

Summary
In mouse proximal tubular epithelial cells, high glucose (HG) for 24 h increases VEGF expression at the protein and the mRNA level, suggesting a transcriptional mechanism. HG-induced VEGF synthesis is prevented by captopril, an inhibitor of angiotensin-converting enzyme, and, by losartan, a specific antagonist of angiotensin type 1 receptor (AT1), suggesting that VEGF synthesis is mediated by Ang II. Synthesis of angiotensinogen (AGT), a precursor of Ang II, is induced by HG. Although renin and ACE expression is not affected, their activity is increased by HG. Ang I and Ang II synthesis is also increased by HG, and captopril prevents increased Ang II, but not Ang I, synthesis. AT1 activation is increased by with HG, and its activation is prevented by captopril and losartan. The ERK pathway is activated by HG within minutes of stimulation and lasting for up to 24h. The initial phase of ERK activation is due to HG itself and leads to AGT upregulation and the sustained phase is mediated for the most part by Ang II-activated AT1 receptor and leads to increased VEGF synthesis. In conclusion, our study shows that MCTs express an endogenous renin-angiotensin system that is activated by HG to stimulate VEGF synthesis, through activation of the ERK pathway.
Angiotensin II (Ang II) and vascular endothelial growth factor (VEGF) are important mediators of kidney injury in diabetes. VEGF expression is increased in proximal tubules of mice with type 1 diabetes. In mouse proximal tubular epithelial cells (MCT) cultured with 30 mM glucose (HG) for 24 h, VEGF expression is increased at the protein and the mRNA level, suggesting a transcriptional mechanism. HG stimulation of VEGF synthesis is prevented by captopril, an inhibitor of angiotensin-converting enzyme, and, by losartan, a specific antagonist of angiotensin type 1 receptor (AT1), suggesting that VEGF synthesis is mediated by Ang II. Synthesis of angiotensinogen (AGT), a precursor of angiotensin II, is increased in MCTs cultured in HG. Although synthesis of renin and ACE is not affected by HG, their activity is increased in the conditioned medium. Concentrations of Ang I and Ang II are also increased in conditioned medium from HG-treated MCTs and captopril prevents increased Ang II, but not Ang I, synthesis. Finally, AT1 is activated in MCTs treated with HG, and its activation is prevented by captopril and losartan. The ERK pathway is activated by HG within minutes of stimulation and lasting for up to 24h. The initial phase of ERK activation is due to HG itself and leads to AGT upregulation and the sustained phase is mediated for the most part by Ang II-activated AT1 receptor and leads to increased VEGF synthesis.
These data show that: 1) HG increases AGT synthesis and activation of renin and ACE by MCTs, leading to local production of Ang I and Ang II. 2) Ang II activates endogenous AT1 and stimulates synthesis of VEGF. 3) HG activation of ERK starts within minutes and lasts for up to 24h. Early ERK activation is involved in AGT upregulation and sustained ERK activation, mediated via AT1, is responsible for VEGF synthesis.
In conclusion, our study shows that MCTs express an endogenous renin-angiotensin system that is activated by high glucose to stimulate the synthesis of VEGF, through activation of the ERK pathway.
Keywords: Renin-angiotensin system, VEGF, glucose, epithelial cells, kidney
Introduction
The renin-angiotensin-aldosterone system (RAS) is a major humoral system involved in the control of blood pressure and volume (Campbell, 1987). This system produces angiotensin II (Ang II) from a precursor, angiotensinogen (AGT), produced by the liver (Freeman and Rostorfer, 1972), through sequential activation of two proteases, renin and angiotensin-converting enzyme (ACE). Renin, produced in the kidney by the afferent arteriolar myoepithelial cells and released into the circulation, cleaves circulating AGT to yield a decapeptide, angiotensin I (Ang I). Ang I is cleaved in the lungs and the vascular endothelium by ACE to produce the octapeptide angiotensin II (Ang II) (Campbell, 1987). Ang II acts on target tissues by binding to two major receptors, AT1 and AT2 (Burns, 2000).
Renin is synthesized as an inactive proenzyme which is activatated by cleavage by trypsin to produce the active enzyme (Persson, 2003) or without proteolysis by binding to the (pro)renin receptor (Nguyen, 2006). Unlike other proteases from its class, renin has a strict specificity for its substrate AGT (Persson, 2003). ACE, a transmembrane glycoprotein, is a nonspecific carboxypeptidase that cleaves dipeptides from a variety of substrates (Lee et al., 1971, Soffer et al., 1974). The enzyme has a greater affinity for bradykinin than for Ang I, and in addition to generating Ang II, ACE has been shown to inactivate bradykinin (Scherf et al., 1986, Kramer et al., 1990).
Since systemic Ang II levels are decreased in diabetes, local RAS has been implicated in diabetic nephropathy (Kennefick and Anderson, 1997, Lai et al., 1998, Carey and Siragy, 2003). All the components of the RAS are present within the kidney (Burns et al., 1993, Navar et al., 1996). AGT mRNA and protein are localized in proximal tubular cells, suggesting that tubular AGT provides the substrate for tubular Ang I and Ang II formation (Navar et al., 1996).
Ang II receptors belong to the G-protein-coupled receptor family and have a similar affinity for Ang II (Berry et al., 2001). AT1 is widely distributed throughout the kidney, and in vascular smooth muscle cells throughout the renal vasculature, including the afferent and efferent arterioles (Miyata et al., 1999). Angiotensin receptors are also found on proximal tubular and thick ascending limb epithelia, distal tubules, cortical collecting ducts, glomerular podocytes and macula densa cells (Miyata et al., 1999). AT2 expression is high in fetal kidney, but decreases markedly during the neonatal period (Ozono et al., 1997). In the adult kidney, it is expressed in the afferent arteriole, glomerular endothelial and mesangial cells, proximal tubular epithelial cells and interstitial cells (Miyata et al., 1999, Ozono et al., 1997).
Locally-produced Ang II contributes to hemodynamic and cellular processes involved in renal matrix expansion, proteinuria and kidney failure in type 1 diabetes (Kagami et al., 1994, Leehey et al., 2000, Remuzzi and Bertani, 1998, Wolf et al., 1993, Wolf and Neilson, 1990). Reduction of renal hypertrophy and matrix expansion by ACE inhibitors and Ang II receptor blockers attest to the critical role of Ang II in kidney disease (Kohzuki et al., 1995, Lewis et al., 1993). Ang II effects are partly mediated by TGFβ (Wolf et al., 1993, Wolf and Neilson, 1990) and other growth factors (Kagami et al., 1994). Hyperglycemia and Ang II increase VEGF synthesis in renal cells (Feliers et al., 2005, Cha et al., 2000, Pupilli et al., 1999, Kim et al., 2005), and neutralization of VEGF ameliorates proteinuria in diabetic rats (De Vriese et al., 2001, Flyvbjerg et al., 2002). Although hyperglycemia is known to activate intra-renal RAS in rats (Lansang et al., 2002), it is not known whether high glucose can activate it in the proximal tubules; whether hyperglycemia recruits RAS in proximal tubular epithelial cells to promote VEGF expression is also not known.
In view of the importance of Ang II as a mediator of hyperglycemia-associated renal injury in diabetes, we examined the role of proximal tubular epithelial cell renin-angiotensin system in the regulation of VEGF expression by high glucose.
Materials & Methods
Materials
Antibodies directed against VEGF and AGT were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against renin and ACE were from Abcam (Cambridge, MA). Conformation-specific antibodies against activated AT1 and AT2 were from Assay Designs (Ann Arbor, MI), and antibodies against AT1 and AT2 were from Alomone Labs (Jerusalem, Israel).
Cell Culture
SV40-immortalized murine proximal tubular epithelial cells (MCT) were provided by Dr. Eric Neilson (Vanderbilt University, Nashville, TN). MCTs in culture express in vivo characteristics of proximal tubular epithelial cells (Haverty et al., 1988). Cells were grown in Dulbecco’s minimal essential medium (DMEM) containing 5 mM glucose and 10% FBS (Feliers et al., 2007, Sataranatarajan et al., 2008). Monolayers of MCTs were grown to 60 to 70 % confluence and serum-deprived overnight before treatment, and were barely confluent at the time of treatment.
Immunoblotting experiments were performed as previously described (Feliers et al., 2007, Sataranatarajan et al., 2008). MCTs were homogenized in lysis buffer (50 mM Tris.HCl, pH 7.4, 150 mM KCl, 1 mM EDTA, 50 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM EGTA, 0.5% Nonidet P40, and protease inhibitor mix [Sigma, St Louis, MO]). Protein concentration was measured and 10–20 μg of whole-cell lysates were separated on SDS-PAGE, transferred to nitrocellulose membranes and probed with various primary antibodies, and IRDye800- or IRDye700-coupled secondary antibodies were used for detection using Odyssey Infrared Imaging System (LiCor Biosciences, Lincoln, NE). For detection of secreted VEGF, 1 ml of conditioned medium (CM) was collected and subjected to TCA precipitation of proteins (15% TCA for 2 h at 4°C). Precipitated proteins were pelleted by centrifugation (10 min at 14,000 rpm at 4°C), and remaining TCA was extracted with ether. Pellets were suspended in 20 μl of Laemmli sample buffer and subjected to immunoblot as described above.
Concentration of Ang I and II was measured in 100 μl of unconcentrated CM using immunofluorescent kits from Bachem (San Carlos, CA), according to the manufacturer’s instructions.
Renin and ACE activity. Renin activity was measured in 100 μl of unconcentrated CM using the fluorometric Sensolyte Renin assay kit (Anaspec, San Jose, CA), according to the manufacturer’s instructions. ACE activity was measured in the same CM using the ACE colorimetric enzymatic assay (Alpco Diagnostics, Salem, NH).
Quantitative RT-PCR was performed as follows. RNA was extracted from treated cells using Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Quantitative RT-PCR amplification of VEGF or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript was performed using the Superscript One-Step RT-PCR kit from Invitrogen and the following primers:
VEGF sense (5′-ACATCTTCAAGCCGTCCTGTGTGC-3′),
VEGF antisense (5′-AAATGGCGAATCCAGTCCCACGAG-3′),
GAPDH sense (5′-CGATGCTGGCGCTGAGTAC-3′),
GAPDH antisense (5′-CGTTCAGCTCAGGGATGACC-3′) (Feliers et al., 2005).
SYBR Green PCR master (Applied Biosystems, Foster City, CA) mix was added and PCR amplification was performed using a RealPlex4 Mastercycler (Eppendorf, Westbury, NY). Dissociation curve analysis was performed after PCR amplification to confirm the specificity of the primers. Relative mRNA expression was calculated using the ΔΔCt method as reported by us previously (Sataranatarajan et al., 2007).
Statistics
Data from a minimum of 3 experiments were expressed as mean±SEM and analyzed by ANOVA for comparison among multiple groups using Newman-Keuls post-test analysis (GraphPad Prizm®); p<0.05 was considered significant.
Results
High glucose increases VEGF synthesis
VEGF expression was measured in quiescent MCTs cultured in the presence of 5 mM (normal glucose, NG) or 30 mM glucose (high glucose, HG) for 24 h. HG significantly increased VEGF protein expression (267±35% of control, p<0.01) in MCT lysates (Fig. 1A). Equimolar mannitol had no effect on VEGF expression. In the same cells, VEGF mRNA was also significantly increased (433±18% of control, p<0.001 by ANOVA), which largely accounted for the increment in VEGF protein (Fig. 1B). These data suggest that HG stimulates VEGF synthesis through a transcriptional mechanism in MCTs and that it is not due to an osmotic effect. VEGF secretion was assessed by measuring VEGF in the CM by immunoblot. Figure 1C shows that HG (152.1±11.6% of control, p<0.01), but not equimolar mannitol (100.2±2.3% of control, NS), increased VEGF secretion by MCTs.
Figure 1. HG augments VEGF protein and mRNA expression.
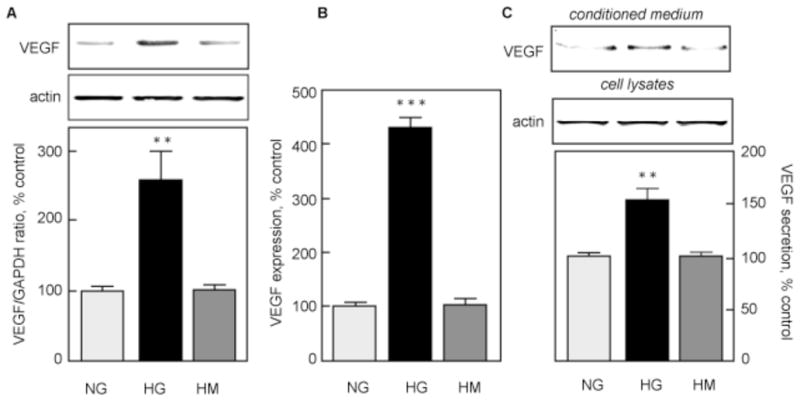
MCTs were incubated in normal glucose (5 mM, NG), high glucose (30 mM, HG) or high mannitol (5 mM glucose + 25 mM mannitol, HM) for 24 h. A. VEGF expression was measured by immunoblot, and actin was used as a loading control. The lower panel shows combined data from 4 independent experiments. **p<0.01 by ANOVA. B. VEGF mRNA expression was measured by qRT-PCR as described in the Methods section. Actin was used as a loading control. The lower panel shows data from 3 independent experiments. ***p<0.001 by ANOVA. C. VEGF secretion was measured by immunoblot as described in the Methods section. Cellular actin was used as a loading control. The lower panel shows combined data from 4 independent experiments. **p<0.01 by ANOVA.
Involvement of the renin-angiotensin system
To test the possibility that activation of an endogenous RAS mediates hyperglycemia-induced VEGF synthesis, MCTs were pretreated with captopril, a specific ACE inhibitor, or losartan, a specific AT1 antagonist, for 1 h before incubation with HG for 24 h. HG stimulation of VEGF synthesis was prevented by captopril (110±12% of control, p<0.01 vs HG, NS vs NG; Fig. 2A) and by losartan (90±16% of control, p<0.001 vs HG, NS vs NG; Fig. 2B). These data show that ACE and AT1 mediate the effect of HG on VEGF synthesis, suggesting involvement of RAS in MCTs.
Figure 2. HG stimulation of VEGF synthesis is due to RAS activation.
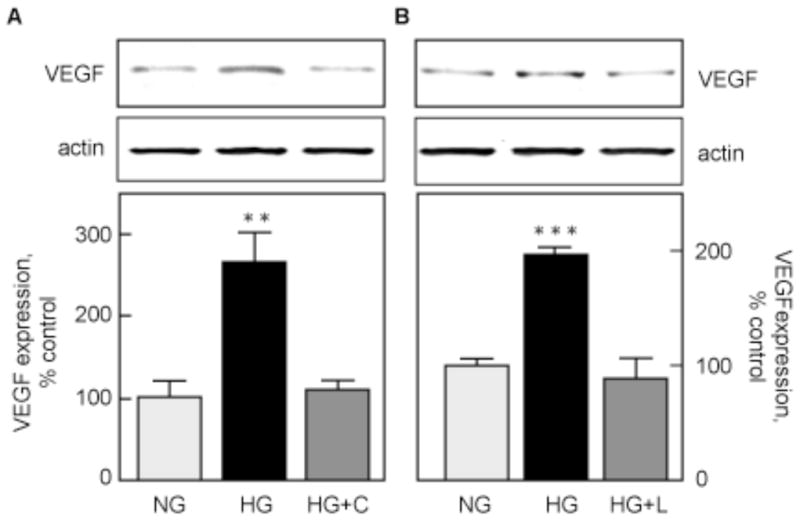
A. VEGF protein expression was measured by immunoblotting in MCTs incubated with NG or HG for 24 h, with or without a 1 h pre-incubation with 100 μM captopril (HG+C). B. The same experiment was repeated with 100 μM losartan (HG+L). The lower panels show data from 3 independent experiments. **p<0.01,***p<0.001 by ANOVA.
To assess RAS recruitment by HG, we first measured AGT synthesis. Figure 3 shows that HG significantly upregulated AGT protein at 24h (160.7±4.0 % of control, p<0.05 vs NG) and that this upregulation was not affected by inhibition of ACE (152.8±7.6 % of control, p<0.05 vs NG, NS vs HG by ANOVA).
Figure 3. AGT synthesis.
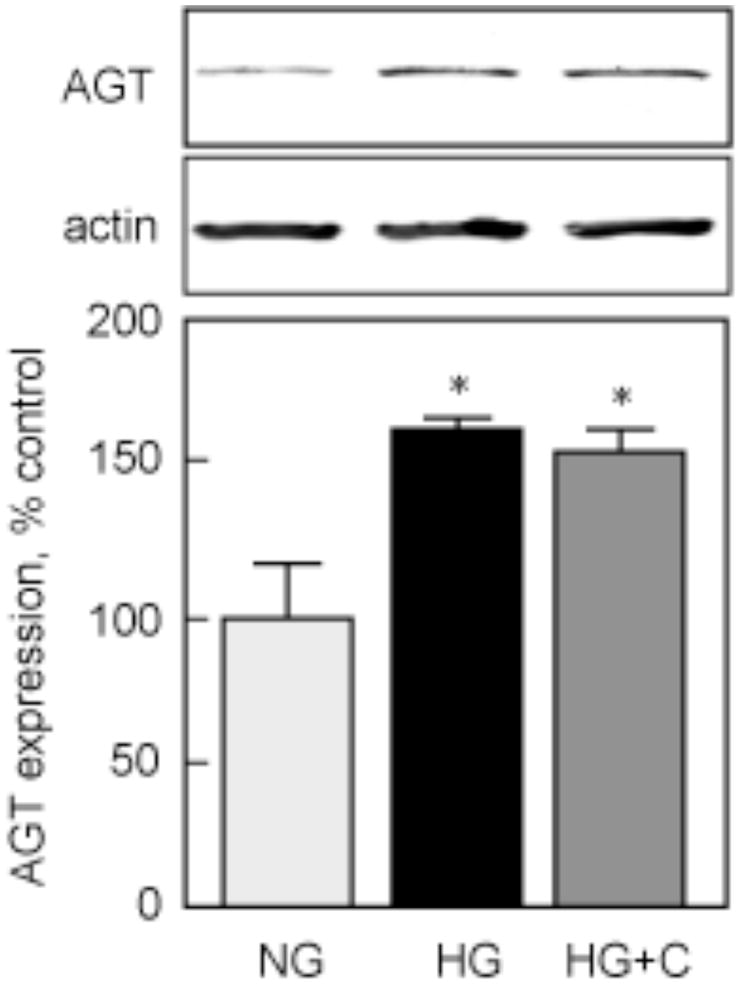
MCTs were incubated with NG or HG for 24 h, with or without 1 h pre-incubation with 100 μM captopril (HG+C). AGT protein expression was measured by immunoblot, and GAPDH expression was used as a loading control. The lower panels show data from 3 independent experiments. *p<0.05 by ANOVA.
Renin activity was measured in CM from MCTs incubated with HG for 24 h. Basal renin activity (31.1±0.7 mU/mg protein) was significantly increased by HG (36.4±0.4 mU/mg protein, p<0.05 vs NG; Fig. 4A). ACE inhibition by captopril did not inhibit HG stimulation of renin activity (36.6±0.7 mU/mg protein, NS vs HG, p<0.05 vs NG; Fig. 4A). Renin activity was not affected by incubation of MCTs with equimolar mannitol (32.7±0.3 mU/mg protein, p<0.05 vs HG, NS vs NG; Fig. 4A). Renin protein expression was measured in the corresponding cell lysates by immunoblot (Fig. 4B). The antibody recognized a ~45 kDa band that could be pro-renin, as renin has an estimated molecular weight of 37 kDa (Nguyen, 2006). HG did not significantly modify renin expression; however, in the presence of captopril, renin expression was significantly increased by HG (214±30 % of control, p<0.01 vs NG; Fig. 4B).
Figure 4. HG activates renin.

MCTs were incubated in normal glucose (NG), high glucose (HG), high glucose + captopril (HG+C) or high mannitol (HM) for 24 h. Renin activity (A) was measured as described in the Methods section in 100 μl of non-concentrated CM from cells incubated as indicated. Results were derived from 4 independent experiments with duplicates for each condition. *p<0.05, NS = not significant by ANOVA. Renin expression (B) was measured by immunoblot on cell lysates corresponding to the CM used in A. GAPDH expression was used as a loading control. The lower panels show data from 4 independent experiments. *p<0.05, **p<0.01 by ANOVA.
We next measured ACE activity in the same CM. Figure 5A shows that basal ACE activity (77.2±6.9 mU/mg protein) was significantly increased by HG (131.0±12.8 mU/mg protein, p<0.01 vs NG). Captopril prevented HG-induced stimulation of ACE activity (63.2±8.8 mU/mg protein, NS vs NG, p<0.01 vs HG). Similar to renin, equimolar mannitol did not stimulate ACE activity (77.3±12.5 mU/mg protein, NS vs NG, p<0.01 vs HG). ACE protein expression, measured in the corresponding cell lysates by immunoblot, showed that incubation of MCT with HG, with or without captopril, did not significantly modify ACE expression (Fig. 5B).
Figure 5. HG activates ACE.
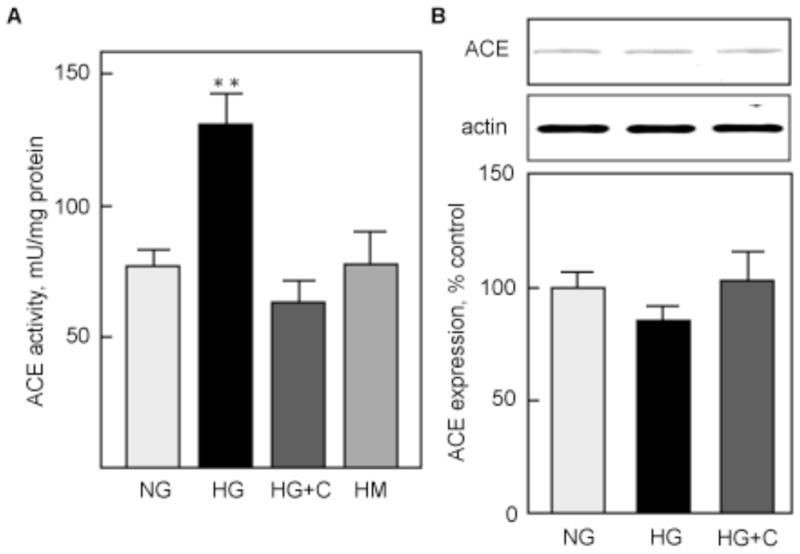
ACE activity (A) was measured under the same conditions as renin activity (Fig. 4), and results are expressed identically. *p<0.05, NS = not significant by ANOVA. ACE protein expression (D) was measured by immunoblot on cell lysates corresponding to the CM used in A. GAPDH expression was used as a loading control. The lower panels show data from 4 independent experiments. *p<0.05, **p<0.01 by ANOVA.
Concentration of Ang I and Ang II was measured in the same CM. Figure 6A shows that basal Ang I concentration (2.6 pg/mg protein) was significantly increased by HG (16.1±1.4 pg/mg protein, p<0.01 vs NG). Captopril did not affect HG stimulation of Ang I concentration (16.9±1.5 pg/mg protein, p<0.01 vs NG, NS vs HG). Equimolar mannitol did not increase Ang I concentration (2.0±0.4 pg/mg protein, NS vs NG, p<0.01 vs HG). Figure 6B shows that basal Ang II concentration (2.5 pg/mg protein) was significantly increased by HG (15.9±1.2 pg/mg protein, p<0.01 vs NG, NS vs HG), but not by equimolar mannitol (1.9±0.5 pg/mg protein, NS vs NG, p<0.01 vs HG). Captopril prevented HG-induced increased in Ang II concentration (16.9±1.5 pg/mg protein, NS vs NG, p<0.01 vs HG; Fig. 6B).
Figure 6. HG increases concentration of Ang I and Ang II in CM.
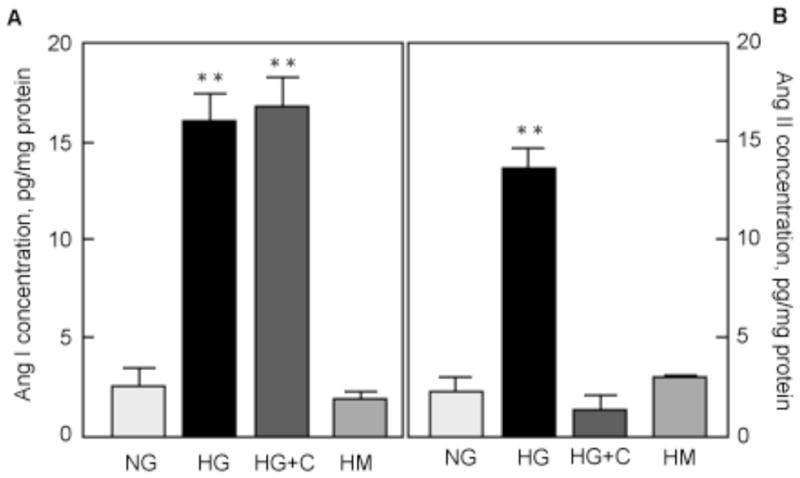
MCTs were incubated in normal glucose (NG), high glucose (HG), high glucose + captopril (HG+C) or high mannitol (HM) for 24 h. Ang I (A) and Ang II (B) concentrations were measured as described in the Methods section in 100 μl of non-concentrated CM. Results were derived from 4 independent experiments with duplicates for each condition. *p<0.05, NS = not significant by ANOVA.
AT1 receptor activation
Ang II acts on cells by binding to receptors on the cell surface, AT1 and AT2. As MCTs express only the AT1, but not the AT2 receptor (data not shown), we assessed AT1 activation by immunoblot using a conformation-specific antibody that reacts only with the ligand-bound receptor (Gupta et al., 2007). Figure 7A shows that AT1 is activated by HG in MCTs in a time-dependent manner, starting at 1 h and reaching a maximum at 2–4 h (~200 % of control, p<0.01 vs NG). Figure 7B shows that at 24 h, AT1 remained significantly activated by HG (174.1±15.3 % of control, p<0.05 vs NG). Captopril prevented AT1 activation at 24 h (89.8±4.7 % of control, NS vs NG, p<0.05 vs HG), indicating that AT1 activation at 24 h requires ACE activity and endogenous production of Ang II. Losartan, the specific AT1 antagonist, inhibited activation of AT1 by HG (88.1±25.3 % of control, NS vs NG, p<0.05 vs HG; Fig. 7C).
Figure 7. HG induces AT1 activity.
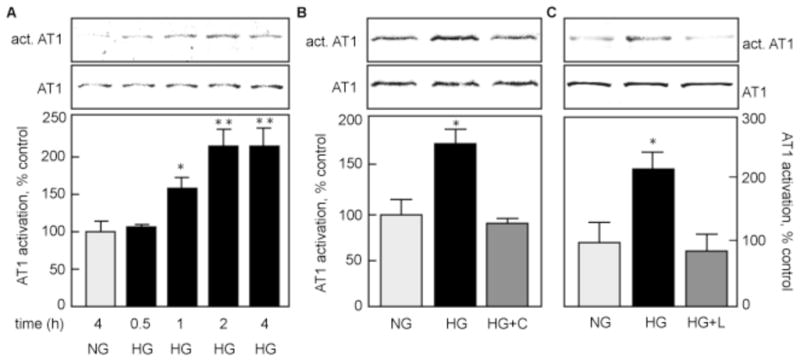
AT1 activation was assessed by immunoblot with conformation-specific AT1 antibody (act. AT1), AT1 expression was measured with an antibody against total AT1. A. MCTs were incubated with HG for up to 4 h, and compared to cells incubated with NG for 4 h. B and C. Cells were incubated with HG with or without pre-incubation with 100 μM captopril (HG+C; B) or 100 μM losartan (HG+L; B). The lower panel shows data from 3 independent experiments. *p<0.05, **p<0.01 by ANOVA.
Role of extracellular-activated kinase (ERK)
We then sought to study signaling events leading to VEGF synthesis. We focused on the extracellular-regulated kinase (ERK) pathway because it is activated by HG in MCT cells (Mariappan et al., 2007), and because it is involved in stimulation of VEGF synthesis (Fukuda et al., 2002, Salem et al., 2005). Figure 8A shows that ERK is activated by HG in a time-dependent manner, starting at 5 min and reaching a maximum at 15–60 min. To examine the role of the ERK pathway in VEGF synthesis, we pre-incubated MCT cells with 10 μM of U0126, an inhibitor of MEK1 - the kinase that activates ERK (Mariappan et al., 2007) - for 30 min prior to stimulation with HG for 24 h. Figure 8B shows that HG-induced VEGF synthesis (180.3±4.1 % of control, p<0.01 vs NG) was prevented by inhibition of the ERK pathway (109.3±7.4 % of control, NS vs NG, p<0.01 vs HG). Our data showed that AGT is upregulated by HG (Fig. 3). Inhibition of the ERK pathway by U0126 prevented upregulation of AGT by HG at 4h (135.1±36.4 vs 291.0±25.3 % of control, p<0.01 by ANOVA). Because Ang II can also activate the ERK pathway (Duff et al., 1995) through the AT1 receptor (Ahn et al., 2004) and HG activates AT1 through generation of Ang II (Figs 6 & 7), we examined whether HG-induced ERK activation was mediated by AT1. Figure 8D shows that HG strongly activated ERK at 24 h (411.0±83.1 % of control, p<0.01 vs NG) and that this activation was significantly, albeit incompletely, inhibited by the AT1 antagonist (172.7±19.3 % of control, p<0.05 vs NG, p<0.01 vs HG). Together, these data show that HG activates ERK early (minutes) and late (24h). The early activation of ERK is responsible for upregulation of AGT and the late activation is mediated for the most part via Ang II-activated AT1 receptor.
Figure 8. HG activation of ERK is required for VEGF synthesis.
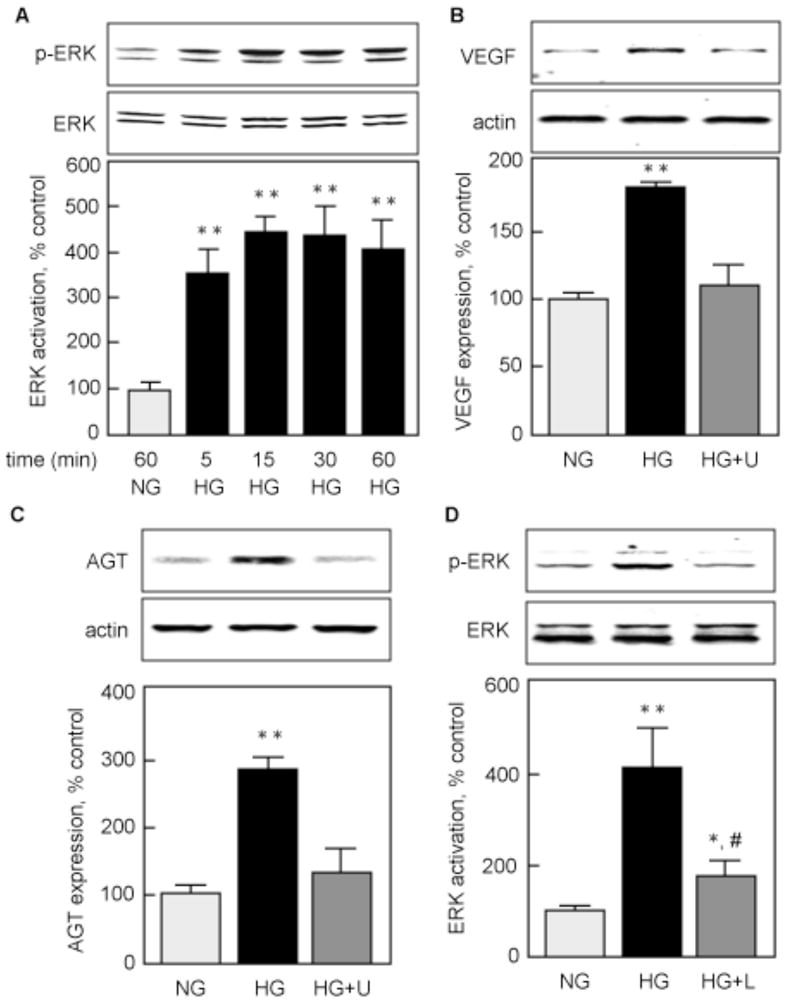
A) MCTs were incubated with high glucose (HG) for 5 to 60 min, and ERK activation was assessed by measuring phosphorylation on Thr202/Tyr204 with a specific antibody. MCTs incubated in normal glucose (NG) for 60 min were used as control. B) VEGF protein expression was measured by immunoblotting in MCTs incubated with NG or HG for 24 h, with or without a 1 h pre-incubation with 10 μM U0126 (HG+U). C) AGT protein expression was measured by immunoblotting in cells incubated with NG or HG for 4 h, with or without a 1 h pre-incubation with 10 μM U0126 (HG+U). D) ERK activation was measured in MCTs incubated with NG or HG for 24 h, with or without a 1 h pre-incubation with 100 losartan (HG+L).
Discussion
Our study shows that incubation of MCTs with HG increases synthesis of AGT through activation of the ERK pathway, and induces activation of renin and ACE. Renin cleaves AGT to produce Ang I, which is then processed to Ang II by ACE. Ang II then binds to and activates AT1 to increase transcription of the vegf gene, leading to increased VEGF synthesis through activation of the ERK pathway (Fig. 9).
Figure 9. HG activates an endogenous RAS in MCTs to induced VEGF synthesis.
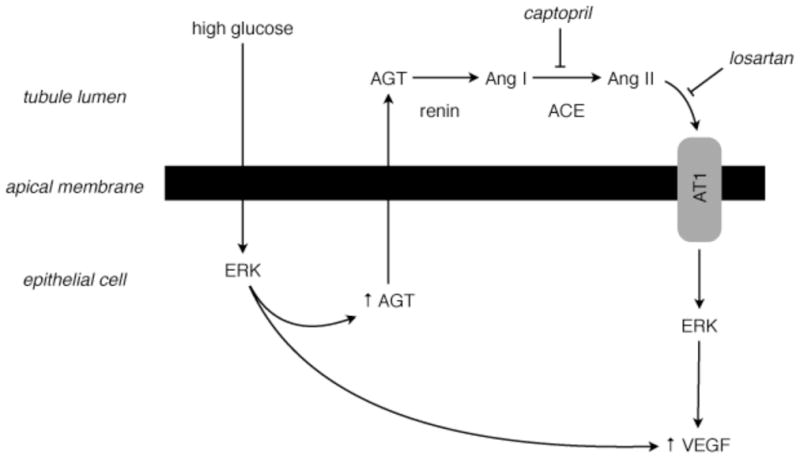
We propose that glucose present in the tubular lumen is taken up by proximal tubular epithelial cells and stimulates synthesis and secretion of angiotensinogen (AGT), as well as secretion and activation of renin and angiotensin-converting enzyme (ACE). In the tubular space, AGT is converted to Ang I by renin, and Ang I to Ang II by ACE, a step inhibited by captopril. Locally formed Ang II then binds to and activates AT1 (a step blocked by losartan), which leads to stimulation of VEGF synthesis.
It is known that the kidney expresses a self-contained RAS that acts in a paracrine fashion. AGT is expressed predominantly in the proximal renal tubule (Ingelfinger et al., 1990), renin and ACE in the juxtaglomerular apparatus, and in tubular epithelial cells (Lai et al., 1998). In diabetic nephropathy, intra-renal RAS is activated and Ang II is increased (Mezzano et al., 2003) in spite of suppression of the systemic RAS (Carey and Siragy, 2003). Locally produced Ang II contributes to hemodynamic and cellular processes involved in matrix expansion, proteinuria and kidney failure in type 1 diabetes (Leehey et al., 2000). The non-hemodynamic actions of Ang II include augmented release of many growth factors, such as TGFβ and Connective Tissue Growth Factor (Wolf and Ziyadeh, 2007, Ziyadeh and Wolf, 2008). Ang II can also stimulate the synthesis and release of VEGF from many tissues, including renal cells (Feliers et al., 2005, Williams, 1998, Rizkalla et al., 2003). In the kidneys, the major site of VEGF synthesis was long thought to be the podocytes (Simon et al., 1995). However, recent studies have shown that VEGF is also strongly expressed in the proximal tubular compartment in both human (Lindenmeyer et al., 2007) and mouse kidney (Feliers et al., 2005).
The existence of an endogenous RAS that is activated by HG has also been recently uncovered in the podocyte (Durvasula and Shankland, 2008), but this study did not address whether activation of local RAS was involved in VEGF synthesis. It is possible that podocyte-derived Ang II is involved in VEGF synthesis in an autocrine manner, since an AT1 antagonist prevented VEGF synthesis in podocytes in rats with type 1 diabetes (Lee et al., 2004).
Our data showed that ACE inhibition by captopril had no effect on HG-induced renin activity; however, HG augmented renin protein expression only in the presence of captopril. In humans, administration of captopril for 4 weeks resulted in increased pro-renin expression and renin activity (Sealey et al., 1981). As captopril inhibits Ang II synthesis, this upregulation of pro-renin is likely due to disruption of the negative feedback exerted by Ang II on its synthesis (Campbell, 1987). The fact that HG increases renin activity without changing pro-renin expression suggests HG acts at the level of activation. However, addition of captopril did not increase renin activity beyond that induced by HG, despite upregulation of pro-renin. This suggests that captopril fails to stimulate activation of pro-renin in MCTs. In vitro, pro-renin can be activated by various proteases, including plasmin, cathepsin D and kallikrein (Hsueh, 1984). Kallikrein was thought to be the physiological activator of pro-renin because they colocalize in the kidney (Rohrwasser et al., 2003). Captopril could be expected to augment renin activity via kallikrein as it is also an inhibitor of kininase that degrades kallikrein. However, a role for kallikrein in renin activation was disproved by use of specific inhbitors (De Vito et al., 1996) and of knockout mice (Rohrwasser et al., 2003). Instead, it seems that in the kidney, pro-renin is activated by binding to a specific receptor (Nguyen et al., 2002), which induces a conformational change of pro-renin that allows activation without proteolytic cleavage (Nguyen, 2006). It is possible that Ang II exerts a negative feedback on pro-renin synthesis (this may not be happening as renin protein is unchanged from control in HG-treated cells) but not on the (pro)renin receptor, so that inhibition of Ang II synthesis by captopril results in upregulation of pro-renin which cannot be activated.
Our data show activation of an endogenous RAS in MCTs leads to increased extracellular synthesis of Ang II followed by autocrine action of Ang II to generate VEGF. However, our data do not rule out intracellular synthesis of Ang II, which has been demonstrated in other cell types, such as cardiomyocytes (Singh et al., 2007), mesangial cells (Vidotti et al., 2004), podocytes (Yoo et al., 2007) and vascular smooth muscle cells (Freeman and Rostorfer, 1972). The intracellular RAS (iRAS) differs from the extracellular RAS in that the conversion of Ang I to Ang II is mediated by chymase instead of ACE (Lavrentyev et al., 2007, Singh and Leehey, 2007). Intracellular Ang II translocates to the nucleus (Robertson and Khairallah, 1971), binds to chromatin-associated AT1 receptors (Booz et al., 1992, Re et al., 1984) and regulates the transcription of several genes, including those of the RAS, such as AGT and renin (Kumar et al., 2008). An important role for iRAS in hypertension-induced cardiac fibrosis (Ichihara et al., 2006) and hypertrophy (Baker et al., 2004, Mazzolai et al., 1998) has been demonstrated. Although presence of intracellular Ang II and nuclear AT1 has been demonstrated in the cortex from rat kidneys (Pendergrass et al., 2006), the role of iRAS in renal tubular injury is yet to be established.
Acknowledgments
This work was supported by grants from the American Heart Association (DF), RO1 DK077295 (BSK), and VA Research Service Merit Review Grant (BSK). We thank Dr E. Neilson for mouse proximal tubular epithelial cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AHN S, WEI H, GARRISON TR, LEFKOWITZ RJ. Reciprocal Regulation of Angiotensin Receptor-activated Extracellular Signal-regulated Kinases by -Arrestins 1 and 2. J Biol Chem. 2004;279:7807–7811. doi: 10.1074/jbc.C300443200. [DOI] [PubMed] [Google Scholar]
- BAKER KM, CHERNIN MI, SCHREIBER T, SANGHI S, HAIDERZAIDI S, BOOZ GW, DOSTAL DE, KUMAR R. Evidence of a novel intracrine mechanism in angiotensin II-induced cardiac hypertrophy. Regul Pept. 2004;120:5–13. doi: 10.1016/j.regpep.2004.04.004. [DOI] [PubMed] [Google Scholar]
- BERRY C, TOUYZ R, DOMINICZAK AF, WEBB RC, JOHNS DG. Angiotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide. Am J Physiol Heart Circ Physiol. 2001;281:H2337–65. doi: 10.1152/ajpheart.2001.281.6.H2337. [DOI] [PubMed] [Google Scholar]
- BOOZ GW, CONRAD KM, HESS AL, SINGER HA, BAKER KM. Angiotensin-II-binding sites on hepatocyte nuclei. Endocrinology. 1992;130:3641–9. doi: 10.1210/endo.130.6.1597161. [DOI] [PubMed] [Google Scholar]
- BURNS KD. Angiotensin II and its receptors in the diabetic kidney. Am J Kidney Dis. 2000;36:449–67. doi: 10.1053/ajkd.2000.16192. [DOI] [PubMed] [Google Scholar]
- BURNS KD, HOMMA T, HARRIS RC. The intrarenal renin-angiotensin system. Semin Nephrol. 1993;13:13–30. [PubMed] [Google Scholar]
- CAMPBELL DJ. Circulating and tissue angiotensin systems. J Clin Invest. 1987;79:1–6. doi: 10.1172/JCI112768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAREY RM, SIRAGY HM. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab. 2003;14:274–81. doi: 10.1016/s1043-2760(03)00111-5. [DOI] [PubMed] [Google Scholar]
- CHA DR, KIM NH, YOON JW, JO SK, CHO WY, KIM HK, WON NH. Role of vascular endothelial growth factor in diabetic nephropathy. Kidney Int Suppl. 2000;77:S104–12. doi: 10.1046/j.1523-1755.2000.07717.x. [DOI] [PubMed] [Google Scholar]
- DE VITO E, MARTINEZ DE MELIAN ER, GUARDIA DC. Activation of renal renin by a protein plasma fraction: a novel enzymatic mechanism. Comp Biochem Physiol B Biochem Mol Biol. 1996;113:433–8. doi: 10.1016/0305-0491(95)02065-9. [DOI] [PubMed] [Google Scholar]
- DE VRIESE AS, TILTON RG, STEPHAN CC, LAMEIRE NH. Vascular endothelial growth factor is essential for hyperglycemia-induced structural and functional alterations of the peritoneal membrane. J Am Soc Nephrol. 2001;12:1734–41. doi: 10.1681/ASN.V1281734. [DOI] [PubMed] [Google Scholar]
- DUFF JL, MARRERO MB, PAXTON WG, SCHIEFFER B, BERNSTEIN KE, BERK BC. Angiotensin II signal transduction and the mitogen-activated protein kinase pathway. Cardiovasc Res. 1995;30:511–7. [PubMed] [Google Scholar]
- DURVASULA RV, SHANKLAND SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294:F830–9. doi: 10.1152/ajprenal.00266.2007. [DOI] [PubMed] [Google Scholar]
- FELIERS D, DURAISAMY S, BARNES JL, GHOSH-CHOUDHURY G, KASINATH BS. Translational regulation of vascular endothelial growth factor expression in renal epithelial cells by angiotensin II. Am J Physiol Renal Physiol. 2005;288:F521–9. doi: 10.1152/ajprenal.00271.2004. [DOI] [PubMed] [Google Scholar]
- FELIERS D, LEE MJ, GHOSH-CHOUDHURY G, BOMSZTYK K, KASINATH BS. Heterogeneous nuclear ribonucleoprotein K contributes to angiotensin II stimulation of vascular endothelial growth factor mRNA translation. Am J Physiol Renal Physiol. 2007;293:F607–15. doi: 10.1152/ajprenal.00497.2006. [DOI] [PubMed] [Google Scholar]
- FLYVBJERG A, DAGNAES-HANSEN F, DE VRIESE AS, SCHRIJVERS BF, TILTON RG, RASCH R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–4. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- FREEMAN RH, ROSTORFER HH. Hepatic changes in renin substrate biosynthesis and alkaline phosphatase activity in the rat. Am J Physiol. 1972;223:364–70. doi: 10.1152/ajplegacy.1972.223.2.364. [DOI] [PubMed] [Google Scholar]
- FUKUDA R, HIROTA K, FAN F, JUNG YD, ELLIS LM, SEMENZA GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–11. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- GUPTA A, DECAILLOT FM, GOMES I, TKALYCH O, HEIMANN AS, FERRO ES, DEVI LA. Conformation state-sensitive antibodies to G-protein-coupled receptors. J Biol Chem. 2007;282:5116–24. doi: 10.1074/jbc.M609254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAVERTY TP, KELLY CJ, HINES WH, AMENTA PS, WATANABE M, HARPER RA, KEFALIDES NA, NEILSON EG. Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol. 1988;107:1359–68. doi: 10.1083/jcb.107.4.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HSUEH WA. Potential effects of renin activation on the regulation of renin production. Am J Physiol. 1984;247:F205–12. doi: 10.1152/ajprenal.1984.247.2.F205. [DOI] [PubMed] [Google Scholar]
- ICHIHARA A, KANESHIRO Y, TAKEMITSU T, SAKODA M, SUZUKI F, NAKAGAWA T, NISHIYAMA A, INAGAMI T, HAYASHI M. Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension. 2006;47:894–900. doi: 10.1161/01.HYP.0000215838.48170.0b. [DOI] [PubMed] [Google Scholar]
- INGELFINGER JR, ZUO WM, FON EA, ELLISON KE, DZAU VJ. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J Clin Invest. 1990;85:417–23. doi: 10.1172/JCI114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAGAMI S, BORDER WA, MILLER DE, NOBLE NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor- expression in rat glomerular mesangial cells. J Clin Invest. 1994;93:2431–7. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNEFICK TM, ANDERSON S. Role of angiotensin II in diabetic nephropathy. Semin Nephrol. 1997;17:441–7. [PubMed] [Google Scholar]
- KIM NH, OH JH, SEO JA, LEE KW, KIM SG, CHOI KM, BAIK SH, CHOI DS, KANG YS, HAN SY, HAN KH, JI YH, CHA DR. Vascular endothelial growth factor (VEGF) and soluble VEGF receptor FLT-1 in diabetic nephropathy. Kidney Int. 2005;67:167–77. doi: 10.1111/j.1523-1755.2005.00067.x. [DOI] [PubMed] [Google Scholar]
- KOHZUKI M, YASUJIMA M, KANAZAWA M, YOSHIDA K, FU LP, OBARA K, SAITO T, ABE K. Antihypertensive and renal-protective effects of losartan in streptozotocin diabetic rats. J Hypertens. 1995;13:97–103. [PubMed] [Google Scholar]
- KRAMER HJ, GLANZER K, MEYER-LEHNERT H, MOHAUPT M, PREDEL HG. Kinin- and non-kinin-mediated interactions of converting enzyme inhibitors with vasoactive hormones. J Cardiovasc Pharmacol. 1990;15(Suppl 6):S91–8. [PubMed] [Google Scholar]
- KUMAR R, SINGH VP, BAKER KM. The intracellular renin-angiotensin system: implications in cardiovascular remodeling. Curr Opin Nephrol Hypertens. 2008;17:168–73. doi: 10.1097/MNH.0b013e3282f521a8. [DOI] [PubMed] [Google Scholar]
- LAI KN, LEUNG JC, LAI KB, TO WY, YEUNG VT, LAI FM. Gene expression of the renin-angiotensin system in human kidney. J Hypertens. 1998;16:91–102. doi: 10.1097/00004872-199816010-00014. [DOI] [PubMed] [Google Scholar]
- LANSANG MC, OSEI SY, COLETTI C, KRUPINSKI J, HOLLENBERG NK. Hyperglycaemia-induced intrarenal RAS activation: the contribution of metabolic pathways. J Renin Angiotensin Aldosterone Syst. 2002;3:19–23. doi: 10.3317/jraas.2002.003. [DOI] [PubMed] [Google Scholar]
- LAVRENTYEV EN, ESTES AM, MALIK KU. Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. Circ Res. 2007;101:455–64. doi: 10.1161/CIRCRESAHA.107.151852. [DOI] [PubMed] [Google Scholar]
- LEE EY, SHIM MS, KIM MJ, HONG SY, SHIN YG, CHUNG CH. Angiotensin II receptor blocker attenuates overexpression of vascular endothelial growth factor in diabetic podocytes. Exp Mol Med. 2004;36:65–70. doi: 10.1038/emm.2004.9. [DOI] [PubMed] [Google Scholar]
- LEE HJ, LARUE JN, WILSON IB. Human plasma converting enzyme. Arch Biochem Biophys. 1971;142:548–51. doi: 10.1016/0003-9861(71)90518-2. [DOI] [PubMed] [Google Scholar]
- LEEHEY DJ, SINGH AK, ALAVI N, SINGH R. Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl. 2000;77:S93–8. doi: 10.1046/j.1523-1755.2000.07715.x. [DOI] [PubMed] [Google Scholar]
- LEWIS EJ, HUNSICKER LG, BAIN RP, ROHDE RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–62. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- LINDENMEYER MT, KRETZLER M, BOUCHEROT A, BERRA S, YASUDA Y, HENGER A, EICHINGER F, GAISER S, SCHMID H, RASTALDI MP, SCHRIER RW, SCHLONDORFF D, COHEN CD. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol. 2007;18:1765–76. doi: 10.1681/ASN.2006121304. [DOI] [PubMed] [Google Scholar]
- MARIAPPAN MM, FELIERS D, MUMMIDI S, CHOUDHURY GG, KASINATH BS. High Glucose, High Insulin, and Their Combination Rapidly Induce Laminin- 1 Synthesis by Regulation of mRNA Translation in Renal Epithelial Cells. Diabetes. 2007;56:476–85. doi: 10.2337/db05-1334. [DOI] [PubMed] [Google Scholar]
- MAZZOLAI L, NUSSBERGER J, AUBERT JF, BRUNNER DB, GABBIANI G, BRUNNER HR, PEDRAZZINI T. Blood pressure-independent cardiac hypertrophy induced by locally activated renin-angiotensin system. Hypertension. 1998;31:1324–30. doi: 10.1161/01.hyp.31.6.1324. [DOI] [PubMed] [Google Scholar]
- MEZZANO S, DROGUETT A, BURGOS ME, ARDILES LG, FLORES CA, AROS CA, CAORSI I, VIO CP, RUIZ-ORTEGA M, EGIDO J. Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int Suppl. 2003:S64–70. doi: 10.1046/j.1523-1755.64.s86.12.x. [DOI] [PubMed] [Google Scholar]
- MIYATA N, PARK F, LI XF, COWLEY AW., JR Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol. 1999;277:F437–46. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- NAVAR LG, INSCHO EW, MAJID SA, IMIG JD, HARRISON-BERNARD LM, MITCHELL KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- NGUYEN G. Renin/prorenin receptors. Kidney Int. 2006;69:1503–6. doi: 10.1038/sj.ki.5000265. [DOI] [PubMed] [Google Scholar]
- NGUYEN G, DELARUE F, BURCKLE C, BOUZHIR L, GILLER T, SRAER JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OZONO R, WANG ZQ, MOORE AF, INAGAMI T, SIRAGY HM, CAREY RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30:1238–46. doi: 10.1161/01.hyp.30.5.1238. [DOI] [PubMed] [Google Scholar]
- PENDERGRASS KD, AVERILL DB, FERRARIO CM, DIZ DI, CHAPPELL MC. Differential expression of nuclear AT1 receptors and angiotensin II within the kidney of the male congenic mRen2.Lewis rat. Am J Physiol Renal Physiol. 2006;290:F1497–506. doi: 10.1152/ajprenal.00317.2005. [DOI] [PubMed] [Google Scholar]
- PERSSON PB. Renin: origin, secretion and synthesis. J Physiol. 2003;552:667–71. doi: 10.1113/jphysiol.2003.049890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUPILLI C, LASAGNI L, ROMAGNANI P, BELLINI F, MANNELLI M, MISCIGLIA N, MAVILIA C, VELLEI U, VILLARI D, SERIO M. Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells. J Am Soc Nephrol. 1999;10:245–55. doi: 10.1681/ASN.V102245. [DOI] [PubMed] [Google Scholar]
- RE RN, VIZARD DL, BROWN J, BRYAN SE. Angiotensin II receptors in chromatin fragments generated by micrococcal nuclease. Biochem Biophys Res Commun. 1984;119:220–7. doi: 10.1016/0006-291x(84)91641-3. [DOI] [PubMed] [Google Scholar]
- REMUZZI G, BERTANI T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339:1448–56. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- RIZKALLA B, FORBES JM, COOPER ME, CAO Z. Increased renal vascular endothelial growth factor and angiopoietins by angiotensin II infusion is mediated by both AT1 and AT2 receptors. J Am Soc Nephrol. 2003;14:3061–71. doi: 10.1097/01.asn.0000099374.58607.c9. [DOI] [PubMed] [Google Scholar]
- ROBERTSON AL, JR, KHAIRALLAH PA. Angiotensin II: rapid localization in nuclei of smooth and cardiac muscle. Science. 1971;172:1138–9. doi: 10.1126/science.172.3988.1138. [DOI] [PubMed] [Google Scholar]
- ROHRWASSER A, ISHIGAMI T, GOCIMAN B, LANTELME P, MORGAN T, CHENG T, HILLAS E, ZHANG S, WARD K, BLOCH-FAURE M, MENETON P, LALOUEL JM. Renin and kallikrein in connecting tubule of mouse. Kidney Int. 2003;64:2155–62. doi: 10.1046/j.1523-1755.2003.00302.x. [DOI] [PubMed] [Google Scholar]
- SALEM Y, SHPUNGIN S, PASDER O, POMP O, TALER M, MALOVANI H, NIR U. Fer kinase sustains the activation level of ERK1/2 and increases the production of VEGF in hypoxic cells. Cell Signal. 2005;17:341–53. doi: 10.1016/j.cellsig.2004.08.001. [DOI] [PubMed] [Google Scholar]
- SATARANATARAJAN K, LEE MJ, MARIAPPAN MM, FELIERS D. PKC regulates the stimulation of vascular endothelial factor mRNA translation by angiotensin II through hnRNP K. Cell Signal. 2008;20:969–977. doi: 10.1016/j.cellsig.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATARANATARAJAN K, MARIAPPAN MM, LEE MJ, FELIERS D, CHOUDHURY GG, BARNES JL, KASINATH BS. Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am J Pathol. 2007;171:1733–42. doi: 10.2353/ajpath.2007.070412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHERF H, PIETSCH R, LANDSBERG G, KRAMER HJ, DUSING R. Converting enzyme inhibitor ramipril stimulates prostacyclin synthesis by isolated rat aorta: evidence for a kinin-dependent mechanism. Klin Wochenschr. 1986;64:742–5. doi: 10.1007/BF01734341. [DOI] [PubMed] [Google Scholar]
- SEALEY JE, OVERLACK A, LARAGH JH, STUMPE KO, ATLAS SA. Effect of captopril and aprotinin on inactive renin. J Clin Endocrinol Metab. 1981;53:626–30. doi: 10.1210/jcem-53-3-626. [DOI] [PubMed] [Google Scholar]
- SIMON M, GRONE HJ, JOHREN O, KULLMER J, PLATE KH, RISAU W, FUCHS E. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol. 1995;268:F240–50. doi: 10.1152/ajprenal.1995.268.2.F240. [DOI] [PubMed] [Google Scholar]
- SINGH R, LEEHEY DJ. Effect of ACE inhibitors on angiotensin II in rat mesangial cells cultured in high glucose. Biochem Biophys Res Commun. 2007;357:1040–5. doi: 10.1016/j.bbrc.2007.04.038. [DOI] [PubMed] [Google Scholar]
- SINGH VP, LE B, BHAT VB, BAKER KM, KUMAR R. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H939–48. doi: 10.1152/ajpheart.00391.2007. [DOI] [PubMed] [Google Scholar]
- SOFFER RL, REZA R, CALDWELL PR. Angiotensin-converting enzyme from rabbit pulmonary particles. Proc Natl Acad Sci U S A. 1974;71:1720–4. doi: 10.1073/pnas.71.5.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIDOTTI DB, CASARINI DE, CRISTOVAM PC, LEITE CA, SCHOR N, BOIM MA. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am J Physiol Renal Physiol. 2004;286:F1039–45. doi: 10.1152/ajprenal.00371.2003. [DOI] [PubMed] [Google Scholar]
- WILLIAMS B. A potential role for angiotensin II-induced vascular endothelial growth factor expression in the pathogenesis of diabetic nephropathy? Miner Electrolyte Metab. 1998;24:400–5. doi: 10.1159/000057401. [DOI] [PubMed] [Google Scholar]
- WOLF G, MUELLER E, STAHL RA, ZIYADEH FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor. J Clin Invest. 1993;92:1366–72. doi: 10.1172/JCI116710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLF G, NEILSON EG. Angiotensin II induces cellular hypertrophy in cultured murine proximal tubular cells. Am J Physiol. 1990;259:F768–77. doi: 10.1152/ajprenal.1990.259.5.F768. [DOI] [PubMed] [Google Scholar]
- WOLF G, ZIYADEH FN. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol. 2007;106:p26–31. doi: 10.1159/000101797. [DOI] [PubMed] [Google Scholar]
- YOO TH, LI JJ, KIM JJ, JUNG DS, KWAK SJ, RYU DR, CHOI HY, KIM JS, KIM HJ, HAN SH, LEE JE, HAN DS, KANG SW. Activation of the renin-angiotensin system within podocytes in diabetes. Kidney Int. 2007;71:1019–27. doi: 10.1038/sj.ki.5002195. [DOI] [PubMed] [Google Scholar]
- ZIYADEH FN, WOLF G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. doi: 10.2174/157339908783502370. [DOI] [PubMed] [Google Scholar]