

Abstract
Whereas recognition of LPS by the MD-2—TLR4 receptor complex is important for triggering protective inflammatory responses in animals, terminating many of these responses requires LPS inactivation by a host lipase, acyloxyacyl hydrolase (AOAH). To test if endogenously-produced recombinant AOAH can modulate responses to LPS and Gram-negative bacteria, we engineered transgenic mice that overexpress AOAH in dendritic cells and macrophages, cell types that normally produce it. Transgenic mice deacylated LPS more rapidly than did wildtype controls. They were also protected from LPS-induced hepatosplenomegaly, recovered more quickly from LPS-induced weight loss, and were more likely to survive when challenged with live E. coli. Constitutive overexpression of AOAH in vivo hastened recovery from LPS exposure without interfering with the normal acute inflammatory response to this important microbial signal molecule. Our results suggest that the extent to which macrophages and dendritic cells produce AOAH may influence the outcome of many Gram-negative bacterial diseases.
Keywords: lipopolysaccharide, LPS, acyloxyacyl hydrolase, AOAH, Gram-negative bacteria, deacylation, dendritic cell, macrophage, hepatosplenomegaly, CD68
INTRODUCTION
Monocyte-macrophages can sense minute quantities of Gram-negative bacterial LPS and activate inflammatory responses that provide effective host defense. Responses to LPS can also be harmful, however, and several mechanisms are known to limit them. Some involve proteins that directly bind LPS and prevent its interaction with MD-2--TLR4, others inactivate LPS by rapidly removing it from the circulation, while others diminish or extinguish cellular pathways that have been activated by LPS [1].
Another important mechanism that limits responses to LPS is enzymatic degradation of lipid A, the moiety that is sensed by MD-2—TLR4. Two inactivating enzymatic reactions are known. In zebrafish, an intestinal alkaline phosphatase can inactivate LPS within the gut by dephosphorylating it [2]. Although endogenous phosphatases might also inactivate LPS within the internal organs or cells of fish and other vertebrates [3;4], at this time the importance of this phenomenon is uncertain. The second known inactivating reaction is carried out by acyloxyacyl hydrolase (AOAH), a highly conserved host lipase [5] that selectively removes the secondary acyl chains from lipid A [6]. The resulting deacylated (tetraacylated) LPS (dLPS) is 0.2–1% as potent as fully (hexa)acylated LPS [7]. Furthermore, AOAH-treated LPS may be an antagonist, competing with LPS for binding to the MD-2—TLR4 complex [8;9]. A host defense role for AOAH was suggested by observing that AOAH-deficient mice developed long-lasting hepatosplenomegaly, exaggerated polyclonal antibody production, and prolonged immunosuppression after they were exposed to LPS or Gram-negative bacteria [10–12]. Enzymatic deacylation of LPS may thus be necessary for animals to regain fitness following infections with Gram-negative bacteria synthesizing LPS that can activate MD-2—TLR4 [13].
Previously, we found that providing recombinant AOAH can restore normal recovery responses to Aoah−/− mice [14;15]. Here we tested the hypothesis that increased constitutive expression of AOAH by cells that normally produce it, macrophages and dendritic cells, hastens recovery from LPS exposure in vivo. We produced transgenic mice in which the human macrophage-selective CD68 promoter drives high-level AOAH expression. We then tested CD68p-AOAH transgenic mice for their ability to deacylate LPS and resist challenge with LPS or live E. coli. Our results suggest that devising methods to increase endogenous AOAH activity might benefit animals with many Gram-negative bacterial diseases.
RESULTS
LPS induces hepatic AOAH activity in vivo
When we injected wildtype C57Bl/6 mice intraperitoneally with 25 μg LPS and harvested their livers at different time points, we found that AOAH activity increased approximately 6-fold, peaking on day 3 post-challenge and returning almost to baseline levels by day 9 (Fig. 1A). We also observed a corresponding increase in serum AOAH activity (Fig. 1B). LPS thus induces a significant but transient increase in AOAH activity in wildtype mice, in agreement with previous findings [16]. The increase in AOAH activity roughly paralleled the time-course of hepatic LPS deacylation, which occurs gradually over 1 to 3 days [17].
Figure 1. LPS induces AOAH activity in vivo.

(A) C57Bl/6 mice were injected i.p. with either PBS or 25 μg LPS. Livers were harvested at the times indicated and AOAH activity was measured. (B) AOAH activity in serum, measured in 10 μl samples as described in Methods. Data are expressed as mean ± SD, n=3. *P<0.05; **P<0.01 (Student’s t-test). Statistical analyses were performed using GraphPad Prism 5 for Windows.
Generation of CD68p-AOAH transgenic mice and detection of AOAH activity in serum
To study the effects of AOAH overexpression in vivo, we produced transgenic mice that constitutively produce high levels of AOAH in macrophages and dendritic cells. To distinguish transgenic AOAH from endogenous AOAH, a FLAG sequence was added to the carboxyl terminus of murine AOAH. The FLAG-tagged murine AOAH cDNA sequence was inserted into an expression vector downstream of human CD68 promoter sequences, the 83bp first intron of the CD68 gene (IVS-1), and a polyadenylation signal (Fig. 2A). The IVS-1 sequence has been shown to act as a macrophage-selective enhancer when cloned downstream of the human CD68 promoter sequence [18]. When we transfected this construct into RAW264.7 macrophages, we found that the expression of AOAH from the CD68 promoter was of similar magnitude to that found using the early CMV promoter (data not shown). Two transgenic founder lines were obtained; mice in each line appeared normal and produced viable offspring. Serum obtained from CD68p-AOAH transgenic mice had AOAH activity that was considerably higher than that found in wildtype C57Bl/6 mice (Fig. 2B).
Figure 2. CD68p-AOAH transgene construct.
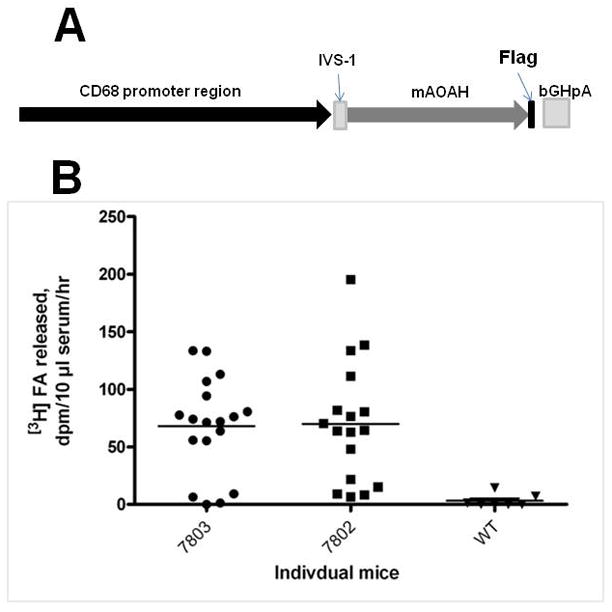
(A) DNA construct showing the human CD68 promoter, the 86bp first intron of the CD68 gene (IVS-), and murine AOAH cDNA. (B) Deacylation of LPS by mouse serum. Serum was obtained from the offspring of F1 matings in two founder lines (7803 and 7802) and assayed for its ability to release 3H labeled fatty acids from [3H/14C]LPS. WT = wildtype.
CD68 promoter sequences drive transgene expression in macrophages and dendritic cells
We assessed transgene mRNA expression in thioglycollate-elicited peritoneal macrophages, bone marrow-derived macrophages, and dendritic cells. Both AOAH and the transgene-specific FLAG region were measured by quantitative real-time PCR. As expected, there was little or no FLAG message detected in cells from wildtype mice (data not shown). In transgenic mice, the pattern of FLAG expression correlated with that of AOAH mRNA (r2=0.74, p= 0.0003, n = 6 samples of liver or spleen; linear regression, GraphPad Prism 5). Dendritic cells and peritoneal macrophages from transgenic mice had approximately 16-fold and 26-fold higher AOAH mRNA expression, respectively, than did wildtype mice, whereas bone marrow macrophages had 37-fold higher expression (Fig. 3A). Most of the AOAH mRNA measured in the transgenic mice was thus due to the transgene. We also measured AOAH enzymatic activity in cell lysates. Transgenic macrophages and, to a lesser extent, dendritic cells had significantly higher AOAH activity than did their wildtype counterparts (Fig. 3B). We conclude that the CD68 promoter-driven transgene achieves high-level, constitutive expression of AOAH in macrophages and dendritic cells in vivo.
Figure 3. Increased AOAH expression in monocytes-macrophages, dendritic cells and phagocyte-rich tissues.

(A and C) AOAH mRNA abundance was assessed using quantitative PCR. Total RNA was isolated from thioglycollate-elicited macrophages (IP macs), bone marrow-derived macrophages (BMDM) and dendritic cells (BMDC), liver, spleen, lung and kidney. Means and 1 S.D. are shown. (B and D) AOAH activity was estimated in cell and tissue lysates. n=4–6, data combined from two experiments. *p<0.05, **p<0.01, ***p<0.001 (Student’s t-test). (E) Correlation of plasma and liver AOAH activity in wildtype and CD68p-AOAH transgenic mice. r2 = 0.92, p < 0.0001 (linear regression). WT = wild-type; TG = AOAH transgenic.
Increased AOAH expression in phagocyte-rich organs of CD68p-AOAH transgenic mice
Based on the known expression of CD68 in macrophages, we predicted that the macrophage-rich tissues of CD68p-AOAH mice would have elevated AOAH expression. Liver, spleen, and lung had greatly increased AOAH mRNA abundance, with liver being the lowest and lung the highest (Fig. 3C). This is similar to the pattern observed by others for macrosialin (CD68) expression [19].
In wildtype mice, AOAH is produced most abundantly by renal cortical epithelial cells [20], which secrete it into the urine. We found high levels of AOAH mRNA expression in the kidneys of both transgenic and wildtype mice. Measurement of transgene-specific FLAG mRNA confirmed very low expression of the transgene in the kidneys of CD68p-AOAH transgenic mice (data not shown) in keeping with the conclusion that transgene expression occurs principally in tissues rich in macrophages and dendritic cells. We also compared tissue AOAH activity in wildtype and transgenic mice and found significant increases in transgenic liver, spleen and lung, but not in kidney (Fig. 3D). The liver had the highest AOAH activity and the lung had the lowest. We also observed a strong correlation between liver and plasma AOAH activity in transgenic and wildtype mice (Fig. 3E).
AOAH overexpression in macrophages allows more rapid LPS deacylation
We next measured the rate of LPS deacylation by explanted macrophages ex vivo. Double-radiolabeled LPS was utilized to assay the deacylation rate by measuring release of 3H fatty acyl chains from the 14C labeled backbone. Thioglycollate-elicited peritoneal macrophages were incubated with radiolabeled LPS for 6 or 12 hours. At the end of the incubation period, the media were harvested and the cells were washed twice before their LPS content was measured as described in Methods. Whereas the amount of cell-associated LPS was similar in both transgenic and wildtype macrophages, the transgenic macrophages had deacylated 19% and 44% of the LPS by 6 and 12 hours, respectively, while the wildtype macrophages had deacylated approximately 5% and 25% by these time points (Fig. 4A).
Figure 4. Rapid in vitro and in vivo LPS deacylation in transgenic mice.

(A) Thioglycollate-elicited peritoneal macrophages were incubated with radiolabeled LPS for 6 or 12 hours before deacylation was calculated as described in Methods. Percent deacylation = percent loss of secondary acyl chains. Equivalent numbers of wildtype and transgenic mouse macrophages were used (~0.15 mg protein/well). (B) 5μg radiolabeled LPS were injected intravenously into mice, livers were harvested 4 hours later, and LPS deacylation was determined as described in Methods. n = 4 mice/group. Data are representative of 2 independent experiments. For comparisons between wildtype and transgenic macrophages at each time point: * p<0.05, *** p < 0.001 (Student’s t-test). WT = wild-type; TG = AOAH transgenic.
To determine if transgenic mice also have a higher rate of LPS deacylation in vivo, we injected mice i.v. with 5 μg of radiolabeled LPS and harvested their livers 4 hours later. Deacylation of the LPS, which was rapidly taken up by the liver [21;22], was measured as previously described [23]. By 4 hours, CD68p-AOAH mice had released approximately 55% of the 3H radioactivity from liver-associated LPS while the wildtype mice had only released an average of 31% (Fig. 4B). We analyzed the fatty acid composition of the LPS recovered from the livers and observed that the secondary fatty acids (laurate and myristate) were less abundant in LPS isolated from transgenic mice, confirming more rapid removal of these acyl chains with increased AOAH expression by liver macrophages (data not shown).
Increased AOAH expression in transgenic mice hastens recovery from LPS challenge
AOAH-mediated conversion of fully (hexaacylated) LPS to partially deacylated (tetraacylated) LPS (dLPS) occurs slowly, over a period of many hours, both in vivo and in explanted dendritic cells and macrophages [24]. It was therefore unlikely that enhanced constitutive AOAH expression would protect mice from the rapid inflammatory response that is observed within minutes of LPS exposure. Indeed, at 1 and 4 hours after an intraperitoneal dose of 10 μg of O14 E.coli LPS, CD68p-AOAH transgenic mice had plasma levels of IL-6 and TNF-α that were equivalent to those of wildtype mice (Fig. 5A & B).
Figure 5. Increased AOAH expression shortens recovery time from LPS challenge.

Mice were injected i.p. with 10 μg of E.coli O14 LPS and bled 1 hr (A) or 4 hrs (B) later to measure serum TNF-α and IL-6, respectively. (C) E.coli O14 LPS (10 mg/kg body weight) was injected i.p into wildtype and transgenic mice. Mice were weighed daily for 7 days. *p <0.05, **p<0.01 (Student’s t-test). Data are combined from two experiments, n=9/group. (D) Constitutive overproduction of AOAH does not protect mice from a non-LPS agonist. Weights were monitored daily for 7 days after i.p. injection of 1mg/kg UT 12 antibody. Data were combined from two independent experiments, each with n = 4. WT = wild-type; TG = AOAH transgenic.
In contrast, AOAH prevents several long-term responses to LPS in vivo [11;25;26]. We thus hypothesized that transgenic mice expressing large amounts of AOAH would recover more quickly from LPS challenge. We first challenged wildtype and CD68p-AOAH transgenic mice intraperitoneally with 10 mg/kg E. coli O14 LPS. Both transgenic and wildtype mice showed significant weight loss one day after injection (Fig. 5C). The transgenic mice began to regain weight more quickly, however, and they regained their original weights more rapidly than did the wildtype mice.
AOAH does not protect mice from challenge with an agonistic monoclonal antibody to MD-2--TLR4
It is conceivable that the ability of CD68p-AOAH mice to recover more quickly from LPS challenge is conferred by an activity of the enzyme that is unrelated to its ability to deacylate LPS. To explore this possibility, we injected wildtype and transgenic mice intraperitoneally with 20μg of UT12, an agonistic monoclonal antibody to the TLR4-MD—2 complex [27]. UT12 induces stimulatory signals downstream of MD-2—TLR4 that are similar to those induced by LPS [28;29]. We monitored the weights of mice after injection for seven days and observed similar changes in weight in both wildtype and transgenic mice (Fig. 5D). Overproduction of AOAH thus enables more rapid weight gain following LPS challenge but has no such effect when the same intracellular signaling pathway is activated by a non-LPS agonist.
CD68p-AOAH mice are less susceptible to LPS-induced hepatomegaly
In the liver, LPS deacylation is carried out mainly by Kupffer cells [30]. Mice deficient in AOAH were unable to deacylate LPS and developed hepatomegaly within 7 days after a single i.v. dose of 5 μg E.coli O14 LPS (0.25 mg/kg body weight) [31]. Much higher doses of LPS could induce hepatomegaly in wildtype mice, however, indicating that the increase in liver size is a dose-dependent response to LPS [32]. We hypothesized that an increase in hepatic AOAH would prevent or reduce the development of hepatomegaly when LPS was administered intravenously. We challenged transgenic and wildtype mice with PBS or 30 μg (1.5 mg/kg) E. coli O14 LPS intravenously and weighed their livers 7 days after injection. Livers from wildtype mice were significantly larger than those from transgenic mice (Fig. 6A).
Figure 6.

CD68p-AOAH transgenic mice are less susceptible to LPS-induced hepatosplenomegaly and death. Wildtype and transgenic mice were challenged i.v. with either PBS or 30 μg E. coli O14 LPS. (A) Livers were harvested 7 days after challenge and weighed. The liver weight/body weight fraction (%) is plotted. *** p<0.001 (Student’s t-test). (B, C) Mice were challenged i.p. with 4–5 × 108 cfu of E.coli O14 bacteria. The spleens and livers were harvested and weighed 9 days after challenge. ***p<0.005 (Student’s t-test). (D) Mice received 2 – 4.5 × 109 cfu E. coli O14 i.p. and were followed for 7 days. n = 11/group, p < 0.03 (Mantel-Cox test). WT = wild-type; TG = AOAH transgenic.
Mice overexpressing AOAH are less susceptible to hepatosplenomegaly and death induced by Gram-negative bacteria
Animals are usually exposed to LPS in its natural setting, the Gram-negative bacterial cell wall. Therefore we wanted to find out if AOAH-overexpressing mice would be protected from a Gram-negative bacterial challenge. Mice were injected i.p. with a low dose of an avirulent strain of E.coli (1.5–3 × 108 colony forming units) and their livers and spleens were weighed 9 days later. We observed that spleens from wildtype mice were approximately 70% larger than spleens from their transgenic counterparts (Fig. 6B). Livers from wildtype mice also tended to be larger than those from transgenic mice (Fig. 6C) (p=0.055; Student’s t-test). Whereas all of the 9 transgenic mice survived this challenge, 2 of the 9 wildtype mice did not. We then injected mice with a larger dose (1–4.5 × 109) of live E. coli O14 and followed them for one week. AOAH-overexpressing mice were much more likely to survive than were wildtype mice (Fig. 6D) (p < 0.03; Mantel-Cox).
DISCUSSION
The last decade brought great progress in understanding how animals defend themselves from both commensal and pathogenic microbes. A key element of this progress has been the identification of mechanisms through which animal cells sense, and respond to, particular microbial molecules. Although often referred to as molecular “patterns”, in fact the microbial molecular structures that are sensed by most of the known host receptors (TLRs, NODs) are quite specific: MD-2—TLR4 best recognizes LPS molecules that have 6 saturated acyl chains, for example, whereas TLR2/TLR1 and TLR2/TLR6 can distinguish between lipopeptides that have two or three fatty acyl chains. NOD1 and NOD2 likewise show specificity for muramyl peptides with certain amino acid residues. It is thus of interest that vertebrates also have a highly conserved enzyme that attacks LPS molecules in such a way that only the key ‘signal’ structures, the secondary acyl chains, are removed. AOAH can partially deacylate, and thus inactivate, LPSs that have the optimally stimulatory (hexaacyl) lipid A structure [5;33]. To date, the evidence suggests that inactivation by AOAH is required for recovery from exposure to LPS, as none of the other known mechanisms for neutralizing LPS (see Introduction) seems to compensate fully for the enzyme’s absence, at least in mice.
Cody et al. showed that LPS upregulates AOAH mRNA expression in vivo [16]. Here we found that a single low dose of LPS is able to augment hepatic AOAH activity by approximately sixfold, with increased levels for up to five days after challenge (Fig. 1). Since the majority of the LPS that enters the bloodstream is cleared by the liver, the increase in hepatic AOAH expression (which could be from either resident Kupffer cells or from neutrophils or monocytes that are recruited to the liver following LPS stimulation) may be very important for enhancing LPS deacylation in vivo. We found previously that providing recombinant human AOAH in an adenoviral vector restored LPS deacylation and prevented both LPS-induced hepatomegaly and prolonged endotoxin tolerance in Aoah−/− mice [34]. Since intravenously-administered adenovirus vectors largely produce their cargo transgenes in hepatocytes [35], we wanted to know if increasing constitutive AOAH expression in cells that normally make it, such as macrophages and dendritic cells, would also enable wildtype mice to resist LPS challenge. Accordingly, we generated transgenic mice expressing the murine AOAH cDNA under the control of the CD68p-IVS-1 expression cassette that was known to drive expression of other transgenes in macrophages in vivo [36–39]. These transgenic mice had high AOAH expression in macrophages and dendritic cells and secreted the enzyme into the blood. We also observed high AOAH expression in macrophage-rich tissues. There was an unexpected discrepancy between AOAH mRNA expression and activity in these tissues, however. Whereas the liver had the highest AOAH activity, its AOAH mRNA abundance was lower than that of spleen and lung. This difference may reflect, at least in part, the extent to which the AOAH precursor is processed to the more active mature form in different organs [20;40]. Alternatively, post-transcriptional regulation may prevent translation in these tissues. Importantly, constitutive overexpression did not interfere with the initial pro-inflammatory response to LPS (Fig. 5A & B), in keeping with previous observations that AOAH-mediated deacylation/inactivation occurs over many hours and does not diminish acute reactions to LPS in vivo. In contrast, the ability of elevated constitutive AOAH expression to protect mice from LPS challenge was evident in two different test systems. First, after LPS challenge, transgenic mice returned to their pre-challenge weights more quickly than did wildtype mice (Fig. 5C). Second, mice with high constitutive AOAH activity were protected from developing LPS and Gram-negative bacteria-induced hepatosplenomegaly and death (Fig. 6A & 7). Importantly, AOAH overexpression did not hasten recovery from weight loss induced by a potent non-LPS MD-2—TLR4 agonist, confirming that the enzyme’s ability to deacylate LPS is required for its beneficial effect.
Our findings raise the possibility that overexpressing AOAH in macrophages and/or dendritic cells could hasten recovery from many Gram-negative diseases in other animals, including humans. Drugs that increase constitutive AOAH activity might therefore warrant investigation. Furthermore, the findings reported here suggest that genetic variability in AOAH expression might affect recovery from diseases caused by Gram-negative bacteria that produce hexaacyl LPS [13].
METHODS
Construction of CD68p-AOAH Transgenic Mice
Murine AOAH cDNA was amplified using primers 5′-TCTAGACACCATGAAGTTTCCCTGGAAAGTCTTCAAGACC-3 and 5′-CTACTTATCGTCGTCATCCTTGTAATCGTGTCCTCCTTGGTCTC-3′, resulting in addition of a 5′ XbaI site and a 3′ Flag epitope (underlined bases). The PCR product was cloned into pCR 2.1, introducing a second XbaI site downstream of the Flag sequence. The mAOAH-Flag fragment was released from the plasmid by XbaI digestion and inserted into pcDNA3 vector containing the human CD68 promoter and 83bp of the first intron of the human CD68 gene [41]. The construct was isolated from this plasmid (pCDNA3-hCD68p-IVS-mAOAHFlag) and purified. Injection into C576BL/6 eggs and insertion into pseudopregnant foster mothers was carried out by the UT-Southwestern transgenic mouse core. Two founder lines (7802 and 7803) were confirmed by PCR analysis of genomic DNA extracted from the tails of mice; the primers amplified a 548 bp region from the distal portion of the CD68 promoter to the proximal portion of the AOAH gene: 5′-GAAGCAGGGCCAACAGTCCCC T-3′ and 5′-CTCTCCATCGCCACCTGGACT-3′. These primers were also used for genotyping offspring.
LPS Preparations
Double-radiolabeled Rc Salmonella typhimurium PR122 LPS that had 175,000 3H dpm in the fatty acyl chains and 10,000 14C dpm in the glucosamine backbone per microgram was prepared as previously described [42]. Unlabeled LPS from E. coli O14 was purified by phenol-chloroform-petroleum ether extraction [42]. The purity of the LPS was confirmed by silver-stained SDS-PAGE [43] and by its failure to stimulate IL-6 release from Tlr4−/− macrophages in vitro.
Cell Culture and transfection
Peritoneal macrophages, bone marrow-derived dendritic cells (BMDC), and macrophages (BMDM) were generated as described previously [44], [45]. RAW264.7 cells were grown in DMEM and transfected (2–4 μg) using Superfect (Qiagen) with pCDNA3-hCD68p-IVS-1mAOAHFlag, pRC.CMV.mAOAHFlag (AOAH Flag expressed from the CMV promoter), or a GFP-expressing control plasmid.
AOAH activity assays
Mice (6–8 weeks old) were deeply anesthetized with isofluorane before the liver, kidney, spleen and lung were harvested, weighed and sonicated in lysis buffer (0.1% Triton X-100 in PBS pH 7.2). AOAH activity in the tissues and in serum or plasma obtained from tail vein bleeds was measured as described previously [42].
Quantitative PCR
Total RNA was isolated from snap-frozen tissues and reverse-transcribed to cDNA using the iScript™ cDNA synthesis kit (Bio-Rad laboratories, USA). Real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) in an ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA). The following primers were used for AOAH mRNA amplification: 5-′GCCATGATTGGAAACGATGTT-3 and5′-GTTGGCGTA CATTTGTTCAGG A-3′. Transgene mRNA was quantitated by amplifying the FLAG region using the primers 5′-ACCAAGGAGGACACGATTACA AG-3′ and 5′-CCGCCAGTGTGATGGATA TCT-3.′ Dose-response curves were produced using plasmids expressing known amounts of 18rRNA, AOAH, and FLAG. The reaction conditions used were 50°C for 2min; 95°C for 10min; and 95°C for 15sec, 60°C for 1min for 40 cycles. All sample and standard reactions were done in duplicate. CTs for all reported values were within the range from 20 to 25.6.
Estimation of LPS Deacylation
In vivo LPS deacylation was measured by injecting 5 μg of radiolabeled LPS into the lateral tail vein and harvesting organs 4 hours later Approximately 100 mg of tissue were sonicated in lysis buffer (0.1% Triton X-100 in PBS pH 7.2) and deacylation was measured as described. [46]. To measure in vitro deacylation, thioglycollate-elicited peritoneal macrophages (wildtype, CD68p-AOAH, or Aoah−/−) were plated at 1.5 × 106 cells/ml in 6 well plates. Double-labeled Salmonella typhimurium LPS (1μg/ml) was added and media and cells harvested 6 and 12 hours later. The dpm in 100μl of the cell lysate were counted. The 3H dpm fatty acid released into cells and media were determined by precipitating 500μl of both media and cell lysates with 1ml ethanol and incubation at −20°C for 30 minutes. The 3H dpm in the ethanol supernatant were measured and the dpm in the wildtype and transgenic cells were corrected for non-specific deacylation by subtracting the dpm released by the Aoah−/− cells. The percent deacylation for each time point was estimated using the formula: [ethanol soluble 3H dpm released in cells + supernatant]/[total 3H dpm in cells + supernatant] × 100.
In vivo LPS challenge
Protocols for animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center. Wildtype mice were injected i.p. with 25 μg of E.coli O14 LPS and the AOAH activity in liver lysates was measured at sequential time points. To evaluate the impact of AOAH on LPS-induced cytokine release, wildtype and transgenic mice were injected i.p. with 5μg E.coli 014 LPS. Mice were bled 1 and 4 hours post-challenge and serum levels of TNF-α and IL-6 were measured using ELISA (BD Biosciences). To assess clinical responses to LPS, mice were i.p. injected with 10 mg/kg E.coli O14 LPS and weighed daily. For hepatomegaly studies, wildtype and CD68p-AOAH mice were challenged with 30μg of E.coli 014 LPS or E.coli O14 bacteria (1.5–3 × 108 cfu) intravenously. Livers were harvested after 7 days and the liver weight/body weight fraction (%) was calculated [47]. In other experiments, mice were given an i.p. injection of 1–4 × 109 cfu E. coli O14 and followed for 7 days.
Acknowledgments
We thank Shoichiro Ohta (Saga, Japan) for generously providing UT12 antibody.
This study was funded by grant AI18188 from the National Institute of Allergy and Infectious Diseases and by the Jan and Henri Bromberg Chair in Internal Medicine, UT-Southwestern Medical Center.
The findings of this study have not been presented, in full or in part, at any meeting.
Footnotes
Conflict of Interest Statements: Noredia Ojogun: No conflict.
Tang-Yong Kuang. No conflict.
Baomei Shao: No conflict.
David Robert Greaves. No conflict.
Robert S. Munford: No conflict.
Alan W. Varley: No conflict.
Reference List
- 1.Munford RS. Detoxifying endotoxin: time, place, person. J Endotoxin Res. 2005 doi: 10.1179/096805105X35161. In press. [DOI] [PubMed] [Google Scholar]
- 2.Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007 Dec 13;2(6):371–82. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson AA, Munford RS. Dephosphorylation of the lipid A moiety of Escherichia coli lipopolysaccharide by mouse macrophages. Infect Immun. 1987;55:974–8. doi: 10.1128/iai.55.4.974-978.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaishnava S, Hooper LV. Alkaline phosphatase: keeping the peace at the gut epithelial surface. Cell Host Microbe. 2007 Dec 13;2(6):365–7. doi: 10.1016/j.chom.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Munford RS, Varley AW. Shield as signal: lipopolysaccharides and the evolution of immunity to Gram-negative bacteria. PLoS Pathog. 2006;2:e67. doi: 10.1371/journal.ppat.0020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munford RS, Hall CL. Detoxification of bacterial lipopolysaccharides (endotoxins) by a human neutrophil enzyme. Science. 1986;234:203–5. doi: 10.1126/science.3529396. [DOI] [PubMed] [Google Scholar]
- 7.Kitchens RL, Ulevitch RJ, Munford RS. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J Exp Med. 1992;1760:485–94. doi: 10.1084/jem.176.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitchens RL, Munford RS. Enzymatically deacylated lipopolysaccharide (LPS) can antagonize LPS at multiple sites in the LPS recognition pathway. J Biol Chem. 1995;270:9904–10. doi: 10.1074/jbc.270.17.9904. [DOI] [PubMed] [Google Scholar]
- 9.Teghanemt A, Zhang D, Levis EN, Weiss JP, Gioannini TL. Molecular basis of reduced potency of underacylated endotoxins. J Immunol. 2005 Oct 1;175(7):4669–76. doi: 10.4049/jimmunol.175.7.4669. [DOI] [PubMed] [Google Scholar]
- 10.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 11.Lu M, Zhang M, Takashima A, et al. Lipopolysaccharide deacylation by an endogenous lipase controls innate antibody responses to Gram-negative bacteria. Nat Immunol. 2005;6:989–94. doi: 10.1038/ni1246. [DOI] [PubMed] [Google Scholar]
- 12.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008 Sep 11;4(3):293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munford RS. Sensing Gram-negative bacterial lipopolysaccharides: a human disease determinant? Infect Immun. 2008;76:454–65. doi: 10.1128/IAI.00939-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 15.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008 Sep 11;4(3):293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cody MJ, Salkowski CA, Henricson BE, Detore GR, Munford RS, Vogel SN. Effect of inflammatory and anti-inflammatory stimuli on acyloxyacyl hydrolase gene expression and enzymatic activity in murine macrophages. J Endotoxin Res. 1997;4:371–9. [Google Scholar]
- 17.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 18.Greaves DR, Quinn CM, Seldin MF, Gordon S. Functional comparison of the murine macrosialin and human CD68 promoters in macrophage and nonmacrophage cell lines. Genomics. 1998 Nov 15;54(1):165–8. doi: 10.1006/geno.1998.5546. [DOI] [PubMed] [Google Scholar]
- 19.Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001 Jul;103(3):351–61. doi: 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feulner JA, Lu M, Shelton JM, Zhang M, Richardson JA, Munford RS. Identification of acyloxyacyl hydrolase, a lipopolysaccharide-detoxifying enzyme, in the murine urinary tract. Infect Immun. 2004;72:3171–8. doi: 10.1128/IAI.72.6.3171-3178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coulthard MG, Swindle J, Munford RS, Gerard RD, Meidell RS. Adenovirus-mediated transfer of a gene encoding acyloxyacyl hydrolase (AOAH) into mice increases tissue and plasma AOAH activity. Infect Immun. 1996;64:1510–5. doi: 10.1128/iai.64.5.1510-1515.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 23.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 24.Katz SS, Weinrauch Y, Munford RS, Elsbach P, Weiss J. Deacylation of lipopolysaccharide in whole Escherichia coli during destruction by cellular and extracellular components of a rabbit inflammatory peritoneal exudate. J Biol Chem. 1999;274:36579–84. doi: 10.1074/jbc.274.51.36579. [DOI] [PubMed] [Google Scholar]
- 25.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 26.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008 Sep 11;4(3):293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta S, Bahrun U, Shimazu R, Matsushita H, Fukudome K, Kimoto M. Induction of Long-Term Lipopolysaccharide Tolerance by an Agonistic Monoclonal Antibody to the Toll-Like Receptor 4/MD-2 Complex. Clin Vaccine Immunol. 2006 Oct 1;13:1131–6. doi: 10.1128/CVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohta S, Bahrun U, Shimazu R, Matsushita H, Fukudome K, Kimoto M. Induction of Long-Term Lipopolysaccharide Tolerance by an Agonistic Monoclonal Antibody to the Toll-Like Receptor 4/MD-2 Complex. Clin Vaccine Immunol. 2006 Oct 1;13:1131–6. doi: 10.1128/CVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008 Sep 11;4(3):293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem . 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 31.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 32.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 33.Erwin AL, Munford RS. Deacylation of structurally diverse lipopolysaccharides by human acyloxyacyl hydrolase. J Biol Chem. 1990;265:16444–9. [PubMed] [Google Scholar]
- 34.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 35.Kopfler WP, Willard M, Betz T, Willard JE, Gerard RD, Meidell RS. Adenovirus mediated transfer of a gene encoding human apolipoprotein A-1 into normal mice increases circulating high density lipoprotein cholesterol. Circulation. 1994;90:1319–27. doi: 10.1161/01.cir.90.3.1319. [DOI] [PubMed] [Google Scholar]
- 36.Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001 Jul 1;103:351–61. doi: 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine Deactivation of Macrophages in Transgenic Mice Constitutively Overexpressing IL-10 Under Control of the Human CD68 Promoter. The Journal of Immunology. 2002 Apr 1;168:3402–11. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 38.Shi C, Sakuma M, Mooroka T, et al. Downregulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008 Sep 17; doi: 10.1182/blood-2008-01-137018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006 Jul;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staab JF, Ginkel DL, Rosenberg GB, Munford RS. A saposin-like domain influences the intracellular localization, stability, and catalytic activity of human acyloxyacyl hydrolase. J Biol Chem. 1994 Sep 23;269(38):23736–42. [PubMed] [Google Scholar]
- 41.Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001 Jul 1;103:351–61. doi: 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munford RS, Erwin AL. Eucaryotic lipopolysaccharide deacylating enzyme. Meth Enzymol. 1992;209:485–92. doi: 10.1016/0076-6879(92)09059-c. [DOI] [PubMed] [Google Scholar]
- 43.Tsai C-M, Frasch CE. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal Biochem. 1982;119:115–9. doi: 10.1016/0003-2697(82)90673-x. [DOI] [PubMed] [Google Scholar]
- 44.Lu M, Zhang M, Kitchens RL, Fosmire S, Takashima A, Munford RS. Stimulus-dependent deacylation of bacterial lipopolysaccharide by dendritic cells. J Exp Med. 2003;197:1745–54. doi: 10.1084/jem.20030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schook LB, Allen PM, Niederhuber JE. Bone marrow-derived macrophage as accessory cells in antigen-induced T cell proliferation. H-2I region requirements for L-glutamic60-L-alanine30-L-tyrosine10 response. J Immunol. 1983 Feb;130(2):661–4. [PubMed] [Google Scholar]
- 46.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 47.Shao B, Lu M, Katz SC, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–35. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]