DMSO-treated control. (C) Flow cytometric analysis of Ser10 phosphorylated histone H3 in wild-type HCT-116 cells and p53−/− HCT-116 cells following 8 h treatment with DMSO (control) or 40 μM DATS. Results are mean ± SE (n=3). *, P < 0.05, significantly different compared with corresponding DMSO-treated control by t-test. The results were consistent in two independent experiments.
DISCUSSION
We have shown previously that DATS treatment triggers mitochondria (intrinsic pathway)-mediated apoptosis in cancer cells characterized by induction of Bax/Bak, mitochondrial swelling, collapse of mitochondrial membrane potential, release of apoptogenic molecules from mitochondria to the cytosol, and activation of caspase-9 and caspase-3 [17,24,28]. Previous studies from our laboratory have also shown that the DATS-treated cells are arrested in mitosis in an Chk1 dependent manner [20,23]. The present study builds upon these observations and demonstrates that the DATS-mediated mitotic arrest is not unique to the prostate cancer cells because HCT-116 human colon cancer cell line behaves similarly. One of the objectives of the present study was to gain insights into the fate of the cells arrested in mitosis after DATS exposure. We found that the cells arrested in mitosis are driven to apoptotic DNA fragmentation because knockdown of Chk1 confers protection against mitotic arrest as well as apoptotic DNA fragmentation. However, it is likely that DATS also triggers apoptosis independently of the Chk1-mediated mitotic arrest because knockdown of Chk1 does not fully rescue DNA fragmentation resulting from DATS exposure especially in the LNCaP cell line. The relationship between DATS-mediated cell cycle arrest and apoptosis is summarized in Figure 7.
Figure 7.

Schematic presentation of the mechanistic relationship between diallyl trisulfide (DATS)-induced cell cycle arrest and apoptosis.
The DATS-mediated G2/M arrest in both LNCaP and HCT-116 cells correlates with Tyr15 hyperphosphorylation of Cdk1 suggesting inactivation of the Cdk1/cyclinB1 kinase complex. The DATS-mediated accumulation of P-Cdk1 occurs at both 20 and 40 μM concentrations in both cell lines especially at the 8 h time point. At the same time abrogation of the Cdk1 phosphorylation at the 16 h time point suggests that the cells arrested in G2 upon DATS exposure are able to reactivate Cdk1 and enter mitosis but again get arrested in mitosis as judged by hyperphosphorylation of P-histone H3 lasting for 16 h.
We found that the Chk1-dependence of DATS-induced mitotic arrest in the LNCaP as well as HCT-116 cell line is not influenced by p53, a downstream target of Chk1 [30], but correlates with inactivation of APC/C. This conclusion stems from the following observations: (a) the DATS-induced mitotic arrest is significantly reversed by knockdown of Chk1 but not p53 protein in both cell lines, (b) the mitotic arrest resulting from DATS exposure is observed in wild-type as well as p53−/− HCT116 cells, and (c) the DATS treatment causes accumulation and/or hyperphosphorylation of APC/C substrates cyclinB1 and securin in both cell lines. The activity of APC/C is regulated by reversible phosphorylation of its components as well as interaction between APC/C core and the regulatory subunits Cdc20 and Cdh1, which are responsible for substrate recognition by the holoenzyme [39,40]. For example, phosphorylation of Cdc20 has been shown to inhibit the activity of APC/C during spindle checkpoint induction [42–44]. In addition, Cdh1 phosphorylation inhibits its binding to APC/C core and Cdh1 is dephosphorylated before mitotic exit [45]. It is of note that DATS treatment causes electrophoretic mobility retardation of Cdh1 suggesting phosphorylation in both LNCaP and HCT-116 cells. However, further studies are needed to firmly establish phosphorylation of Cdh1 in DATS-treated cells and to explore if phosphorylation of Cdh1 is mediated by Chk1 kinase.
The DATS-mediated cell cycle arrest in androgen-responsive LNCaP cell line, colon carcinoma cell line HCT-116, and PC-3 and DU145 androgen-independent prostate cancer cells are observed at 20 and 40 μM concentrations [20,23, and present study], which are within the pharmacologically achievable range. The peak plasma concentration of DATS in rats following treatment with 10 mg DATS is about 31 μM [46]. The pharmacokinetic parameters for DATS in humans are yet to be determined, but 200 mg of synthetic DATS in combination with 100 μg selenium has been given to human subjects every other day for one month without any evidence of harmful side effects [47]. It is therefore possible that the plasma concentrations of DATS required for cancer cell growth inhibition and cell cycle arrest may be achievable in humans. It is important to point out that DATS-mediated mitotic arrest is also observed in vivo [29]. Specifically, we have shown previously that oral gavage of 1 and 2 mg DATS/day, three times per week, to male TRAMP mice not only significantly inhibits incidence and burden of poorly-differentiated carcinoma but also results in increased expression of securin and cyclinB1 [29].
In summary, the present study indicates that DATS-mediated and Chk1-dependent mitotic arrest in cancer cells is neither a cell line specific response nor influenced by the p53 status. We also demonstrate, for the first time, that DATS-mediated mitotic arrest is linked to apoptosis.
Acknowledgments
This investigation was supported by USPHS grant CA113363 awarded by the National Cancer Institute. Wild-type HCT-116 and its p53−/− variant were a generous gift from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD)
Abbreviations
- OSCs
organosulfides
- DAS
diallyl sulfide
- DADS
diallyl disulfide
- DATS
diallyl trisulfide
- Chk1
checkpoint kinase 1
- FBS
fetal bovine serum
- DAPI
4′,6-diamidino-2-phenylindole
- Cdk1
cyclin-dependent kinase 1
- Cdc
cell division cycle
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- BSA
bovine serum albumin
- BSA buffer
PBS containing 0.5 % (w/v) BSA and 0.15 % (w/v) glycine
- APC/C
anaphase promoting complex/cyclosome
References
- 1.You WC, Blot WJ, Chang YS, et al. Allium vegetables and reduced risk of stomach cancer. J Natl Cancer Inst. 1989;18:162–164. doi: 10.1093/jnci/81.2.162. [DOI] [PubMed] [Google Scholar]
- 2.Gao CM, Takezaki T, Ding JH, Li MS, Tajima K. Protective effect of allium vegetables against both esophageal and stomach cancer: a simultaneous case-referent study of a high-epidemic area in Jiangsu Province, China. Jpn J Cancer Res. 1999;90:614–621. doi: 10.1111/j.1349-7006.1999.tb00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsing AW, Chokkalingam AP, Gao YT, et al. Allium vegetables and risk of prostate cancer: a population-based study. J Natl Cancer Inst. 2002;94:1648–1651. doi: 10.1093/jnci/94.21.1648. [DOI] [PubMed] [Google Scholar]
- 4.Block E. The organosulfur chemistry of the genus Allium- implications for the organic chemistry of sulfur. Angew Chem Int Ed Engl. 1992;31:1135–1178. [Google Scholar]
- 5.Sparnins VL, Barany G, Wattenberg LW. Effects of organosulfur compounds from garlic and onions on benzo[a]pyrene-induced neoplasia and glutathione S-transferase activity in the mouse. Carcinogenesis. 1988;9:131–134. doi: 10.1093/carcin/9.1.131. [DOI] [PubMed] [Google Scholar]
- 6.Wargovich MJ, Woods C, Eng VWS, Stephens LC, Gray K. Chemoprevention of N-nitrosomethylbenzylamine-induced esophageal cancer in rats by the naturally occurring thioether, diallyl sulfide. Cancer Res. 1988;48:6872–6875. [PubMed] [Google Scholar]
- 7.Hong JY, Wang ZY, Smith TJ, et al. Inhibitory effects of diallyl sulfide on the metabolism and tumorigenicity of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in A/J mouse lung. Carcinogenesis. 1992;13:901–904. doi: 10.1093/carcin/13.5.901. [DOI] [PubMed] [Google Scholar]
- 8.Reddy BS, Rao CV, Rivenson A, Kelloff G. Chemoprevention of colon carcinogenesis by organosulfur compounds. Cancer Res. 1993;53:3493–3498. [PubMed] [Google Scholar]
- 9.Hu X, Benson PJ, Srivastava SK, et al. Glutathione S-transferases of female A/J mouse liver and forestomach and their differential induction by anti-carcinogenic organosulfides from garlic. Arch Biochem Biophys. 1996;336:199–214. doi: 10.1006/abbi.1996.0550. [DOI] [PubMed] [Google Scholar]
- 10.Singh SV, Pan SS, Srivastava SK, et al. Differential induction of NAD(P)H:quinone oxidoreductase by anti-carcinogenic organosulfides from garlic. Biochem Biophys Res Commun. 1998;244:917–920. doi: 10.1006/bbrc.1998.8352. [DOI] [PubMed] [Google Scholar]
- 11.Brady JF, Ishizaki H, Fukuto JM, et al. Inhibition of cytochrome P-450 2E1 by diallyl sulfide and its metabolites. Chem Res Toxicol. 1991;4:642–647. doi: 10.1021/tx00024a008. [DOI] [PubMed] [Google Scholar]
- 12.Herman-Antosiewicz A, Singh SV. Signal transduction pathways leading to cell cycle arrest and apoptosis induction in cancer cells by Allium vegetable-derived organosulfur compounds: a review. Mutation Res. 2004;555:121–131. doi: 10.1016/j.mrfmmm.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Herman-Antosiewicz A, Powolny AA, Singh SV. Molecular targets of cancer chemoprevention by garlic-derived organosulfides. Acta Pharmacol Sin. 2007;28:1355–64. doi: 10.1111/j.1745-7254.2007.00682.x. [DOI] [PubMed] [Google Scholar]
- 14.Sundaram SG, Milner JA. Diallyl disulfide induces apoptosis of human colon tumor cells. Carcinogenesis. 1996;17:669–673. doi: 10.1093/carcin/17.4.669. [DOI] [PubMed] [Google Scholar]
- 15.Knowles LM, Milner JA. Diallyl disulfide inhibits p34(cdc2) kinase activity through changes in complex formation and phosphorylation. Carcinogenesis. 2000;21:1129–1134. [PubMed] [Google Scholar]
- 16.Filomeni G, Aquilano K, Rotilio G, Ciriolo MR. Reactive oxygen species-dependent c-Jun NH2-terminal kinase/c-Jun signaling cascade mediates neuroblastoma cell death induced by diallyl disulfide. Cancer Res. 2003;63:5940–5949. [PubMed] [Google Scholar]
- 17.Xiao D, Choi S, Johnson DE, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene. 2004;23:5594–5606. doi: 10.1038/sj.onc.1207747. [DOI] [PubMed] [Google Scholar]
- 18.Xiao D, Herman-Antosiewicz A, Antosiewicz J, et al. Diallyl trisulfide-induced G2-M phase cell cycle arrest in human prostate cancer cells is caused by reactive oxygen species-dependent destruction and hyperphosphorylation of Cdc25C. Oncogene. 2005;24:6256–6268. doi: 10.1038/sj.onc.1208759. [DOI] [PubMed] [Google Scholar]
- 19.Hosono T, Fukao T, Ogihara J, et al. Diallyl trisulfide suppresses the proliferation and induces apoptosis of human colon cancer cells through oxidative modification of β-tubulin. J Biol Chem. 2005;280:41487–41493. doi: 10.1074/jbc.M507127200. [DOI] [PubMed] [Google Scholar]
- 20.Herman-Antosiewicz A, Singh SV. Checkpoint kinase 1 regulates diallyl trisulfide-induced mitotic arrest in human prostate cancer cells. J Biol Chem. 2005;280:28519–28528. doi: 10.1074/jbc.M501443200. [DOI] [PubMed] [Google Scholar]
- 21.Xiao D, Singh SV. Diallyl trisulfide, a constituent of processed garlic, inactivates Akt to trigger mitochondrial translocation of BAD and caspase-mediated apoptosis in human prostate cancer cells. Carcinogenesis. 2006;27:533–540. doi: 10.1093/carcin/bgi228. [DOI] [PubMed] [Google Scholar]
- 22.Antosiewicz J, Herman-Antosiewicz A, Marynowski SW, Singh SV. c-Jun NH2-terminal kinase signaling axis regulates diallyl trisulfide-induced generation of reactive oxygen species and cell cycle arrest in human prostate cancer cells. Cancer Res. 2006;66:5379–5386. doi: 10.1158/0008-5472.CAN-06-0356. [DOI] [PubMed] [Google Scholar]
- 23.Herman-Antosiewicz A, Stan SD, Hahm ER, Xiao D, Singh SV. Activation of a novel ataxia-telangiectasia mutated and Rad3 related/checkpoint kinase 1-dependent prometaphase checkpoint in cancer cells by diallyl trisulfide, a promising cancer chemopreventive constituent of processed garlic. Mol Cancer Ther. 2007;6:1249–1261. doi: 10.1158/1535-7163.MCT-06-0477. [DOI] [PubMed] [Google Scholar]
- 24.Kim YA, Xiao D, Xiao H, et al. Mitochondria-mediated apoptosis by diallyl trisulfide in human prostate cancer cells is associated with generation of reactive oxygen species and regulated by Bax/Bak. Mol Cancer Ther. 2007;6:1599–1609. doi: 10.1158/1535-7163.MCT-06-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao D, Li M, Herman-Antosiewicz A, et al. Diallyl trisulfide inhibits angiogenic features of human umbilical vein endothelial cells by causing Akt inactivation and down-regulation of VEGF and VEGF-R2. Nutr Cancer. 2006;55:94–107. doi: 10.1207/s15327914nc5501_12. [DOI] [PubMed] [Google Scholar]
- 26.Powolny AA, Singh SV. Multitargeted prevention and therapy of cancer by diallyl trisulfide and related Allium vegetable-derived organosulfur compounds. Cancer Lett. 2008;269:305–314. doi: 10.1016/j.canlet.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao D, Lew KL, Kim Y, et al. Diallyl trisulfide suppresses growth of PC-3 human prostate cancer xenograft in vivo in association with Bax and Bak induction. Clin Cancer Res. 2006;15:6836–6843. doi: 10.1158/1078-0432.CCR-06-1273. [DOI] [PubMed] [Google Scholar]
- 28.Xiao D, Zeng Y, Hahm E, Kim YA, Ramalingam S, Singh SV. Diallyl trisulfide selectively causes Bax and Bak mediated apoptosis in human lung cancer cells. Env Mol Mut. 2009;50:201–212. doi: 10.1002/em.20431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh SV, Powolny AA, Stan SD, et al. Garlic constituent diallyl trisulfide prevents development of poorly-differentiated prostate cancer and pulmonary metastasis multiplicity in TRAMP mice. Cancer Res. 2008;68:9503–9511. doi: 10.1158/0008-5472.CAN-08-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nature Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 31.Xiao D, Srivastava SK, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24:891–897. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 32.Xiao D, Powolny AA, Singh SV. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger ROS-dependent apoptosis in human breast cancer cells. J Biol Chem. 2008;283:30151–30163. doi: 10.1074/jbc.M802529200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Widrow RJ, Laird CD. Enrichment for submitotic cell populations using flow cytometry. Cytometry. 2000;39:126–130. doi: 10.1002/(sici)1097-0320(20000201)39:2<126::aid-cyto5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 34.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 35.Hendzel MJ, Wei Y, Mancini MA, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 36.Hunter T. Braking the cycle. Cell. 1993;75:839–841. doi: 10.1016/0092-8674(93)90528-x. [DOI] [PubMed] [Google Scholar]
- 37.Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000;33:261–274. doi: 10.1046/j.1365-2184.2000.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–191. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Sudakin V, Ganoth D, Dahan A, et al. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–197. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell. 2002;9:931–943. doi: 10.1016/s1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- 41.Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters JM. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22:6598–6609. doi: 10.1093/emboj/cdg627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yudkovsky Y, Shteinberg M, Listovsky T, Brandeis M, Hershko A. Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem Biophys Res Commun. 2000;271:299–304. doi: 10.1006/bbrc.2000.2622. [DOI] [PubMed] [Google Scholar]
- 43.D’Angiolella V, Mari C, Nocera D, Rametti L, Grieco D. The spindle checkpoint requires cyclin-dependent kinase activity. Genes Dev. 2003;17:2520–2525. doi: 10.1101/gad.267603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Z, Shu H, Oncel D, Chen S, Yu H. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004;16:387–397. doi: 10.1016/j.molcel.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 45.Bembenek J, Yu H. Regulation of the anaphase-promoting complex by the dual specificity phosphatase human Cdc14a. J Biol Chem. 2001;276:48237–48242. doi: 10.1074/jbc.M108126200. [DOI] [PubMed] [Google Scholar]
- 46.Sun X, Guo T, He J, et al. Determination of the concentration of diallyl trisulfide in rat whole blood using gas chromatography with electron-capture detection and identification of its major metabolite with gas chromatography mass spectrometry. Yakugaku Zasshi. 2006;126:521–527. doi: 10.1248/yakushi.126.521. [DOI] [PubMed] [Google Scholar]
- 47.Li H, Li HQ, Wang Y, et al. An intervention study to prevent gastric cancer by micro-selenium and large dose of allitridum. Chinese Med J. 2004;117:1155–1160. [PubMed] [Google Scholar]