

Abstract
Francisella tularensis is a highly virulent bacterial species that causes various forms of tularemia in human beings. The urgency in understanding the pathogenesis of these diseases has stimulated unprecedented interest in this bacterial species over the past few years. Recent findings underscore a number of important distinctions between the Francisella subspecies and emphasize the importance of using type A F. tularensis strains when characterizing pathophysiological responses that are relevant to the lethal forms of human disease. This review focuses on the mediators of cell death induction in infected tissues and the implications of these processes to the pathophysiological changes observed in various host species.
Keywords: Francisella tularensis, caspase, apoptosis, pyroptosis, necrosis
Introduction
Tularemia is most frequently a zoonotic infectious disease in humans that is acquired through the bite of an arthropod vector. Cutaneous transmission of the causative agent, Francisella tularensis, typically presents as an ulceroglandular disease that includes a local necrotic skin lesion, draining lymphadenopathy and high fever. While the disease can present in several other localized forms and may be challenging to diagnose due to the variation in presenting symptoms, expeditious antibiotic treatment generally results in a favorable outcome (Matyas, et al., 2007). Disseminated tularemia poses a more serious clinical challenge, due to the potential for rapid growth and dispersion of the pathogen, substantial tissue damage within the lungs, liver and spleen, and the impairment of vital organ functions by unknown mechanisms. Systemic disease can follow virtually any route of transmission (cutaneous, respiratory or gastrointestinal) and is thought to reflect the hematogenous dissemination of the infection after a period of pathogen replication at the initial infection site. The most severe form of systemic disease follows respiratory transmission of the pathogen. Inhalation of live organisms is also thought to be the most likely route of transmission in the event that F. tularensis were to be used as a biological weapon. Because the microbe can survive in small droplet aerosols and shows a very low infectious dose, the pathogen has been designated a Category A Select Agent of Biowarfare. Research with the pathogen requires CDC registration to assure that appropriate biosafety procedures (Biosafety Level 3) are followed, biosecurity is assured, and the risk of exposure to the public and laboratory personnel is minimized. There is currently no approved Francisella vaccine for general use.
Only two of the four subspecies of Francisella (subsp. tularensis, holarctica, novicida and mediasiatica) cause significant disease in immunocompetent humans. Infections with F. tularensis subsp. tularensis (also known as type A) are associated with the highest mortality rates (Dennis,et al., 2001, Farlow,et al., 2005, Staples,et al., 2006, Matyas,et al., 2007) and are predominantly seen in North America. F. tularensis subsp. holarctica causes serious illness in Europe, Asia and North America, but outbreaks are not typically associated with lethal disease. The other Francisella subspecies only rarely cause disease and then only in immunocompromised individuals (Dennis,et al., 2001, Tarnvik,et al., 2004, Farlow,et al., 2005, Staples,et al., 2006, Matyas,et al., 2007). There are a number of reasons that F. novicida and an attenuated type B F. tularensis strain, the Live Vaccine Strain (LVS), have been so often used as experimental surrogates for the more virulent subspecies. These model organisms pose a lower risk to laboratory personnel, induce diseases in mice and other laboratory animals that are similar to those seen in humans (Anthony & Kongshavn, 1987, Conlan,et al., 2002, Bokhari,et al., 2008), and were the first to be manipulated genetically to define determinants of pathogenicity. They can also infect a range of host cells in vitro, which has aided in the definition of potentially important immune responses to F. tularensis. In this regard, one of the most widely used experimental models for studying disease pathogenesis and immunity to Francisella has been the infection of mice with LVS (Anthony & Kongshavn, 1987, Conlan,et al., 2002, Lyons & Wu, 2007). However, studies conducted during the past few years have demonstrated important differences between LVS and F. novicida on the one hand and the pathogenic type A and type B Francisella strains. In this review, we will consider some of these differences, focusing on laboratory animal models of disease pathogenesis.
Pathophysiology of tularemia in humans and nonhuman primates
Tularemia in human beings is an acute febrile disease that shows cutaneous, oculoglandular, pneumonic, gastrointestinal, or septic features (Dennis,et al., 2001, Feldman,et al., 2001, Tarnvik,et al., 2004, Matyas,et al., 2007). Patients generally present with a history of high fever, headache and signs of toxicity (myalgia, anorexia, prostration) of several days duration. Pneumonia with nonproductive cough and liver damage indicated by elevated liver transaminases often follow. Overall case fatality rates in the U.S. are under 2% (Dennis,et al., 2001, Matyas,et al., 2007), but these rates can vary depending on the F. tularensis subspecies (type A versus type B) and the geographic distribution of the cases (Farlow,et al., 2005, Staples,et al., 2006). Primary pneumonic tularemia caused by type A F. tularensis has been reported to have mortality rates in untreated patients as high as 60% (McCrumb, 1961, Dennis,et al., 2001, Feldman,et al., 2001). The clinical response to aminoglycoside therapy (eg., streptomycin or gentamicin) is generally rapid and leads to complete recovery in most cases (Hassoun,et al., 2006).
One of the most complete descriptions of the histopathology of human tularemia was reported by Lamps et al (2004), who examined autopsy or biopsy samples from 10 confirmed cases. While the largest proportion of these infections began with cutaneous transmission, all involved disseminated disease. Lymph node involvement was seen in all cases and characterized by inflammatory cell infiltration and various degrees of necrosis in the outer cortex. Most cases showed pulmonary involvement characterized by necrotizing pneumonia. Histological examination of lung tissues demonstrated neutrophilic infiltration, fibrin deposition and the presence of cellular debris, hemorrhage and edema. Pleuritis was seen in three cases. Gross pathology also included hepatosplenomegaly with small nodules visible in both organs. Hepatic granulomas and microabscesses were a common finding and pyogranulomatous foci in the spleen, lung and lymph nodes often showed central necrosis. Thus, the histological appearance of tularemia in humans is characterized by irregular microabscesses, (pyo)granulomas in the liver, spleen, and lymph nodes and necrotizing pneumonia. These features indicate a highly cytotoxic process that results from damage to both inflammatory and parenchymal cells.
Experimental tularemia has also been characterized in a number of nonhuman primate species following infection with either type A (White,et al., 1964, Twenhafel,et al., 2009) and type B strains (Hall,et al., 1972, Schricker,et al., 1972). These studies indicate that diseases caused by type A F. tularensis progress more rapidly than type B tularemia and result in significant morbidity and mortality. Infection with type B organisms produced a milder disease with a prolonged course and survival. In one study involving aerosol delivery of type B F. tularensis to Rhesus macaques, inflammatory lesions developed in the lungs over 6-21 days, beginning as bronchiolitis and spreading to the surrounding alveolar walls (Hall,et al., 1972). Lesions also developed in tracheobronchial lymph nodes and the liver, but began to resolve during the observation period.
Studies of pneumonic tularemia induced by aerosol challenge of Rhesus macaques with the type A F. tularensis strain SCHU S4 (White,et al., 1964) indicated that the rate of disease progression depended on the size of the aerosol particle. Early bronchiolitis, which was centered on the terminal and respiratory bronchioles, developed within 24 hours and progressed to bronchopneumonia in 72-96 hours. Lymphadenitis, splenitis and hepatitis developed at 24-72 hours and were characterized by infiltrating neutrophils and macrophages.
A more recent study using the SCHU S4 strain was performed in African green monkeys infected by low dose aerosol exposure (Twenhafel,et al., 2009). The SCHU S4 strain rendered primates moribund by 7-11 days post infection (p.i.) and produced necrotic inflammatory foci in the lungs, spleen, liver and lymph nodes that were initially comprised of neutrophils and macrophages. Lesions were not seen in the kidneys, despite the presence of Francisella antigens in glomeruli. The authors also described necrotizing vasculitis, a feature not previously reported in human infections.
Thus, the nonhuman primate models recapitulate the pathogenic differences between type A and type B F. tularensis strains that have been reported for human infections. The presence of organisms and lesions in lymph nodes, liver and spleen following respiratory infection is consistent with a rapid dispersion of the pathogen to secondary infection sites. Cellular infiltrates and tissue damage were also similar to those seen in human tularemia. The morphologic data provide important information about the location of the earliest lesions in smaller airways and the nature of early infiltrating inflammatory cells, data not generally available from human studies.
Pathogenesis of disease in the mouse caused by F. tularensis subsp. holarctica LVS and F. novicida
Infection of mice with LVS has been an enduring animal model of tularemia. This bacterium was initially attenuated by repeated passage on agar and was used for many years for immunizing at-risk individuals. LVS has retained partial virulence in mice compared to clinical type B strains and can establish infection when given by the intradermal (i.d.), intranasal (i.n.) or intraperitoneal (i.p.) routes. However, the lethal dose-50% (LD50) by these different routes varies considerably (i.e., approximately 10 CFU for i.p., 103 CFU for i.n. and >106 CFU for i.d. challenge) (Fortier,et al., 1991, Sjostedt,et al., 1994, Conlan,et al., 2002). Disseminated infections in mice can follow any route of transmission and are characterized by high bacterial burdens in the lungs, liver and spleen during the first week p.i. Death of the host typically occurs 6 – 10 d after respiratory challenge, depending on the challenge dose. Hepatosplenomegaly, pneumonia, and systemic toxicity are all common clinical features of advanced disease in mice. Organisms can be detected within tissue macrophages, neutrophils, dendritic cells (DC), hepatocytes and type II pulmonary epithelial cells of infected mice (Conlan & North, 1992, Conlan,et al., 2003, Bosio & Dow, 2005, Rasmussen,et al., 2006, Hall,et al., 2007, Bokhari,et al., 2008, Hall,et al., 2008). A recent study by Hall et al (2008) indicated that diverse types of lung cells contain LVS following respiratory challenge in mice. The pattern of uptake of LVS was similar to that seen in mice infected with the type A SCHU S4 strain, but differed somewhat from the early pattern of uptake of the F. novicida U112 strain. Whereas the predominant cell type containing each of these pathogens on day 1 was the alveolar macrophage, infiltrating neutrophils constituted the predominant Francisella-containing cell in the lungs by day 3 post-infection.
The tissue responses of mice to infection with LVS are similar to those seen early in type A F. tularensis infections in primates and includes the formation of focal inflammatory infiltrates with high microbial burdens (Anthony & Kongshavn, 1987, Fortier,et al., 1991, Conlan & North, 1992, Conlan,et al., 2002, Conlan,et al., 2003). Conlan and colleagues (Conlan & North, 1992, Conlan,et al., 2002) reported that multifocal hepatic microgranulomas and splenic pyogranulomatous infiltrates formed in response to LVS in several inbred strains of mice. These cellular infiltrates were later shown to be comprised of macrophages, myeloid precursor cells and T lymphocytes (Rasmussen,et al., 2006, Bokhari,et al., 2008). Francisella antigens are detected at this stage within hepatic and splenic inflammatory cells as well as occasional hepatocytes, which are highly permissive for the intracellular replication of the organism (Conlan & North, 1992, Rasmussen,et al., 2006, Bokhari,et al., 2008). Whereas the spleens of LVS-infected mice showed congestion and pyogranulomatous infiltrates in the red pulp, infected lungs retained a fairly normal appearance. The alveolar septa were comparatively devoid of inflammation or parenchymal tissue damage, and only occasionally were peribronchial or perivascular pyogranulomas observed.
The histopathological changes induced by LVS in mice are also noteworthy for the apparent viability of the histiocytic, neutrophillic and lymphoid cells that comprise these structures during most of the first week (Fig. 1A). Inflammatory foci grow in size and frequency with time, and most of the cells that comprise these infiltrates, even as late as day 5 p.i., appear viable. Less than 20% of the cells show evidence of cell death (double strand DNA breaks or the expression of activated caspases) at any given time (Rasmussen,et al., 2006, Bokhari,et al., 2008)(Wickstrum, J.R., Bokhari, S.M., Fischer, J.L., Pinson, D.M., Yeh, H.W., Horvat, R.T., Parmely, M.J., in press). In the liver, Francisella antigens are primarily restricted to the granulomas, further suggesting that cells within these foci are viable and immunologically active (Rasmussen,et al., 2006, Bokhari,et al., 2008) (Fig. 1B). During this period, the liver, spleen and lungs of LVS-infected mice are also active sites for the expression of a range of proinflammatory and anti-inflammatory cyokines, including IL-1β, IL-6, IL-12p40, TNF-α, IFN-γ, IP-10, MCP-1 and IL-10 (Golovliov,et al., 1995, Cole,et al., 2006, Wickstrum,et al., 2007, Bokhari,et al., 2008). Inducible nitric oxide synthase is expressed on days 4 – 6 p.i. within hepatic granulomas (Cole,et al., 2006, Bokhari,et al., 2008). Thus, LVS induces a diverse array of innate immune responses during the first week of infection in mice attesting to the nontoxic nature of the infection during this time despite high organ burdens. Indeed, evidence exists that the immune response to LVS may actually contribute to the pathological changes that develop during infection. Only small numbers of undersized hepatic granulomas form in LVS-infected IFN-γ-deficient mice, and in situ cell death is significantly diminished in these animals compared to infected wild type mice (Bokhari,et al., 2008). However, whereas tissue damage induced by LVS and F. novicida may have an immune component (Chen,et al., 2004, Metzger,et al., 2007), there is little comparable evidence that immune-mediated tissue injury contributes to the pathological changes associated with type A F. tularensis infections.
Fig. 1.
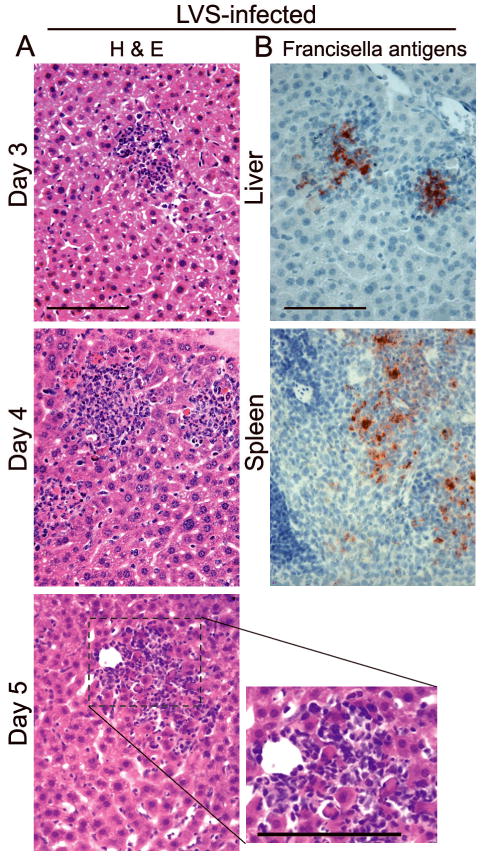
Histopathological changes in mice challenged i.n. with a minimum lethal dose of LVS (3,000 CFU) (Bokhari,et al., 2008). (A) Note that inflammatory infiltrates induced by LVS continue to grow over time with only minimum cell death and little apparent necrosis. (B) Francisella antigens are localized within inflammatory infiltrates in the liver and spleen on day 4 p.i. Bars, 100 μm.
F. novicida, which is essentially nonpathogenic in immunocompetent humans, causes lethal tularemia in mice when administered at low challenge doses via respiratory challenge (Owen,et al., 1964, Chen,et al., 2004, Mares,et al., 2008). Like LVS, F. novicida induces a highly proinflammatory disease. In a recent study by Mares et al (2008), death occurred in mice within 6 days after i.n. challenge with only 200 CFU of F. novicida strain U112. Significant cytokine and chemokine expression as well as inflammasome activation was observed, but not until day 3 p.i. Proinflammatory host constituents containing damage-associated molecular patterns, such as high-mobility group box-1 (HMGB-1) protein, were also expressed at this time. This constellation of responses suggests that F novicida, like LVS, causes tissue injury primarily by recruiting and activating inflammatory cells at sites of infection.
Pathophysiological response of mice to infection with type A and type B F. tularensis
Conlan et al (2003) provided the first detailed description of the histopathological changes occurring in mice infected with natural isolates of type A and type B F. tularensis. In BALB/c and C57BL/6 mice challenged by aerosol inhalation with 10 – 20 CFU of the pathogens, the livers and spleens were often colonized by the second day of infection and showed significant pathological changes by day 4 p.i. Tissue damage closely mimicked the features of tularemia in humans. Although the nature of these pathological responses to the two subspecies were similar, the severity at a given time p.i. was greater for the type A strain, which may have reflected its slightly higher rate of growth in vivo. Gross changes were apparent by day 4 p.i. and included a friable, pallid appearance to the liver. Microscopic hepatic granulomas were present as early as day 2, but by day 4 p.i. were replaced by multiple foci of necrosis, which coalesced as they grew over time. There was little evidence of an in situ inflammatory response to tissue damage, and the parenchyma of the organ outside these necrotic areas retained a fairly normal appearance. The gross pathology of the spleens of infected mice included either splenomegaly or splenic atrophy with hemorrhagic infarction. Early histopathological changes included red pulp congestion and an infiltration of granulocytes, and progressed on day 4 to extensive cellular pyknosis in the red pulp with large areas of necrosis.
We have recently confirmed these findings with a number of type A F. tularensis clinical isolates as well as SCHU S4 (Wickstrum, et al., in press)(Fig. 2). In the liver, multifocal microgranulomas formed by day 3 following respiratory challenge, but nearly all of the cells in these inflammatory infiltrates on the following day became pyknotic and showed double strand DNA breaks (TdT-mediated dUTP-nick end labeling or TUNEL). As foci of necrosis replaced viable granulomas, Francisella antigens, which weree initially associated with the inflammatory infiltrates, became widely distributed throughout the liver parenchyma (Fig. 2). This suggests that the normal barrier function of the granulomas (Bokhari,et al., 2008) was lost with the death of inflammatory cells within these structures.
Fig. 2.
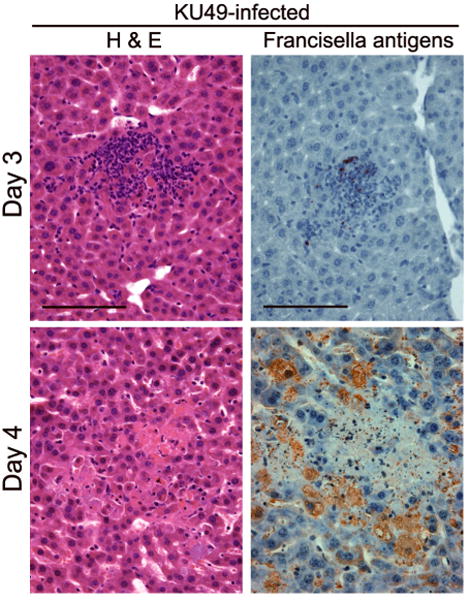
Hepatic microgranulomas form 3 days after i.n. challenge with 25 CFU of the type A F. tularensis KU49 (Wickstrum, et al., in press), and these structures are soon replaced by foci of necrosis with little inflammatory cell infiltration in the tissue parenchyma. Details of this respiratory challenge model have been published previously (Bokhari,et al., 2008). Concomitant with the death of the cells in the granulomas on day 4, bacterial antigens, detected by immunoperoxidase techniques (Bokhari,et al., 2008), become distributed throughout the liver, especially within hepatocytes, which become greatly enlarged. Bars, 100 μm.
Cells infiltrating the splenic red pulp by day 3 p.i. included numerous F4/80+ cells and Ly-6G+ cells (Fig. 3A). The former, which are probably macrophages (Rasmussen,et al., 2006), were abundant throughout the red pulp, while the latter, which are probably myeloid progenitor cells (Rasmussen,et al., 2006), were localized in discrete pyogranulomas. By day 4 p.i., pyknotic TUNEL+ cells were seen in the red pulp and the pyogranulomas, and cell death overtook the white pulp follicles the following day. While cells expressing F4/80 were essentially absent and necrosis replaced much of the red pulp on day 4 (Fig. 3B), pyknotic cells within the pyogranulomas retained the Ly-6G antigen. Immunoperoxidase staining revealed an initial focal accumulation of Francisella antigens, which changed to a more extensive distribution throughout the red pulp by day 4. Bacterial burdens within the organ increased 100-fold between day 3 and day 4. Thus, within 4 days of respiratory challenge with a minimum lethal dose of type A F. tularensis, the spleens of mice showed cell death among both F4/80+ cells and Ly-6G+ cells, extensive red pulp necrosis and microbial dissemination throughout the organ.
Fig. 3.
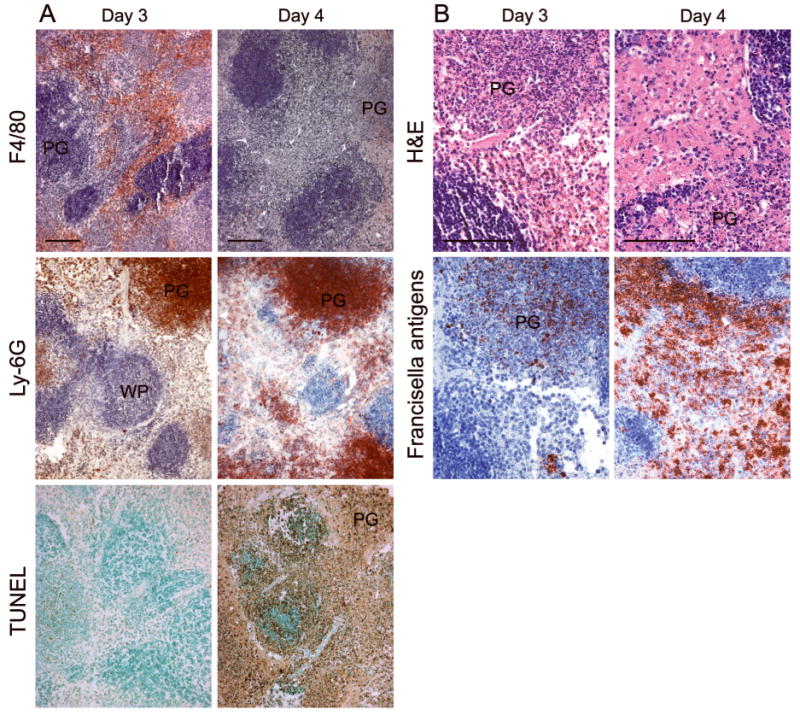
Segregated spatial distribution of cells bearing F4/80 and Ly-6G markers within the spleen of a mouse infected with the type A F. tularensis strain KU49. Infiltrating F4/80+ cells are diffusely distributed throughout the red pulp on day 3, whereas Ly-6G+ cells are predominantly localized within pyogranulomas (PG). By day 4, extensive cell death and necrosis are present throughout the red pulp, rather than the white pulp (WP). Francisella antigens are seen in both the diffuse red pulp areas and the pyogranulomas. Bars, 100 μm.
Much of the lung tissue of mice challenged with type A F. tularensis by aerosol inhalation or i.n. injection did not show extensive pathological changes, suggesting that pneumonia in mice is an unlikely cause of death (Conlan,et al., 2003). However, despite their low frequencies, peribronchial and perivascular pyogranulomas were present and become necrotic by day 4 p.i. (Fig. 4). The principal cells detected in these lesions were Ly-6G-positive, and nearly all of these became TUNEL+ by day 4. Very few F4/80+ cells were detected in these large inflammatory infiltrates of the lungs, although infected F4/80+ cells appear to be present elsewhere in the lungs following challenge with type A strains (Hall,et al., 2008). In contrast with the localized pyogranulomas, the alveolar septae had a fairly normal appearance, and only modest cellular infiltration or debris was seen in the alveolar spaces. Thus, the composition of inflammatory infiltrates differed by organ, with the lung lesions being comprised of large numbers of Ly-6G+ cells. By comparison, the liver and spleen contained mixed infiltrates consisting of both Ly-6G+ and F4/80+ cells, and the two cell types were spatially segregated within the spleen.
Fig. 4.
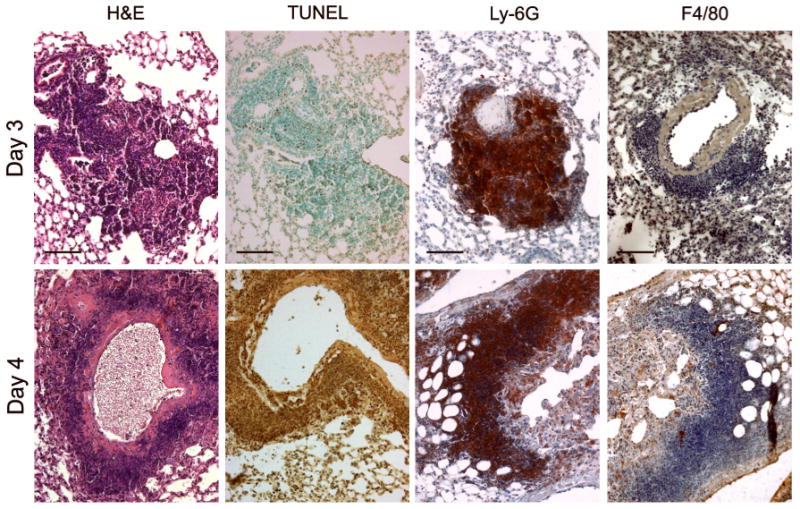
Histopathological changes in the lungs of mice infected by the i.n. route with a minimum lethal dose of type A F. tularensis KU49. Note the perivascular and peribronchial pyogranulomatous infiltrates on day 3 and day 4 and the extensive cell death (TUNEL) seen on day 4 p.i. Most of the cells in areas of cell death and necrosis bear the surface marker Ly-6G. The F4/80 cell surface marker was present on only a few infiltrating cells. Bars, 100 μm.
Considered together, these findings indicate that the mouse is an appropriate model of disseminated tularemia as long as one uses fully virulent type A and type B F. tularensis strains for challenge. The pathological changes induced by these subspecies are quite similar to those seen in humans suffering from naturally-acquired F. tularensis infections or nonhuman primates challenged with pathogenic F. tularensis strains (e.g., SCHU S4).
The mouse thymus may represent an exception to these overall conclusions. Chen et al (Chen,et al., 2005) reported a significant depletion of CD4+CD8+ cortical thymocytes in mice infected by aerosol inhalation with low doses (10 – 20 CFU) of type A, but not type B, F. tularensis. While the basis for these subspecies differences is not clear, adrenalectomized or TNF receptor-deficient mice showed significantly decreased infection-induced thymic atrophy. Neither thymocyte depletion nor peripheral lymphopenia has been consistently reported in F. tularensis infections in humans, and most previous studies have emphasized the similar pathologic features of type A and type B F. tularensis infections in a variety of animal models.
Mechanisms of cell death in tularemia
The morphological finding of necrosis in diseased tissues merely describes the appearance of the tissues after dead cells have reached equilibrium with their surrounding environment (Majno & Joris, 1995). The term necrosis does not imply a single cellular or molecular mechanism for initiating cell death. Pathogen-induced cell death signaling can take many forms, including apoptosis, pyroptosis, pyronecrosis, oncosis and autophagy-associated death (Labbe & Saleh, 2008, Kroemer,et al., 2009). Apoptotic cell death is mediated by a group of cysteine-dependent aspartate-specific proteases (caspases) whose activation can be induced by a variety of signals. For example, TNF receptors recruit and activate procaspase-8, an initiator caspase of the extrinsic pathway of apoptosis. The intrinsic apoptosis pathway is activated when cytochrome c is released from mitochondria, which promotes the activation of procaspase-9. These two pathways can interact with one another through the cleavage of Bid and ultimately activate shared effector or executioner caspases, such as caspase-3, 6, and 7, which cleave multiple cellular protein substrates. The morphological features of apoptosis include cytoplasmic condensation, cell membrane blebbing, and chromatin condensation (pyknosis), fragmentation (karyorrhexis) and extensive degradation (karyolysis). Apoptosis is an inherently non-inflammatory, immunologically silent process characterized by nuclear condensation, chromatin cleavage, and nonlytic outer cell membrane changes.
A caspase-3-independent, caspase-1-dependent pathway of rapid cell death induction, called pyroptosis, is associated with proinflammatory signaling (Fink & Cookson, 2007, Weiss,et al., 2007). Caspase-1 was first described as a protease that processes pro-IL-1β and is also known to cleave pro-IL-18 prior to its release from cells. Procaspase-1 is recruited to multi-protein complexes known as inflammasomes where it is activated by autocatalytic proteolysis. Inflammasomes often contain a nucleotide oligomerization domain (NOD)-like receptor family member, which can serve as a ligand sensor (microbial pattern or stress recognition molecule), and may also contain an adaptor molecule, such as apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). The latter facilitates the binding of procaspase-1 to the complex. Thus, multifunctional caspase-1 coordinates the simultaneous processing and release of proinflammatory cytokines with the induction of cell death. In addition, pyroptosis can be further distinguished from apoptosis by the loss of plasma membrane integrity and the release of highly inflammatory cytoplasmic constituents into the extracellular environment in the former process (Brennan & Cookson, 2000, Weiss,et al., 2007, Labbe & Saleh, 2008).
Both apoptotic and pyroptotic pathways of cell death have been shown to be activated in F. tularensis-infected cells. Lai et al (Lai,et al., 2001, Lai & Sjostedt, 2003) reported that infection of the mouse J774.1 macrophage-like cells with LVS at high multiplicities of infection (MOI) activated intrinsic pathway apoptosis commencing at 24 h p.i. Infected cells showed release of mitochondrial cytochrome c and the activation of procaspase-9 and procaspase-3. Procaspase-1 cleavage was not seen under these conditions, but this may reflect a deficiency in the caspase-1-dependent pathway in this cell line (Henry & Monack, 2007). Subsequent efforts have been made by several laboratories to detect the activation of procaspase-3 in LVS-infected tissues, but the frequencies of such cells have been reported to be quite low (Rasmussen,et al., 2006, Bokhari,et al., 2008).
Mariathasan (2005) reported that caspase-3-independent, caspase-1-dependent cell death occurred in mouse thioglycollate-elicited peritoneal macrophages within a few hours of challenge with F. novicida U112 or LVS at comparatively low MOIs. Both the escape of U112 from the phagosome and type I IFN receptor signaling appeared essential for caspase-1 activation, IL-1β and IL-18 release, and cell death induction (Henry & Monack, 2007). F. novicida-infected caspase-1-deficient macrophages did undergo cell death, but required longer periods of time, suggesting that both pyroptosis and apoptosis can mediate cell death induction by this Francisella species. When challenged subcutaneously with 105 CFU F. novicida U112, caspase-1-deficient or ASC-deficient mice showed greater than 1000-fold increases in bacterial burdens in their lungs, livers and spleens 2 days p.i., suggesting that the pyroptotic pathway of cell death normally contributes to the early elimination of F. novicida-infected cells. More recently, Mares et al (2008) reported that caspase-1 activation in the lungs of mice challenged by the respiratory route with a 100 CFU of F. novicida U112 was delayed for several days. Caspase activation was accompanied by high level expression of a number of proinflammatory chemokines and cytokines and the release of HMGB-1. These findings indicate that the novicida and holarctica subspecies induce an inflammatory form of cell death in the mouse, the timing of which is dependent on the challenge route and dose.
In marked contrast to the caspase-1-associated cell death reported for the novicida and holarctica subspecies, infection of mice with < 25 CFU of type A F. tularensis results in significant levels of expression of cleaved caspase-3 in infected tissues (Fig. 5) (Wickstrum, et al., in press). In the spleen, cells expressing activated caspase-3 were found throughout the red pulp on day 4 p.i. By contrast, cells expressing cleaved caspase-1 were predominantly located within the marginal zone between the red and white pulp, a distribution that mimicked that seen in the LVS-infected spleen. Caspase-1 deficient mice that were challenged with type A F. tularensis showed frequencies of TUNEL+ cells and patterns of tissue necrosis that were indistinguishable from those of wild type infected mice, indicating a limited role for the pyroptosis in cell death induction by this subspecies (Wickstrum, et al., in press).
Fig. 5.
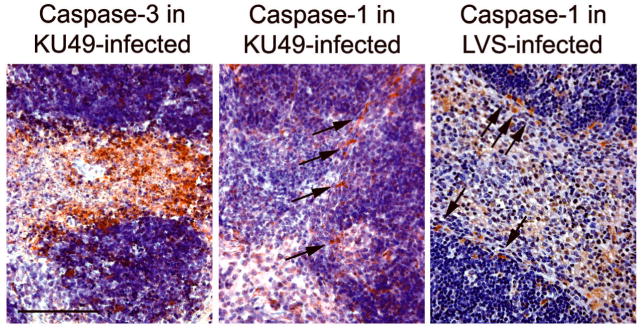
Programmed cell death pathways activated during infection with either type A F. tularensis strain KU49 or LVS. Strain KU49 has been described previously (Wickstrum,et al., 2007). Arrows indicate the expression of cleaved caspase-1, detected by immunoperoxidase techniques (Bokhari,et al., 2008) (antibody from Santa Cruz), in marginal zone macrophages. Bar, 100 μm.
Infected caspase-3-deficient mice differed from infected wild type mice in several important ways. First, they had less apparent necrosis and significantly increased frequencies of F4/80+ cells in their spleens (Fig. 6A), implying that apoptosis normally mediates death induction in macrophages. The death of Ly-6G+ spleen cells was not similarly decreased in caspase-3-deficient mice, suggesting that these cells die by a caspase-3-independent mechanism. Second, the frequency of TNF-α+ and iNOS+ cells was significantly increased in infected caspase-3-deficient mice (Wickstrum, et al., in press), implying that the maintenance of macrophage viability in the caspase-deficient mice preserved functions shown previously to mediate host defenses against other Francisella subspecies (Elkins,et al., 2007, Metzger,et al., 2007). Third, the ability of cells within hepatic granulomas to spatially retain Francisella antigens was lost with the disintegration of these structures (Fig. 2), and this effect was caspase-3-dependent (Fig. 6B). Thus, in the absence of caspase-3, viability of inflammatory cells appeared to be higher, and the antimicrobial function of hepatic granulomas was maintained. This indicates that caspase-3-mediated cell death favors the dissemination of the infection throughout the liver.
Fig. 6.
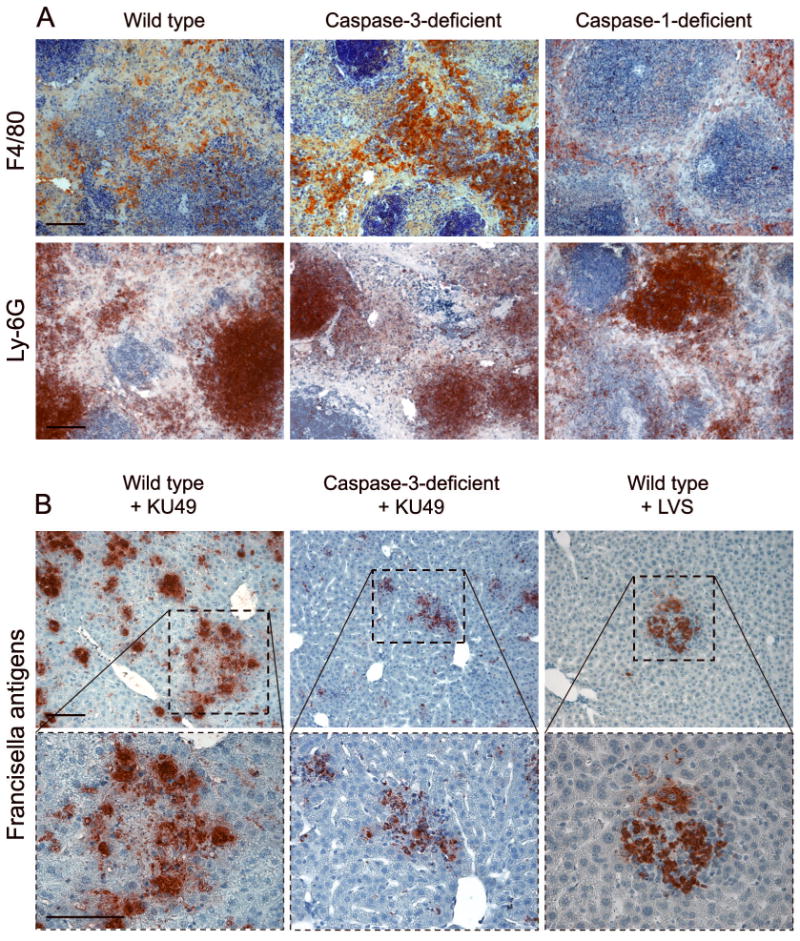
(A) Depletion of F4/80+ cells in the spleens of wild type mice is caspase-3-dependent. Caspase-1-deficient mice do not show a similar effect. Cells expressing Ly-6G become pyknotic in all three mouse strains. (B) The distribution of Francisella antigens in the livers of wild type and caspase-3-deficient mice infected with KU49. Higher power images of the boxed areas are shown below. Note the similar localization of microbial antigens within granulomas in KU49-infected caspase-3-deficient mice and wild type mice challenged with LVS, which does not induce necrosis of the granulomas (Fig. 1A). Bars, 100 μm.
The function of caspase-3 in mediating cell death in tularemia may be restricted to tissues in which macrophages play a central role in pathogen uptake and killing. Bosio et al (2007) reported that the cells infiltrating peribronchial pyogranulomas in the lungs of SCHU S4-infected mice only infrequently expressed cleaved caspase-3. We too have been unable to detect significant numbers of cells expressing activated caspase-3 in lung infiltrates. This may reflect the paucity of macrophages present in these inflammatory foci and the limited role that caspase-3 plays in programmed cell death among Ly-6G+ cells, which constitute the majority of cells in lung pyogranulomas (Fig. 4).
Potential sequelae of programmed cell death in tularemia
Generally, two outcomes have been associated with the death of cells infected with intracellular pathogens. In the first, cell death can provide the host with an early advantage by eliminating infected cells and removing a replication niche for the microbe. This appears to be the case in F. novicida infections in mice, as evidenced by the higher early tissue burdens and earlier deaths seen in caspase-1-deficient mice that are challenged with the pathogen. Alternatively, cell death may serve as an immune evasion mechanism for the microbe by eliminating immune cells, such as macrophages, that have been recruited to sites of infection. Extensive death of macrophages is a hallmark feature of type A F. tularensis infection, and the findings reviewed here suggest that apoptotic death of these cells diminishes potentially important innate immune responses. This hypothesis predicts different roles for caspase-1 and caspase-3 during Francisella infections. The former appears to mediate host resistance to the novicida and holarctica subspecies, while the latter may provide an immune evasion mechanism for type A F. tularensis. Some cells, such as hepatocytes and dendritic cells, may support significant intracellular replication of F. tularensis without undergoing rapid-onset pyroptosis or apoptosis. This finding further emphasizes the ability of F. tularensis to regulate cell death induction to its own advantage, apparently delaying cell demise until exit from its intracellular environment benefits the pathogen. How these findings impact the design of preemptive vaccines or novel post-exposure therapeutics is as yet unknown. However, they do again underscore the importance of testing countermeasures in animal models that reproduce human disease as closely as possible.
Acknowledgments
We thank Rebecca Horvat for her assistance in maintaining and characterizing type A F. tularensis strains. This work was supported by a grant from the National Institutes of Health (R21 AI062939) and a bridging grant from the University of Kansas Medical Center Research Institute.
References
- Anthony LS, Kongshavn PA. Experimental murine tularemia caused by Francisella tularensis, live vaccine strain: a model of acquired cellular resistance. Microb Pathog. 1987;2:3–14. doi: 10.1016/0882-4010(87)90110-0. [DOI] [PubMed] [Google Scholar]
- Bokhari SM, Kim KJ, Pinson DM, Slusser J, Yeh HW, Parmely MJ. NK cells and gamma interferon coordinate the formation and function of hepatic granulomas in mice infected with the Francisella tularensis live vaccine strain. Infect Immun. 2008;76:1379–1389. doi: 10.1128/IAI.00745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio CM, Dow SW. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J Immunol. 2005;175:6792–6801. doi: 10.4049/jimmunol.175.10.6792. [DOI] [PubMed] [Google Scholar]
- Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- Chen W, KuoLee R, Shen H, Conlan JW. Susceptibility of immunodeficient mice to aerosol and systemic infection with virulent strains of Francisella tularensis. Microb Pathog. 2004;36:311–318. doi: 10.1016/j.micpath.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Chen W, Kuolee R, Austin JW, Shen H, Che Y, Conlan JW. Low dose aerosol infection of mice with virulent type A Francisella tularensis induces severe thymus atrophy and CD4+CD8+ thymocyte depletion. Microb Pathog. 2005;39:189–196. doi: 10.1016/j.micpath.2005.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole LE, Elkins KL, Michalek SM, et al. Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J Immunol. 2006;176:6888–6899. doi: 10.4049/jimmunol.176.11.6888. [DOI] [PubMed] [Google Scholar]
- Conlan JW, North RJ. Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect Immun. 1992;60:5164–5171. doi: 10.1128/iai.60.12.5164-5171.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlan JW, KuoLee R, Shen H, Webb A. Different host defences are required to protect mice from primary systemic vs pulmonary infection with the facultative intracellular bacterial pathogen, Francisella tularensis LVS. Microb Pathog. 2002;32:127–134. doi: 10.1006/mpat.2001.0489. [DOI] [PubMed] [Google Scholar]
- Conlan JW, Chen W, Shen H, Webb A, KuoLee R. Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb Pathog. 2003;34:239–248. doi: 10.1016/s0882-4010(03)00046-9. [DOI] [PubMed] [Google Scholar]
- Dennis DT, Inglesby TV, Henderson DA, et al. Tularemia as a biological weapon: medical and public health management. JAMA. 2001;285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- Elkins KL, Cowley SC, Bosio CM. Innate and adaptive immunity to Francisella. Ann N Y Acad Sci. 2007;1105:284–324. doi: 10.1196/annals.1409.014. [DOI] [PubMed] [Google Scholar]
- Farlow J, Wagner DM, Dukerich M, et al. Francisella tularensis in the United States. Emerg Infect Dis. 2005;11:1835–1841. doi: 10.3201/eid1112.050728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman KA, Enscore RE, Lathrop SL, et al. An outbreak of primary pneumonic tularemia on Martha's Vineyard. N Engl J Med. 2001;345:1601–1606. doi: 10.1056/NEJMoa011374. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect Immun. 1991;59:2922–2928. doi: 10.1128/iai.59.9.2922-2928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovliov I, Sandstrom G, Ericsson M, Sjostedt A, Tarnvik A. Cytokine expression in the liver during the early phase of murine tularemia. Infect Immun. 1995;63:534–538. doi: 10.1128/iai.63.2.534-538.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JD, Craven RR, Fuller JR, Pickles RJ, Kawula TH. Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect Immun. 2007;75:1034–1039. doi: 10.1128/IAI.01254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall WC, Kovatch RM, Schricker RL. Tularaemic pneumonia: Pathogenesis of the aerosol-induced disease in monkeys. The Journal of Pathology. 1972;110:193–201. doi: 10.1002/path.1711100302. [DOI] [PubMed] [Google Scholar]
- Hassoun A, Spera R, Dunkel J. Tularemia and once-daily gentamicin. Antimicrob Agents Chemother. 2006;50:824. doi: 10.1128/AAC.50.2.824.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Monack DM. Activation of the inflammasome upon Francisella tularensis infection: interplay of innate immune pathways and virulence factors. Cell Microbiol. 2007;9:2543–2551. doi: 10.1111/j.1462-5822.2007.01022.x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe K, Saleh M. Cell death in the host response to infection. Cell Death Differ. 2008;15:1339–1349. doi: 10.1038/cdd.2008.91. [DOI] [PubMed] [Google Scholar]
- Lai XH, Sjostedt A. Delineation of the molecular mechanisms of Francisella tularensis-induced apoptosis in murine macrophages. Infect Immun. 2003;71:4642–4646. doi: 10.1128/IAI.71.8.4642-4646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai XH, Golovliov I, Sjostedt A. Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect Immun. 2001;69:4691–4694. doi: 10.1128/IAI.69.7.4691-4694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamps LW, Havens JM, Sjostedt A, Page DL, Scott MA. Histologic and molecular diagnosis of tularemia: a potential bioterrorism agent endemic to North America. Mod Pathol. 2004;17:489–495. doi: 10.1038/modpathol.3800087. [DOI] [PubMed] [Google Scholar]
- Lyons RC, Wu TH. Animal models of Francisella tularensis infection. Ann N Y Acad Sci. 2007;1105:238–265. doi: 10.1196/annals.1409.003. [DOI] [PubMed] [Google Scholar]
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Mares CA, Ojeda SS, Morris EG, Li Q, Teale JM. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infect Immun. 2008;76:3001–3010. doi: 10.1128/IAI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyas BT, Nieder HS, Telford SR., 3rd Pneumonic tularemia on Martha's Vineyard: clinical, epidemiologic, and ecological characteristics. Ann N Y Acad Sci. 2007;1105:351–377. doi: 10.1196/annals.1409.013. [DOI] [PubMed] [Google Scholar]
- McCrumb FR. Aerosol Infection of Man with Pasteurella Tularensis. Bacteriol Rev. 1961;25:262–267. doi: 10.1128/br.25.3.262-267.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger DW, Bakshi CS, Kirimanjeswara G. Mucosal immunopathogenesis of Francisella tularensis. Ann N Y Acad Sci. 2007;1105:266–283. doi: 10.1196/annals.1409.007. [DOI] [PubMed] [Google Scholar]
- Owen CR, Buker EO, Jellison WL, Lackman DB, Bell JF. Comparative Studies of Francisella Tularensis and Francisella Novicida. J Bacteriol. 1964;87:676–683. doi: 10.1128/jb.87.3.676-683.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen JW, Cello J, Gil H, Forestal CA, Furie MB, Thanassi DG, Benach JL. Mac-1+ cells are the predominant subset in the early hepatic lesions of mice infected with Francisella tularensis. Infect Immun. 2006;74:6590–6598. doi: 10.1128/IAI.00868-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schricker RL, Eigelsbach HT, Mitten JQ, Hall WC. Pathogenesis of tularemia in monkeys aerogenically exposed to Francisella tularensis 425. Infect Immun. 1972;5:734–744. doi: 10.1128/iai.5.5.734-744.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostedt A, Conlan JW, North RJ. Neutrophils are critical for host defense against primary infection with the facultative intracellular bacterium Francisella tularensis in mice and participate in defense against reinfection. Infect Immun. 1994;62:2779–2783. doi: 10.1128/iai.62.7.2779-2783.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples JE, Kubota KA, Chalcraft LG, Mead PS, Petersen JM. Epidemiologic and molecular analysis of human tularemia, United States, 1964-2004. Emerg Infect Dis. 2006;12:1113–1118. doi: 10.3201/eid1207.051504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnvik A, Priebe HS, Grunow R. Tularaemia in Europe: an epidemiological overview. Scand J Infect Dis. 2004;36:350–355. doi: 10.1080/00365540410020442. [DOI] [PubMed] [Google Scholar]
- Twenhafel N, Alves D, Purcell B. Pathology of Inhalational Francisella tularensis spp tularensis SCHU S4 Infection in African Green Monkeys (Chlorocebus aethiops) Vet Pathol. 2009 doi: 10.1354/vp.08-VP-0302-T-AM. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Henry T, Monack DM. Francisella tularensis: activation of the inflammasome. Ann N Y Acad Sci. 2007;1105:219–237. doi: 10.1196/annals.1409.005. [DOI] [PubMed] [Google Scholar]
- White JD, Rooney JR, Prickett PA, Derrenbacher EB, Beard CW, Griffith WR. Pathogenesis of Experimental Respiratory Tularemia in Monkeys. J Infect Dis. 1964;114:277–283. doi: 10.1093/infdis/114.3.277. [DOI] [PubMed] [Google Scholar]
- Wickstrum JR, Hong KJ, Bokhari S, Reed N, McWilliams N, Horvat RT, Parmely MJ. Coactivating signals for the hepatic lymphocyte gamma interferon response to Francisella tularensis. Infect Immun. 2007;75:1335–1342. doi: 10.1128/IAI.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]