

Abstract
The complexity and heterogeneity of the plasma proteome have presented significant challenges in the identification of protein changes associated with tumor development. We used cell culture as a model system and identified differentially expressed, secreted proteins which may constitute serological biomarkers. A stable isotope labeling by amino acids in cell culture (SILAC) approach was used to label the entire secreted proteomes of the CT26 murine colon cancer cell line and normal young adult mouse colon (YAMC) cell line, thereby creating a stable isotope labeled proteome (SILAP) standard. This SILAP standard was added to unlabeled murine CT26 colon cancer cell or normal murine YAMC colon epithelial cell secreted proteome samples. A multidimensional approach combining isoelectric focusing (IEF), strong cation exchange (SCX) followed by reversed phase liquid chromatography was used for extensive protein and peptide separation. A total of 614 and 929 proteins were identified from the YAMC and CT26 cell lines, with 418 proteins common to both cell lines. Twenty highly abundant differentially expressed proteins from these groups were selected for liquid chromatography-multiple reaction monitoring/mass spectrometry (LC-MRM/MS) analysis in sera. Differential secretion into the serum was observed for several proteins when Apcmin mice were compared with control mice. These findings were then confirmed by Western blot analysis.
Keywords: Colon cancer, secreted proteins, multidimensional method, biomarker, SILAC, SILAP
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and the second leading cause of cancer-related mortality in the U.S., with an estimated 153,760 new cases diagnosed and 52,180 disease related deaths in 2007.1 Early stage of diagnosis is clearly associated with an improved cure rate.2 Screening colonoscopy provides a mechanism for the early diagnosis of CRC, and, in large part due to increased utilization of this tool, the mortality from CRC has slowly but steadily declined since 1980.1 Unfortunately, screening colonoscopy remains underutilized due to the perceived and real inconvenience, potential risks and costs to patients associated with the procedure. Fecal occult blood testing is a non-invasive alternative, however, it lacks the sensitivity or specificity required for an effective screening tool.3 Development of more effective, minimally invasive screening tools, such as serum-based biomarkers, is greatly needed.
The most commonly used biomarker associated with CRC is carcinoembryonic antigen (CEA). This glycoprotein, first identified in 1965,4 is often increased in CRC. Levels are also increased in the majority of gastrointestinal malignancies, however, as well as in other cancers, such as carcinomas of the lung and cervix.5 CEA levels can also be elevated in otherwise healthy patients who are smokers,6 while remaining normal in patients with early CRC.7-9 Thus, CEA also lacks the sensitivity and specificity of a clinically useful screening test.
Developing biomarkers is extremely challenging because of the enormous complexity of biological systems, heterogeneity of human samples and lack of universal quantitative technology. In fact, the number of new biomarkers validated in the past five years has been remarkably small.10-12 MS based proteomics methods have emerged as promising approaches for discovery of diagnostic, prognostic, and therapeutic protein biomarkers. Discovery of better performing biomarkers has proven challenging despite significant advances in proteomic methodology and instrumentation. Although these advances have allowed for the identification of more and lower abundance proteins in complex biological fluids such as serum, they do not address the difficult task of determining biologically relevant candidate biomarkers. Discovery approaches that directly interrogate human serum are also confounded by the presence of proteins that vary from patient to patient as a result of differences in genetic background and environmental exposures, or differential expression of non-specific inflammatory or acute-phase proteins. Finally, traditional immunological approaches to validating biomarker candidates are severely limited by the non-availability of high quality and specific antibodies.
We present an integrated, biologically targeted MS-based discovery approach to address these specific obstacles. Central to this approach is utilization of the secreted proteome of colon derived cell lines. In recent years, study of cell line secreted proteomes has gained interest for several reasons.13 Unlike the intracellular proteome, proteins present in the secreted proteome are logical biomarker candidates for measurement in serum and other biological fluids.14-16 These proteins also play biologically important roles in modulating intercellular communication, cell adhesion, motility and invasion.17 While isolation and identification of proteins secreted by tumor cells in vivo remains difficult, study of a cancer cell line allows for isolation of these proteins, free of proteins produced by other cell types present in tumor tissue such as fibroblasts, inflammatory, or endothelial cells. Another advantage of cell lines is that quantitation can be readily performed using SILAC-based methodology.18 These labeled proteins can subsequently be used as internal standards for relative quantitation in serum, an approach we have termed SILAP standard.19
Biomarker development can be simplified by initial validation in a mouse model, which minimizes both the genetic and environmental variability found in human samples. The Apcmin mouse is a well studied model of human colon carcinogenesis. This mouse harbors a mutation in the adenomatous polyposis coli (APC) gene, a tumor suppressor mutated in 70% of sporadic colon cancers,20 and the causative germ-line mutation in familial adenomatous polyposis (FAP), an autosomal dominant hereditary colorectal cancer syndrome.21-23 In the current study, the secreted proteomes of the murine CT26 colon cancer cell line and the normal murine YAMC colon epithelial cell line were compared using SILAP standard, intact protein isoelectric focusing and two-dimensional liquid chromatography tandem mass spectrometry (IEF-2D-LC/MS/MS). Proteins differentially expressed in the CT26 secreted proteome were identified. From a list of 20 candidates, an LC-MRM/MS assay was developed to monitor 11 of these proteins. Using this assay, differential expression was found in serum from Apcmin mice compared to controls. Selected Western blot analyses were employed to independently confirm these findings.
Experimental Procedures
Chemical Reagents
The following antibodies were purchased: goat polyclonal anti-mouse cathepsin L, cystatin C, secreted phosphoprotein 1, galectin 1 and galectin 3 (all from R&D Systems, Minneapolis, MN); rabbit polyclonal anti-mouse profilin 1, mouse monoclonal anti-mouse vimentin and rabbit polyclonal anti-moue fibronectin 1 (Abcam Inc., Cambridge, MA); HRP-conjugated rabbit anti-goat (Fisher Scientific, Pittsburgh, PA) and goat anti-mouse secondary antibodies (Sigma, St. Louis, MO). All solvents were purchased from Sigma.
Apcmin mouse model
The Apcmin mouse was originally discovered in 1990.24 This mouse model was subsequently found to have a truncated Apc gene in position 850, similar to that in patients with familial adenomatous polyposis (FAP), and in other sporadic cancers. Apcmin mice rapidly develop adenomatous bowel polyps, similar to humans with germline inactivation of one Apc gene. C57BL/6J+/+ (normal, wild-type) and Apcmin mouse tissue and serum were provided by the laboratory of Dr. Anil Rustgi and the Center for Molecular Studies in Digestive and Liver Diseases, the University of Pennsylvania.
General workflow
An overview of the workflow for candidate biomarkers discovery is presented in Figure 1. The SILAP standard was created by mixing SILAC labeled conditioned media derived from CT26 colon cancer and YAMC normal colon epithelial cell lines. Equal amounts of the SILAP standard were added to unlabeled CT26 and YAMC conditioned media samples. Samples were fractionated by IEF and digested with trypsin. Resulting tryptic peptides were further fractionated by SCX chromatography and analyzed by reversed phase LC/MS/MS.
Figure 1.

General workflow for processing of conditioned media samples. Both SILAC labeled (labeled, red) and unlabeled (blue) secreted proteomes were collected from CT26 and YAMC cell lines. A SILAP standard was created by mixing SILAC labeled secreted proteomes from CT26 and YAMC at a 1:1 ratio by mass. An equal amount of the SILAP standard was then added to unlabeled CT26 and unlabeled YAMC secreted proteomes. Individual samples were separated by IEF, trypsin digested, then standard 2D-LC-MS/MS.
Cell culture and labeling
The colon cancer cell line CT26 is an N-nitroso-N-methylurethane-induced, undifferentiated colon carcinoma, cloned to generate the cell line designated CT26 WT (American Type Culture Collection (ATCC), Manassas, VA). CT26 cells were cultured in RPMI 1640 (L-glutamine and 25 mM HEPES) media supplemented with 10 % fetal bovine serum (FBS), glucose (4.5 g/L), sodium bicarbonate (1.5 g/L) and 1 mM sodium pyruvate. Cells were incubated in a humidified incubator at 37 °C and 5 % CO2. Normal murine YAMC colon epithelial cells were the kind gift of Dr. Robert H. Whitehead, Vanderbilt University Medical Center. The murine YAMC epithelial cell line was derived from the colonic mucosa of a transgenic mouse generated by the introduction of thermo labile SV40 T Ag, tsA58.25 YAMC cells were cultured in RPMI 1640 media supplemented with 5 % FBS, murine gamma interferon (5 units/mL) and ITS (insulin, transferrin and selenium) plus Premix (Sigma). YAMC cells were incubated at 33 °C, the permissive temperature for proliferation. YAMC cells cease to proliferate at 37 °C.
Metabolic labeling was performed on both CT26 and YAMC cell lines cultured in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma, St. Louis, MO) deficient in leucine and lysine. The medium was reconstituted according to the manufacturer’s instructions, sterile filtered and stored at 4 °C. [13C6,15N1]-leucine and [13C6,15N2]-lysine were obtained from Cambridge Isotope Laboratories. The powdered amino acids were dissolved in water and required amounts were added (leucine 110 mg/L, lysine 153 mg/L), corresponding to the standard composition of DMEM. Cells were grown for 6-7 passages in order to achieve > 99.0 % labeling. DMEM containing unlabeled leucine and lysine was used to culture unlabeled cells.
Preparation of SILAP standard
SILAC labeled and unlabeled CT26 and YAMC cells at approximately 80 % confluence were washed with PBS to remove serum proteins, and incubated in serum free medium. Conditioned media was collected every day (continuously for three days), pooled, filtered, aliquoted, and stored at −80 °C until needed. The pooled conditioned media was concentrated using a 3 kDa molecular weight cutoff membrane and protein concentration was determined by Coomassie Protein Assay (Pierce Scientific, Milwaukee, WI).
Protein separation by IEF and in-gel trypsin digestion
The SILAP standard was created by mixing 1 mg each of the SILAC labeled CT26 and YAMC conditioned media. 1 mg of the SILAP standard was added to 1 mg of unlabeled CT26 conditioned media and to 1 mg of unlabeled YAMC conditioned media. Protein samples were precipitated using a standard methanol/chloroform protocol. Precipitated proteins were re-suspended in DeStreak Rehydration Solution (GE/Amersham Biosciences, Piscataway, NJ) and 0.5 % immobilized pH gradient (IPG) buffer and focused on 18 cm (pI 3-10) non-linear IPG strips overnight. Each strip was then cut into eight pieces, four 3 cm pieces at the ends and four 1.5 cm pieces in the center of the IPG strip. Each piece was washed successively with 5 % and 1 % TCA, dehydrated with 90 % ACN and allowed to air dry. The approximate pI range of each of these IPG pieces was estimated as follows from information provided by the manufacturer: piece 1 (pI 3.0-4.5), piece 2 (pI 4.5-5.4), piece 3 (pI 5.4-5.7), piece 4 (pI 5.7-5.9), piece 5 (pI 5.9-6.1), piece 6 (pI 6.1-6.4), piece 7 (6.4-8.3) and piece 8 (8.3-10.0). Overnight in-strip digestion was performed for each IPG piece using sequencing grade trypsin (Promega) diluted in 50 mM ammonium bicarbonate at 37 °C. The supernatant was removed and saved. Peptide extraction was performed by adding 1 % TCA in 50 % ACN, and sonicating for 15 min. Extracted peptides were combined with the previously removed supernatant and concentrated by lyophilization.
Peptide separation by LC-SCX
The lyophilized samples were dissolved in SCX mobile phase A (25 mM ammonium formate, 25 % acetonitrile, pH 2.7). SCX chromatography was performed on a PolySulfoethyl A column (100 mm × 2.3 mm, 5 μm, 300 Å, PolyLC, The Nest Group, Inc., Southborough, MA) attached to an 1100 Series HPLC (Agilent, Santa Clara, CA). Samples were loaded for 5 min with mobile phase A, followed by a linear gradient for 45 min to 100 % mobile phase B (500 mM ammonium formate, 25 % acetonitrile, pH 6.8) at a constant flow rate of 0.1 mL/min. Thirty-three 2 min fractions were collected, pooled into 12-15 fractions, lyophilized, and stored at −80 °C awaiting further analysis. In total 102 fractions from CT26 and 105 fractions from YAMC were prepared for reversed phase LC-MS/MS analysis.
Reversed-phase capillary LC-MS/MS analysis
Lyophilized peptides were reconstituted with 0.5 % aqueous acetonitrile containing 0.1 % formic acid for reversed phase separation. A nanoflow high pressure capillary LC system (Eksigent, Dublin, CA) coupled on line to a linear ion trap Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer (LTQ-FT, Thermo Fisher, San Jose, CA) via an in-house-manufactured nanoelectrospray ionization interface was used to analyze peptide samples. The reversed phase capillary column was prepared by slurry-packing Alltech Prosphere C18-AQ, 5 μm, 100 Å into an 18 cm long, 360 μm outer diameter (od) × 75 μm internal diameter (id) fused silica capillary fritted with a polymerized solution containing potassium silicate and formamide. A trap column consisting of Alltech Prosphere C18, 10 μm, 300 Å slurry-packed into a 6 cm long, 360 μm od × 150 μm id fused silica capillary (New Objective, Woburn, MA) was also used. Mobile phases A (0.1 % formic acid in water) and B (0.1 % formic acid in ACN) were used. After loading 10 μl of peptides onto the column, the mobile phase was held at 95 % A for 20 min. A linear gradient to 70 % B was applied over 150 min. To identify the eluting peptides, the linear ion trap mass spectrometer was operated in a data-dependent MS/MS mode (m/z 300–2000) in which each full MS scan in the FT-ICR was followed by 7 MS/MS scans in the ion trap. The seven most intense precursor ions were dynamically selected in order of highest to lowest intensity and then subjected to collision-induced dissociation. The FT-ICR mass resolution was set at 50,000.
Protein identification
Raw data were submitted to Bioworks Browser version 3.2 (Thermo Fisher, San Jose, CA) and batch searched through SEQUEST™ against the NCBI RefSeq database of mouse sequences (version updated 12/06) containing 44,222 total proteins. The database was indexed using the following criteria: strict trypsin cleavage rules with up to two internal cleavage sites; differential modifications of methionine oxidation, carboxyamidomethlyation on cysteine, [13C6,15N1]-leucine and [13C6,15N2]-lysine. All peptides shorter than six amino acids were removed from the data set. The remaining SEQUEST™ output files were further processed using the Trans-Proteomic Pipeline (version 2.8, Institute for Systems Biology, Seattle, WA) for analysis and validation of peptides and proteins using PeptideProphet™ (version 3.0) and ProteinProphet™ (version 2.0), respectively. PeptideProphet™ peptide results were filtered using a minimum peptide probability score of 0.3, translating to a false discovery rate of 9.1 %. ProteinProphet™ protein results were filtered using a minimum probability score of 0.5, translating to a false discovery rate of 3.6 %. All proteins identified by a single unique peptide were eliminated. XPRESS™ software (also from the Trans-Proteomic Pipeline) was originally developed for isotope coded affinity tag (ICAT) labeling experiments,26 but is equally applicable to other differential labeling approaches such as SILAC. Starting with either the unlabeled or SILAC labeled MS/MS spectra, the program reconstructs reversed phase elution profiles for both the unlabeled and SILAC labeled precursor ions. Relative quantitation of unlabeled and SILAC labeled peptides was performed using XPRESS™ with a parent mass tolerance of 0.2 mass units and mass differences of 7.027630 mass units on leucine and 7.93217 lysine, corresponding to [13C6,15N1] leucine and [13C6,15N2] lysine. The elution areas of the unlabeled and labeled precursor ions were determined and a ratio generated. Manual validation by extracted ion monitoring was performed on differentially expressed peptides.
Protein expression analysis
The SILAP standard was used to quantify relative protein levels in secreted proteomes derived from CT26 and YAMC cell lines. The XPRESS™ ratio for proteins common to CT26 and YAMC cell lines were used and their relative expression ratio was calculated as follows. Protein level in CT26 secreted proteome relative to SILAP standard:
Protein level in YAMC secreted proteome relative to SILAP standard:
An expression ratio of 1 indicates a protein present at equal amounts in both cancer and control secreted proteomes. An expression ratio > 1 indicates a protein over-expressed, and conversely an expression ratio < 1 indicates a protein under-expressed in the cancer secreted proteome relative to normal. Normalization of XPRESS™ and Expression ratios was not performed.
Bioinformatics
The proteins identifed from YAMC and CT26 cell line secretomes were compared, and proteins common to both cell lines were used for pathway analysis. To identify pathways, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used as a reference.27 The GI accession numbers from the common proteins were used as a query to the Protein Information Resource (PIR) database (http://pir.georgetown.edu/). All mouse proteins corresponding to each GI accession number were extracted. A significant number of the GI accession numbers had multiple protein entries in the PIR database. Each protein was then used as a query to the UniProt database to extract their corresponding mouse KEGG IDs.28 The individual proteins were used to compensate for any errors in assigning of proteins to GI accession numbers, thereby providing more accurate results. Individual KEGG IDs were then searched against each pathway across the entire KEGG pathway database specific to the species Mus musculus.
Development of LC-MRM/MS assay
Twenty of the most abundant proteins identified in the CT26 secreted proteome, as estimated by means of spectral counting, were chosen for development of an MRM based assay (Table 2). All 20 of these proteins were up-regulated in CT26 compared with YAMC secreted proteome. Using peptides identified in the discovery phase, the MS/MS spectra of the unique unlabeled and stable isotope labeled peptides identified in these 20 candidates were manually inspected, one high quality unique peptide per protein was chosen, and the transition from the precursor ion to the most intense b or y fragment ion for each of these 20 peptides was identified, thus generating a set of 20 MRM transitions for the endogenous tryptic peptides and their corresponding stable isotope labeled internal standards. Theoretical m/z values for each MRM transition were confirmed using ProteinProspector® software (version 4.27, http://prospector.ucsf.edu). Reversed-phase separations were performed using an Everest RP C-18 column (Grace, Deerfield, IL) using a linear gradient (5-50%) over 80 min, using the same reversed phase LC mobile phases as described above. Analysis was performed on a high scan-rate ion trap MS (LTQ, Thermo Fisher). The LC-MRM/MS method was designed to analyze 10-12 transitions (unlabeled and SILAC labeled pairs of 5-6 peptides) simultaneously in a single analysis. Therefore each sample was injected four times to complete the 20 protein analysis in four sets. MRM transitions and retention times were validated in a sample composed of a mixture of unlabeled and SILAC labeled CT26 conditioned media.
Table 2.
Classification expression ratio of proteins common to CT26 and YAMC secreted proteomes. Proteins present at a higher concentration in CT26 have ratios > 1.
| CT26/YAMC ratio |
Number of proteins |
|---|---|
| >9.0 | 138 |
| 6.01 - 9 | 64 |
| 3.01 - 6 | 100 |
| 1.01 - 3 | 77 |
| 0.33 - 1.00 | 27 |
| 0.167 - 0.331 | 3 |
| 0.111 - 0.1671 | 2 |
| <0.110 | 7 |
Preparation of sera samples
Equal amounts of a SILAP standard (500 μg) derived from CT26 conditioned media was added to serum samples pooled from five normal mice and Apcmin mice. Based on protein concentrations, serum volumes used were 70 μL (1.67 mg protein) for normal control mice and 87 μL (1.67 mg protein) for Apcmin mice. The three most abundant serum proteins (albumin, IgG and transferrin) were then removed from both serum samples using a 100 × 4.6 mm Multiple Affinity Removal LC Column (Agilent Technologies, Santa Clara, CA) attached to a Hitachi EZChrome Elite HPLC (Hitachi HTA, San Jose, CA) according to the manufacturer’s instructions. Approximately 70 % of the serum proteins (by mass) were removed by this method, whereas a negligible amount of SILAP standard protein was removed. Samples were concentrated and buffer exchanged into 50 mM ammonium bicarbonate using 3 kDa molecular weight cutoff membranes. Protein concentrations were again determined. Based on these calculations, it was estimated that immunoaffinity processed samples were composed of approximately 500 μg serum protein and 500 μg SILAP standard.
For direct analysis, samples were digested with trypsin, desalted using C-18 SPE columns (MacroSpin™ C18 column, The Nest Group, Inc.), then analyzed by LC-MRM/MS. To improve sensitivity, trypsin digested serum samples were also separated by SCX as described above. Based on overall peptide content as estimated by UV absorbance, the ten major LC-SCX fractions were analysed further by LC-MRM/MS.
Western blot analysis of secreted proteome
Western blot analysis was performed to analyze protein expression in: CT26 and YAMC conditioned media and whole cell lysates; serum pooled from five normal and Apcmin mice; colon tissue from normal and Apcmin mice. Samples were diluted in LDS sample buffer (Invitrogen, Carlsbad, CA), then incubated at 60 °C for 10 min. 10 μg of protein from each conditioned media sample was separated on a NuPAGE 4-12 % Bis-Tris gels (NuPAGE™ Novex gels, Invitrogen). A SeeBlue™ Plus2 (Invitrogen) protein standard was used to estimate molecular weights. For validation in mouse serum, 50 μg of protein from depleted serum samples were similarly analyzed. After gel electrophoresis, samples were transferred to nitrocellulose membrane (Invitrogen) and incubated with individual primary antibodies diluted 1:250 for 1 h at room temperature. Membranes were then incubated for 45 min with the appropriate secondary horseradish peroxidase-conjugated antibody diluted 1:5,000. Protein bands were then visualized by incubating membranes with ECL Plus™ detecting reagents (Amersham Biosciences, Piscataway, NJ).
Extraction of protein from mouse colon tissues
Colon tissues from two control and two Apcmin colon adenoma bearing mice were harvested for Western blot analysis. Tissues were homogenized for 30 sec in lysis buffer (50 mM Tris pH 7, 1 mM EDTA, 1 mM EGTA and 50 mM DTT) using a Brinkman Polytron homogenizer. Tissue lysates were incubated at 4 °C for 15 min on a rocker then centrifuged at 14,000 × g for 10 min to remove debris. Tissue lysates were stored in −80 °C until use. Protein concentrations were estimated in both normal and Apcmin mouse colon tissue lysates by Coomassie Protein Assay (Pierce Scientific). 50 μg of protein was loaded on each lane and Western blot analysis performed as described above.
Results
Cell line secreted proteomes
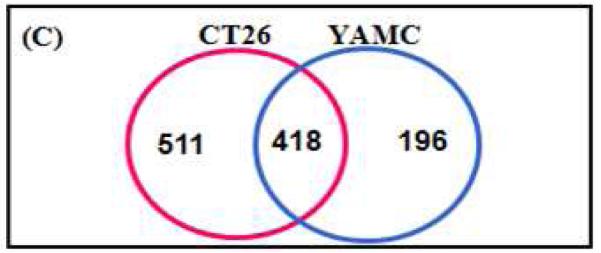
Using a SILAP standard IEF 2D-LC-MS/MS approach (Figure 1), a large number of proteins were identified in the comparison of CT26 and YAMC secreted proteomes. A total of 929 proteins were identified in CT26 and 614 proteins in YAMC secreted proteomes (Supplemental Tables 1 and 2). Taken together a total of 1,125 proteins were identified between the two proteomes, with 418 proteins common to both (Table 1, panel C). Peptides were widely distributed among the 8 IEF pieces (Table 1, panel A). Protein identifications were robust, with 3 or more unique peptides sequenced for the vast majority of proteins (Table 1, panel B).
Table 1.
A) Total number of peptides identified by IPG piece in CT26 and YAMC secreted proteomes. B) Total number of proteins identified by peptide number. C) Venn diagram of proteins identified in CT26 and YAMC secreted proteomes.
| (A) | ||
|---|---|---|
| IPG Pieces |
Total peptiles | |
| YAMC | CT26 | |
| I | 6802 | 10347 |
| II | 6566 | 17242 |
| III | 6571 | 5614 |
| IV | 5571 | 7422 |
| V | 4118 | 7641 |
| VI | 5470 | 5741 |
| VII | 4498 | 6599 |
| VIII | 2561 | 4072 |
| (B) | |||
|---|---|---|---|
| 2 peptide | ≥3 peptide | Total number of proteins |
|
| YANEC | 81 | 533 | 614 |
| CT26 | 139 | 790 | 929 |
 |
Differentially expressed proteins in CT26 versus YAMC secreted proteomes
418 proteins were found in both CT26 and YAMC secreted proteomes (Table 2). 150 of the 418 proteins quantitated (36%) are present at reasonably similar levels in both cancer and normal secreted proteomes, with cancer/normal expression ratios between 0.33 and 3. A large number of proteins are over-expressed by cancer cells, with 202 (48%) proteins present at cancer/normal expression ratios > 6. By contrast, few proteins were found under-expressed by cancer cells with only 12 proteins present at cancer/normal expression ratios < 0.167. To identify an initial set of promising candidate biomarkers for further development, two criteria were established: proteins should be over-expressed in CT26 compared with YAMC secreted proteome, and proteins should be relatively abundant in the CT26 secreted proteome to facilitate LC-MRM/MS assay development. Although the magnitude of differential expression was not used as an initial criteria for candidate selection, certainly, such an approach could be pursued in future work. Using spectral counting, 20 of the most abundant proteins present in the CT26 secreted proteome were chosen for further analysis (Table 3). All 20 proteins were over-expressed in the CT26 secreted proteome when compared to the YAMC secreted proteome. Several of these proteins, particularly cathepsin L and secreted phosphoprotein 1, were abundant in the CT26 secreted proteome, but largely absent in the YAMC secreted proteome. Extracted ion monitoring confirmed differential expression for these proteins (see Figure 2A for representative examples). The retention time of the cathepsin L peptide (ENGGLDSEESYPYEAK) was significantly different in the analysis of the CT26 secreted proteome (84.06 min) when compared with YAMC (51.94 min). This difference is most likely due the different times of analysis and variability of manually packed nanoflow C-18 columns. However, it is noteworthy that the internal standard retention times also changed in a consistent manner and the MS/MS spectra were identical (Figure 2B).
Table 3.
Relative quantitation of 20 abundant proteins by SILAP standard IEF-2D-LC/MS/MS in CT26 and YAMC secreted proteomes.
| Protein description | Expression ratio CT26/YAMC |
YAMC | CT26 | ||||
|---|---|---|---|---|---|---|---|
| total number of peptides |
xpress ratio mean |
xpress stdev |
total number of peptides |
xpress ratio mean |
xpress stdev |
||
| cathepsin L preproprotein | 88.70 | 86 | 0.1 | 0.06 | 9284 | 8.87 | 3.34 |
| lectin, galactose binding, soluble 1 | 14.37 | 384 | 0.68 | 0.08 | 1614 | 9.77 | 3.29 |
| vimentin | 24.65 | 866 | 0.17 | 0.06 | 1464 | 4.19 | 0.93 |
| enolase 1 | 15.68 | 208 | 0.22 | 0.08 | 1425 | 3.45 | 1.02 |
| cystatin C | 15.73 | 39 | 1.73 | 0.6 | 1392 | 27.22 | 13.66 |
| heat shock protein 8 | 5.13 | 444 | 0.7 | 0.35 | 1360 | 3.59 | 0.98 |
| secreted phosphoprotein 1 | 521.00 | 117 | 0.04 | 0.04 | 1181 | 20.84 | 13.24 |
| pyruvate kinase 3 | 29.11 | 185 | 0.09 | 0.03 | 1022 | 2.62 | 1.19 |
| procollagen, type I, alpha 2 | 334.00 | 31 | 0.04 | 0.02 | 979 | 13.36 | 8.38 |
| profilin 1 | 2.58 | 109 | 1.63 | 0.55 | 971 | 4.21 | 2.72 |
| procollagen C-proteinase enhancer protein | 44.53 | 131 | 0.17 | 0.09 | 915 | 7.57 | 1.97 |
| heterogeneous nuclear ribonucleoprotein A2\B1 isoform 1,2 | 3.02 | 39 | 1.77 | 0.67 | 766 | 5.34 | 2.88 |
| S100 calcium binding protein A4 | 178.67 | 54 | 0.03 | 0.02 | 755 | 5.36 | 2.36 |
| nucleobindin 1 | 21.19 | 159 | 0.21 | 0.09 | 649 | 4.45 | 1.47 |
| eukaryotic translation elongation factor 1 alpha 1 | 4.34 | 193 | 0.5 | 0.12 | 638 | 2.17 | 0.76 |
| heat shock protein 1, beta | 9.21 | 665 | 0.29 | 0.07 | 631 | 2.67 | 0.61 |
| lectin, galactoside-binding, soluble, 3 binding protein | 9.31 | 111 | 0.32 | 0.12 | 454 | 2.98 | 0.6 |
| nucleolin | 14.78 | 122 | 0.27 | 0.14 | 418 | 3.99 | 1.12 |
| fibronectin 1 | 7.23 | 197 | 1.21 | 0.43 | 406 | 8.75 | 3.7 |
| heat shock protein 1, alpha | 4.15 | 601 | 0.62 | 0.27 | 405 | 2.57 | 0.46 |
Figure 2.

Examples of extracted ion chromatograms (panel A) for peptides unique to cathepsin L (ENGGLDSEESYPYEAK), secreted phosphoprotein 1 (ESQESADQSDVIDSQASSK) and cystatin C (MLGAPEEADANEEGVR) from SILAP standard containing CT26 and YAMC secreted proteomes. CT26 compared to YAMC peptide levels are calculated by normalizing peak area of unlabeled (top) to labeled (bottom) peptide for each sample, then calculating a ratio across the two samples. The CT26/YAMC expression ratio was 337 for cathepsin L peptide ENGGLDSEESYPYEAK, 8 for secreted phosphoprotein 1 peptide ESQESADQSDVIDSQASSK, and 124 for cystatin C peptide MLGAPEEADANEEGVR. MS/MS spectra (panel B) confirming unlabeled cathepsin L peptide ENGGLDSEESYPYEAK identity in CT26 (top) and YAMC (bottom) samples.
Performance of SILAP standard LC/MRM/MS assay in mouse serum
An MRM based method was developed to monitor 20 biomarker candidates by selecting a single peptide unique to each protein (Supplemental Table 3). Unique peptides were chosen to have more than 10 amino acids, to be doubly or triply charged and to have no internal trypsin cleavage sites. The method was validated in a sample composed of a 1:1 mixture of unlabeled and SILAC labeled CT26 conditioned media. The YAMC secreted proteome was not included in the SILAP for two reasons. First, all proteins studied were over-expressed in the CT26 secreted proteome. Second, by simplifying the sample composition, we hoped to improve our ability to detect the transitions of interest. We could detect 18 of the 20 pairs of MRM transitions from this mixture. We also confirmed selectively by acquisition of a full scan MS/MS spectrum of the precursor ion of each parent peptide. SILAP standard containing normal mouse serum was then analyzed by LC-MRM/MS following immunoaffinity removal of the top three serum proteins.
Unfortunately, we were unable to consistently detect endogenous tryptic peptide- or SILAP standard-derived MRM transitions. When the SILAP standard was added to normal mouse serum samples just prior to LC-MRM/MS analysis rather than at the beginning of sample processing, no improvement was observed, suggesting that ion suppression was occurring as a result of other co-eluting endogenous constituents that were present in the serum.29,30 In order to address this problem, tryptic peptides were purified by LC-SCX prior to LC-MRM/MS analysis. This procedure made it possible to detect all 18 MRM transitions from the stable isotope labeled tryptic peptides. However, seven of the candidate endogenous peptides were below the limit of detection in both samples. For the remaining 11 endogenous tryptic peptides, relative quantitation could be performed in duplicate samples. Cystatin C, secreted phosphoprotein 1, pyruvate kinase 3, procollagen C-proteinase enhancer, nucleobindin 1, heat shock protein 1 alpha, nucleolin and fibronectin 1 were found at higher levels in Apcmin mouse sera. The chromatograms of cystatin C and fibronectin 1 are presented as an example (Figure 3). In contrast, profilin 1 and heat shock protein 8 were found at lower levels in Apcmin mouse sera, with galectin 3 levels similar between the two groups (Table 4).
Figure 3.

Examples of LC-MRM/MS chromatograms for cystatin C (A) and fibronectin 1(B) in normal and Apcmin mouse sera.
Table 4.
Relative quantitation of biomarker candidates in Apcmin (Min) and normal mouse serum using SILAP standard LC-MRM/MS. Mean ratio for each protein and ratios for each of two replicates are presented.
| Proteins | Normal Sera | Min Sera | Min/Normal Ratio |
Normal Sera I | Normal Sera II | Min Sera I | Min Sera II | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean Ratio | Mean Ratio | Light | Heavy | Ratio | Light | Heavy | Ratio | Light | Heavy | Ratio | Light | Heavy | Ratio | ||
| Cystatin C | 0.002 | 0.025 | 9.829 | 187 | 46933 | 0.004 | 60 | 59679 | 0.001 | 981 | 40571 | 0.024 | 628 | 25256 | 0.025 |
| Secreted Phosprotein 1 | 0.002 | 0.003 | 1.316 | 245 | 71620 | 0.003 | 266 | 207778 | 0.001 | 186 | 87598 | 0.002 | 1108 | 272677 | 0.004 |
| Profilin 1 | 0.305 | 0.155 | 0.508 | 1385 | 4146 | 0.334 | 1598 | 5786 | 0.276 | 1351 | 11588 | 0.117 | 939 | 4854 | 0.193 |
| Heat shock protein 8 | 0.147 | 0.086 | 0.583 | 531 | 3473 | 0.153 | 657 | 4629 | 0.142 | 457 | 6383 | 0.072 | 1231 | 12278 | 0.100 |
| Pyruvate Kinase 3 | 0.000 | 0.059 | 0.06/0 | 0 | 5369 | 0.000 | 0 | 5453 | 0.000 | 230 | 4141 | 0.056 | 164 | 2591 | 0.063 |
| Procollagen C- proteinase enhancer |
0.002 | 0.003 | 1.367 | 0 | 11561 | 0.000 | 83 | 20573 | 0.004 | 64 | 12296 | 0.005 | 14 | 45111 | 0.000 |
| Nucleobindin 1 | 0.005 | 0.022 | 4.412 | 0 | 2360 | 0.000 | 65 | 6512 | 0.010 | 212 | 8306 | 0.026 | 312 | 16851 | 0.019 |
| HSP 1, alpha | 0.000 | 0.151 | 0.15/0 | 0 | 2028 | 0.000 | 0 | 1484 | 0.000 | 592 | 4476 | 0.132 | 576 | 3400 | 0.169 |
| Lectin 3 | 0.015 | 0.018 | 1.232 | 359 | 28188 | 0.013 | 269 | 15687 | 0.017 | 229 | 17542 | 0.013 | 342 | 14388 | 0.024 |
| Nucleolin | 0.000 | 0.008 | 0.01/0 | 3051 | 0.000 | 0 | 2888 | 0.000 | 116 | 13736 | 0.008 | 94 | 12026 | 0.008 | |
| Fibronectin 1 | 1.191 | 12.227 | 10.267 | 1421 | 921 | 1.543 | 828 | 987 | 0.839 | 8415 | 754 | 11.160 | 5836 | 439 | 13.294 |
Confirmation of selected candidate biomarkers by Western blot analysis
Western blot analysis was performed to validate the in vitro and LC-MRM/MS findings. Antibodies were available for seven candidate biomarkers. For all seven proteins, over-expression in CT26 when compared to YAMC conditioned media was confirmed (Figure 4). Although these differences were also found when whole cell lysates were compared, cellular levels of cathepsin L, cystatin C, vimentin and secreted phosphoprotein 1 were much lower than in conditioned media. Four of these proteins could also be detected by Western blot analysis in normal and/or Apcmin mouse serum. While cathepsin L, cystatin C and secreted phosphoprotein 1 were present at higher levels in Apcmin mouse compared with normal mouse serum, profilin 1 was present at a lower level in Apcmin mouse serum. Levels of both the pro and mature forms of cathepsin L appeared to be increased in the CT26 secreted proteome and in the Apcmin mouse serum. The pro form was also present at greater amounts than the mature form in both conditioned media and serum. The over-expression of galectin 1, cathepsin L and fibronectin 1 were also observed in the Apcmin mice colon tissue compared to normal mice colon tissue (Figure 4C), whereas galectin 3 showed no difference in protein expression.
Figure 4.

Validation of protein expression by Western blot analysis. A) Protein expression was compared across secreted proteomes (CM) and whole cell lysates were compared for YAMC and CT26 cell lines. Pro-cathepsin L ( ) and mature cathepsin L (
) and mature cathepsin L ( ) forms are denoted. B) Protein expression was compared between pooled normal serum (NS) and Apcmin mouse serum (MS) samples. C) Protein expression in colon tissue from two normal (NT) and two Apcmin (MT) mice.
) forms are denoted. B) Protein expression was compared between pooled normal serum (NS) and Apcmin mouse serum (MS) samples. C) Protein expression in colon tissue from two normal (NT) and two Apcmin (MT) mice.
Bioinformatics analysis
Pathway analysis revealed that of the 418 proteins common to secreted proteomes of CT26 and YAMC cell lines, 171 (~41%) proteins were implicated in at least one biological pathway according to KEGG database. Most classifiable proteins were involved in metabolism. As expected for secreted proteins, none were classified as involved with transcription pathways. The most highly populated pathway was cell communication (Table 5). Other important pathways were cell motility, growth and death, and leukocyte migration. 12 proteins were implicated in various cancers. 11 of these 12 proteins were over-expressed in CT26 compared to YAMC cell line secreted proteome. Only one of the 12 proteins (catenin) was under-expressed in CT26 as compared to YAMC. Out of the 11 proteins which were over-expressed, four proteins have been implicated in colorectal cancer (Table 6). Pathway analysis stratified by differential expression was also performed, however, no specific pathways stood out (data not shown).
Table 5.
KEGG pathway classifications for candidate biomarkers identified common to CT26 and YAMC secreted proteomes.
| Pathways | GI Accession number |
|---|---|
| Metabolism | 59 |
| Genetic Information Processing | |
| Transcription | 0 |
| Translation | 18 |
| Folding, Sorting and Degradation | 23 |
| Proteasome | 16 |
| Replication and Repair | 3 |
| Environmental Information Processing | |
| Membrane Transport | 0 |
| Signal Transduction | 11 |
| MAPK Signaling pathway | 7 |
| Wnt signaling patway | 2 |
| Signaling Molecules and Interaction | 11 |
| Cellular Processes | |
| Cell Motility | 17 |
| Cell Growth and Death | 10 |
| Cell Communication | 31 |
| Focal adhesion | 18 |
| Endocrine System | 10 |
| Insulin signaling pathway | 10 |
| Immune System | 23 |
| Leukocyte transendothelial migration | 8 |
| Nervous System | 5 |
| Sensory System | 1 |
| Development | 0 |
| Behavior | 0 |
| Human Diseases | |
| Cancers | 12 |
| Immune Disorders | 5 |
| Neurodegenerative Diseases | 6 |
| Metabolic Disorders | 2 |
| Infectious Diseases | 0 |
Table 6.
Pathway analysis of candidate biomarkers implicated in cancer. Proteins in bold have been associated with colon cancer.
| Description | Expression ratio range (exact) |
Biological pathways (KEGG Database) |
|---|---|---|
| Transcription elongation factor B (SIII) polypeptide 2 | >9.001 (380) | Renal cell carcinoma |
| Ras related C3 boulinum substrate | >9.001 (13.46) | Adherens junction, Toll like receptor, ALS, Colorectal cancer, renal cell carcinoma. pancreatic cancer, MAPK signaling pathway, Wnt signaling, Axon guidance, VEGF signaling, Focal adhesion, Natural killer cell mediated toxicity, B cell receptor, Fc epsilon RI signaling |
| Platelet derived growth factor | >9.001 (90) |
Colorectal cancer, Glioma. Prostate cancer, Melanoma. MAPK signaling pathway, Calcuim signaling pathway, focal adhesion, Gap |
| Caspase 3, apoptosis related cysteine protease | >9.001 (34.27) | Neurodegenerative disease, MAPK signaling pathway, p53 signaling pathway, apoptosis, Natural killer, Alzhimers, huntington, DRPLA |
| Vcrk sarcom virus CT10 oncogene homolog | >6.001 - 9.0 (6.16 | MAPK signalling pathway, Erb signaling, Focal adhesion, actin cytoskeleton, insulin signaling pathway, renal cell carcinoma |
| Tumor rejection antigen gp96 | >9.001 (26.26) | Prostate cancer |
| Nuclear pore complex associated protein Tpr | >9.001 73.375) | Thyroid cancer |
| Vcrk sarcom virus CT10 oncogene homolog (avian like) | 3.001-6.00 (3.56) | MAPK, Erb, Focal adhesion, actin cytoskeleton, Insulin signaling pathway, renal cell carcinoma. chronic myleoid leukemia |
| Cytochrome C | 3.001 -6.00 (5.5) | p53, apoptosis, Colorectal cancer, Small cell lung cancer |
| Catenin (cadherin associated protein) | 0.33 - 0.99 (1.84) | adherens junction, tight junction, Leukocyte transendothelial |
| Calmodulin | 3.001-6.00 (3.97) | Long-term potentiation, Olfactory transduction, nsulin signaling pathway, nRH signaling pathway, melanogenesis,Huntington’s disease, Glioma. Calcium signaling pathway, Phosphatidylinositol |
Discussion
We present an integrated, biologically targeted approach to protein biomarker discovery and validation. This innovative approach is easily adaptable to studying other diseases, and addresses several major obstacles which have impeded successful protein biomarker development. By using a comparative cell line secreted proteome approach, a large number of biologically relevant candidates have been identified. Integration of a SILAP standard allows for a seamless transition from discovery in vitro to validation in serum. The same peptides used to identify the candidate biomarkers in vitro can be used to perform relative quantitation in serum by SILAP standard LC-MRM/MS. Our approach bypasses the daunting task of characterizing the entire serum proteome, an important advantage, as the vast majority of proteins present in serum are likely to be unrelated to CRC, whereas potential biomarkers are likely to be present at low levels, making direct identification difficult. Validation in a mouse model is another important aspect of our approach, allowing for initial screening of biomarker candidates against a uniform genetic and environmental background. Finally, an LC-MRM/MS approach allows for validation of biomarkers against which high quality antibodies are not readily available.
Using a SILAP standard throughout the discovery and validation phases is crucial to our approach, and offers several advantages to a standard shotgun approach. As with any method integrating a labeled internal standard, the method allows for relative quantitation of the corresponding unlabeled serum proteins, while controlling for nonspecific losses during extensive sample processing.31,32 Limiting the analysis to proteins secreted and over-expressed in colon cancer cells made it possible to exclude acute phase proteins and other abundant serum proteins, while simultaneously focusing on proteins with biological relevance to CRC. A large proportion of the proteins identified in the CT26 secreted proteome were differentially expressed, confirming the findings of other studies investigating secreted proteomes in cancer cell lines.33
To increase the number of candidate biomarkers identified in the discovery phase, IEF was integrated at the intact protein level prior to standard 2D LC-MS/MS analysis of tryptic peptides (Figure 1). IEF represents an attractive, orthogonal approach for deconvoluting complex biological samples. We have described many of its advantages in a recently published study.32 One tradeoff of multiple dimensions of separation is the exponential increase in MS data acquisition time that results from increased fractionation. In this study, the amount time required for data acquisition from a sample processed by IEF-2D-LC/MS/MS was approximately 13 days. Replicate analysis becomes impractical given these long analysis times. This problem only deepens as more layers of fractionation are added. Regardless, our IEF-2D-LC/MS/MS approach allowed for identification of 1125 proteins from both cell lines including 418 common proteins.
Pathway analysis of the common proteins demonstrates a wide variety of important cellular processes such as protein metabolic process, cell motility, cell growth and death and cell communication. Ultimately, our approach should allow for systematic validation of all 418 common proteins identified. For the initial validation, 20 proteins were chosen based on criteria to maximize the likelihood of identifying meaningful biomarkers. These proteins were abundant in the SILAP standard to facilitate MRM development, and they were over-expressed in the CT26 secreted proteome suggesting biological relevance. Eight of the proteins localized in the extracellular compartment, with the other 12 localizing to membrane bound organelles, supporting the possibility that these proteins could be released into serum for detection.
To develop a high throughput clinical assay, it was anticipated that SILAP standard containing serum samples could be analyzed directly by LC-MRM/MS. Even with immunoaffinity removal of the 3 most abundant serum proteins and simplification of the SILAP standard by excluding the YAMC secreted proteome, this was not possible because of ion suppression. However, when incorporated with initial LC-SCX separation, 18 of 20 SILAP standard peptides could be detected in mouse serum together with 11 endogenous tryptic peptides. This made it possible to conduct relative quantitation of these peptides in normal mouse and Apcmin mouse serum. Quantitation by LC-MRM/MS was reproducible and, where antibodies were available, validated by Western blot analysis. Importantly, this approach made it possible to conduct relative quantitation of proteins such as procollagen C-proteinase enhancer and nucleobindin 1 and others where high quality antibodies were not available. We took care to validate the specificity of the MRM transitions by concurrently obtaining a full MS/MS spectrum of each precursor ion. Monitoring of 2 or more peptides/protein could be performed, however, such an approach would severely reduce the number of biomarkers that could be monitored using ion trap methodology. It will be possible in the future to translate this methodology to higher sensitivity and throughput instrumentation, for example by using MRM retention time segmentation in combination with an ultra performance LC instrument coupled to a high sensitivity triple quadrupole mass spectrometer. Taken together, these innovative methods have facilitated the characterization of a large number of proteins secreted by a murine colon cancer cell line when compared with a normal murine colon epithelial cell line. Using the SILAP standard approach, it was possible to interrogate Apcmin and normal mouse serum for 18 of these proteins, a small subset of those identified. Six of the 11 proteins that could be monitored were over-expressed in Apcmin mouse serum by more than 2-fold. Future work to screen a larger number of the candidates identified will yield a large panel of proteins, and will serve as a template for translating our findings to the human disease.
A number of proteins found in this study to be over-expressed in Apcmin mouse serum have been implicated in essential processes responsible for tumor growth and spread. Secreted phosphoprotein 1 (osteopontin, OPN) is a multifunctional, secreted glycoprotein implicated in a number of malignancies including breast, stomach, lung, prostate liver and colon.34 OPN has been implicated in a variety of biological pathways crucial to tumorigenesis, including cell adhesion, chemotaxis, apoptosis, invasion, migration and anchorage-independent growth of tumor cells.35-37 OPN has been previously shown to be increased in serum of patients with CRC and other cancers compared to normal serum.34 Cathepsins are a class of globular proteases, initially described as intracellular peptide hydrolases, although several cathepsins also have extracellular functions. Many cancer cells have been found to secrete cathepsin L to degrade the components of extracellular matrices and basement membranes, thus promoting tumor invasion and metastasis.38-42 Cystatin C is a secreted member of the cystatin superfamily of cysteine protease inhibitors. By inhibiting protease activity, cystatins act to modulate extracellular matrix degradation. Increased levels of cystatin C have been found in several malignancies,43-45 and increased serum levels associated with poorer prognosis in CRC.46
Nucleolin is an abundant RNA- and protein-binding protein. Nucleolin has not been described in the literature as a serum biomarker of CRC. On the cell surface, nucleolin serves as an attachment protein for several ligands from growth factors to virus particles.47-52 Enhanced surface expression of nucleolin has been found in numerous malignancies and on endothelial cells within the tumor vasculature. Interestingly, nucleolin has been shown in CRC cells to modulate cell adhesion and spreading on fibronectin substrates.49 Fibronectin, a multifunctional glycoprotein involved in cell-matrix interactions, is best known as one of the crucial proteins involved with wound healing, however, its expression is also altered during neoplastic transformation.53
Pyruvate kinase 3 is a key enzyme involved with glycolysis and gluconeogenesis, processes often up-regulated in cancer cells. Pyruvate kinase 3 has not been previously associated with CRC, however, a recent study demonstrated that a related pyruvate kinase, M2-PK, could be detected at higher levels in stool samples from patients with large colonic polyps and CRC.54 Procollagen C-proteinase enhancer (PCPE) also has not been previously described in CRC. PCPE is an extracellular matrix glycoprotein which binds to the C-propeptide of procollagen I and acts to enhance procollagen C-proteinase activity.55 PCPE appears important for regulation of extracellular matrix, an important pathway for tumor invasion, angiogenesis and metastasis. Nucleobindin 1 (calnuc) has been described as a calcium binding protein involved with signal transduction events. A recent study demonstrates over-expression of calnuc in colon cancer tissue, and a significant minority of CRC patients with autoantibodies against calnuc.56 Heat shock protein 1 alpha (HSP90) is a molecular chaperone over-expressed in many malignancies57 and has been identified as a therapeutic target in CRC.58 This is the first study to demonstrate an association of increased serum HSP90 levels to CRC.
Only two proteins, profilin 1 and heat shock protein 8, had increased expression in the CT26 secreted proteome but decreased expression in Apcmin mouse serum, providing evidence that such in vitro modeling is a promising strategy for biomarker discovery. This likely reflects proteomic changes induced in cancer cells forced to grow in culture and differences in the genetic background of CT26 cells and Apcmin mice. One example is profilin 1, a widely expressed protein which has been found to act as a tumor suppressor. Interestingly, down-regulation of profilin 1 has been studied in breast cancer cells, and is associated with enhanced motility and invasiveness.59 No studies of profilin 1 in CRC have been published. Proteins under-expressed in disease states are potentially as valuable as their over-expressed counterparts when designing clinically useful biomarker panels.
The integrated MS-based discovery and validation approach presented here provides a workflow for identifying disease biomarkers, and more importantly, a platform for measuring a panel of disease biomarkers. Many CRC candidate biomarkers have been identified. This study has only explored a small fraction of the differentially expressed proteins identified as part of the discovery phase. Current work is focused on systematically characterizing all candidate biomarkers in serum, a process made possible by the SILAP standard and the MRM approach. Obstacles to a high throughput, clinically useful LC-MRM/MS assay remain. Direct analysis of even abundant proteins in serum is difficult without time and labor intensive sample processing. Potential solutions include more extensive immunoaffinity removal of abundant serum proteins or synthesis of heavy isotope peptide analogs for absolute quantitation. Translation of candidate biomarkers identified and validated in our mouse studies would be straightforward. Human cancer cell lines could be rapidly characterized, SILAC labeled and used as an internal standard for interrogating human serum samples. Development of a biomarker panel for the early detection of CRC would lead to an earlier stage of diagnosis, and therefore a greater chance of cure.
Synopsis.
The integrated MS-based discovery and validation approach presented here provides a workflow for identifying disease biomarkers, and more importantly, a platform for measuring a panel of disease biomarkers. Many CRC candidate biomarkers have been identified. This study has only explored a small fraction of the differentially expressed proteins identified as part of the discovery phase. Current work is focused on systematically characterizing all candidate biomarkers in serum, a process made possible by the SILAP standard and the MRM approach.

Supplementary Material
Acknowledgements
We thank Dr. Dinkar Sahal (International Center for Genetic Engineering and Biotechnology) for his insightful comments. Supported by NIH grants P30 ES013508, U54 RR023567, S10 RR019939, R01 DK056645, National Colon Cancer Research Alliance, and the Hansen Foundation.
Abbreviations
- APC
adenomatous polyposis coli
- CEA
carcinoembryonic antigen
- CRC
colorectal cancer
- IEF
isoelectric focusing
- IPG
immobilized pH gradient
- LC
liquid chromatography
- MRM
multiple reaction monitoring
- MS
mass spectrometry
- MS/MS
tandem MS
- SCX
strong cation exchange
- SILAC
stable isotope labeling by amino acids in cell culture
- SILAP
stable isotope labeled proteome
- YAMC
young adult mouse colon
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J. Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst. 2004;96(19):1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 3.Ahlquist DA. Fecal occult blood testing for colorectal cancer. Can we afford to do this? Gastroenterol. Clin. North Am. 1997;26(1):41–55. doi: 10.1016/s0889-8553(05)70282-x. [DOI] [PubMed] [Google Scholar]
- 4.GOLD P, FREEDMAN SO. Demonstration of tumor-specific antigens in human colonic carcinomata by immunological tolerance and absorption techniques. J. Exp. Med. 1965;121:439–462. doi: 10.1084/jem.121.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldenberg DM, Sharkey RM, Primus FJ. Carcinoembryonic antigen in histopathology: immunoperoxidase staining of conventional tissue sections. J. Natl. Cancer Inst. 1976;57(1):11–22. doi: 10.1093/jnci/57.1.11. [DOI] [PubMed] [Google Scholar]
- 6.Stevens DP, Mackay IR. Increased carcinoembryonic antigen in heavy cigarette smokers. Lancet. 1973;2(7840):1238–1239. doi: 10.1016/s0140-6736(73)90975-6. [DOI] [PubMed] [Google Scholar]
- 7.Carriquiry LA, Pineyro A. Should carcinoembryonic antigen be used in the management of patients with colorectal cancer? Dis. Colon Rectum. 1999;42(7):921–929. doi: 10.1007/BF02237104. [DOI] [PubMed] [Google Scholar]
- 8.Herrera MA, Chu TM, Holyoke ED. Carcinoembryonic antigen (CEA) as a prognostic and monitoring test in clinically complete resection of colorectal carcinoma. Ann Surg. 1976;183(1):5–9. doi: 10.1097/00000658-197601000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wanebo HJ, Rao B, Pinsky CM, Hoffman RG, Stearns M, Schwartz MK, Oettgen HF. Preoperative carcinoembryonic antigen level as a prognostic indicator in colorectal cancer. N. Engl. J. Med. 1978;299(9):448–451. doi: 10.1056/NEJM197808312990904. [DOI] [PubMed] [Google Scholar]
- 10.Aebersold R, Anderson L, Caprioli R, Druker B, Hartwell L, Smith R. Perspective: a program to improve protein biomarker discovery for cancer. J. Proteome. Res. 2005;4(4):1104–1109. doi: 10.1021/pr050027n. [DOI] [PubMed] [Google Scholar]
- 11.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 12.Gutman S, Kessler LG. The US Food and Drug Administration perspective on cancer biomarker development. Nat. Rev. Cancer. 2006;6(7):565–571. doi: 10.1038/nrc1911. [DOI] [PubMed] [Google Scholar]
- 13.Kulasingam V, Diamandis EP. Tissue culture-based breast cancer biomarker discovery platform. Int. J. Cancer. 2008;123(9):2007–2012. doi: 10.1002/ijc.23844. [DOI] [PubMed] [Google Scholar]
- 14.Wu CC, Chien KY, Tsang NM, Chang KP, Hao SP, Tsao CH, Chang YS, Yu JS. Cancer cell-secreted proteomes as a basis for searching potential tumor markers: nasopharyngeal carcinoma as a model. Proteomics. 2005;5(12):3173–3182. doi: 10.1002/pmic.200401133. [DOI] [PubMed] [Google Scholar]
- 15.Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, Ward RL, Hawkins NJ, Quinn DI, Russell PJ, Sutherland RL, Breit SN, Moskaluk CA, Frierson HF, Jr., Hampton GM. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc. Natl. Acad. Sci. U. S. A. 2003;100(6):3410–3415. doi: 10.1073/pnas.0530278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah SJ, Yu KH, Sangar V, Parry SI, Blair IA. Identification and Quantification of Preterm Birth Biomarkers in Human Cervicovaginal Fluid by Liquid Chromatography/Tandem Mass Spectrometry. Journal of Proteome Research. 2009 doi: 10.1021/pr8010342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mbeunkui F, Fodstad O, Pannell LK. Secretory protein enrichment and analysis: an optimized approach applied on cancer cell lines using 2D LC-MS/MS. J. Proteome. Res. 2006;5(4):899–906. doi: 10.1021/pr050375p. [DOI] [PubMed] [Google Scholar]
- 18.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 19.Yan Y, Weaver VM, Blair IA. Analysis of protein expression during oxidative stress in breast epithelial cells using a stable isotope labeled proteome internal standard. J. Proteome. Res. 2005;4(6):2007–2014. doi: 10.1021/pr050175d. [DOI] [PubMed] [Google Scholar]
- 20.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359(6392):235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 21.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 22.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 23.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 24.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247(4940):322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 25.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 1993;90(2):587–591. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat. Biotechnol. 2001;19(10):946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 2000;11(11):942–950. doi: 10.1016/S1044-0305(00)00163-X. [DOI] [PubMed] [Google Scholar]
- 30.Bonfiglio R, King RC, Olah TV, Merkle K. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass Spectrom. 1999;13(12):1175–1185. doi: 10.1002/(SICI)1097-0231(19990630)13:12<1175::AID-RCM639>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 31.Oe T, Ackermann BL, Inoue K, Berna MJ, Garner CO, Gelfanova V, Dean RA, Siemers ER, Holtzman DM, Farlow MR, Blair IA. Quantitative analysis of amyloid beta peptides in cerebrospinal fluid of Alzheimer’s disease patients by immunoaffinity purification and stable isotope dilution liquid chromatography/negative electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006;20(24):3723–3735. doi: 10.1002/rcm.2787. [DOI] [PubMed] [Google Scholar]
- 32.Yu KH, Barry CG, Austin D, Busch CM, Sangar V, Rustgi AK, Blair IA. Stable Isotope Dilution Multidimensional Liquid Chromatography-Tandem Mass Spectrometry for Pancreatic Cancer Serum Biomarker Discovery. J. Proteome. Res. 2009 doi: 10.1021/pr800904z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gronborg M, Kristiansen TZ, Iwahori A, Chang R, Reddy R, Sato N, Molina H, Jensen ON, Hruban RH, Goggins MG, Maitra A, Pandey A. Biomarker discovery from pancreatic cancer secretome using a differential proteomics approach. Mol. Cell Proteomics. 2005 doi: 10.1074/mcp.M500178-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Fedarko NS, Jain A, Karadag A, Van Eman MR, Fisher LW. Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin. Cancer Res. 2001;7(12):4060–4066. [PubMed] [Google Scholar]
- 35.Liaw L, Skinner MP, Raines EW, Ross R, Cheresh DA, Schwartz SM, Giachelli CM. The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of alpha v beta 3 in smooth muscle cell migration to osteopontin in vitro. J. Clin. Invest. 1995;95(2):713–724. doi: 10.1172/JCI117718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuck AB, Hota C, Wilson SM, Chambers AF. Osteopontin-induced migration of human mammary epithelial cells involves activation of EGF receptor and multiple signal transduction pathways. Oncogene. 2003;22(8):1198–1205. doi: 10.1038/sj.onc.1206209. [DOI] [PubMed] [Google Scholar]
- 37.Hirama M, Takahashi F, Takahashi K, Akutagawa S, Shimizu K, Soma S, Shimanuki Y, Nishio K, Fukuchi Y. Osteopontin overproduced by tumor cells acts as a potent angiogenic factor contributing to tumor growth. Cancer Lett. 2003;198(1):107–117. doi: 10.1016/s0304-3835(03)00286-6. [DOI] [PubMed] [Google Scholar]
- 38.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer. 2006;6(10):764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 39.Kuester D, Lippert H, Roessner A, Krueger S. The cathepsin family and their role in colorectal cancer. Pathol. Res. Pract. 2008;204(7):491–500. doi: 10.1016/j.prp.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Hashimoto Y, Kondo C, Kojima T, Nagata H, Moriyama A, Hayakawa T, Katunuma N. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother. Radiopharm. 2006;21(3):217–224. doi: 10.1089/cbr.2006.21.217. [DOI] [PubMed] [Google Scholar]
- 41.Adenis A, Huet G, Zerimech F, Hecquet B, Balduyck M, Peyrat JP. Cathepsin B, L, and D activities in colorectal carcinomas: relationship with clinico-pathological parameters. Cancer Lett. 1995;96(2):267–275. doi: 10.1016/0304-3835(95)03930-u. [DOI] [PubMed] [Google Scholar]
- 42.Chauhan SS, Goldstein LJ, Gottesman MM. Expression of cathepsin L in human tumors. Cancer Res. 1991;51(5):1478–1481. [PubMed] [Google Scholar]
- 43.Kos J, Stabuc B, Cimerman N, Brunner N. Serum cystatin C, a new marker of glomerular filtration rate, is increased during malignant progression. Clin. Chem. 1998;44(12):2556–2557. [PubMed] [Google Scholar]
- 44.Kos J, Stabuc B, Schweiger A, Krasovec M, Cimerman N, Kopitar-Jerala N, Vrhovec I. Cathepsins B, H, and L and their inhibitors stefin A and cystatin C in sera of melanoma patients. Clin. Cancer Res. 1997;3(10):1815–1822. [PubMed] [Google Scholar]
- 45.Nakai K, Kikuchi M, Fujimoto K, Kaneko Y, Omori S, Nakai K, Suwabe A. Serum levels of cystatin C in patients with malignancy. Clin. Exp. Nephrol. 2008;12(2):132–139. doi: 10.1007/s10157-008-0043-8. [DOI] [PubMed] [Google Scholar]
- 46.Kos J, Krasovec M, Cimerman N, Nielsen HJ, Christensen IJ, Brunner N. Cysteine proteinase inhibitors stefin A, stefin B, and cystatin C in sera from patients with colorectal cancer: relation to prognosis. Clin. Cancer Res. 2000;6(2):505–511. [PubMed] [Google Scholar]
- 47.Ginisty H, Sicard H, Roger B, Bouvet P. Structure and functions of nucleolin. J. Cell Sci. 1999;112(Pt 6):761–772. doi: 10.1242/jcs.112.6.761. [DOI] [PubMed] [Google Scholar]
- 48.Chen CM, Chiang SY, Yeh NH. Increased stability of nucleolin in proliferating cells by inhibition of its self-cleaving activity. J. Biol. Chem. 1991;266(12):7754–7758. [PubMed] [Google Scholar]
- 49.Reyes-Reyes EM, Akiyama SK. Cell-surface nucleolin is a signal transducing P-selectin binding protein for human colon carcinoma cells. Exp. Cell Res. 2008;314(1112):2212–2223. doi: 10.1016/j.yexcr.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harms G, Kraft R, Grelle G, Volz B, Dernedde J, Tauber R. Identification of nucleolin as a new L-selectin ligand. Biochem. J. 2001;360(Pt 3):531–538. doi: 10.1042/0264-6021:3600531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Semenkovich CF, Ostlund RE, Jr., Olson MO, Yang JW. A protein partially expressed on the surface of HepG2 cells that binds lipoproteins specifically is nucleolin. Biochemistry. 1990;29(41):9708–9713. doi: 10.1021/bi00493a028. [DOI] [PubMed] [Google Scholar]
- 52.Hovanessian AG. Midkine, a cytokine that inhibits HIV infection by binding to the cell surface expressed nucleolin. Cell Res. 2006;16(2):174–181. doi: 10.1038/sj.cr.7310024. [DOI] [PubMed] [Google Scholar]
- 53.Kaspar M, Zardi L, Neri D. Fibronectin as target for tumor therapy. Int. J. Cancer. 2006;118(6):1331–1339. doi: 10.1002/ijc.21677. [DOI] [PubMed] [Google Scholar]
- 54.Koss K, Maxton D, Jankowski JA. Faecal dimeric M2 pyruvate kinase in colorectal cancer and polyps correlates with tumour staging and surgical intervention. Colorectal Dis. 2008;10(3):244–248. doi: 10.1111/j.1463-1318.2007.01334.x. [DOI] [PubMed] [Google Scholar]
- 55.Ricard-Blum S, Bernocco S, Font B, Moali C, Eichenberger D, Farjanel J, Burchardt ER, van der RM, Kessler E, Hulmes DJ. Interaction properties of the procollagen C-proteinase enhancer protein shed light on the mechanism of stimulation of BMP-1. J. Biol. Chem. 2002;277(37):33864–33869. doi: 10.1074/jbc.M205018200. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, Lin P, Qiu S, Peng XX, Looi K, Farquhar MG, Zhang JY. Autoantibodies to Ca2+ binding protein Calnuc is a potential marker in colon cancer detection. Int. J. Oncol. 2007;30(5):1137–1144. [PubMed] [Google Scholar]
- 57.Gooljarsingh LT, Fernandes C, Yan K, Zhang H, Grooms M, Johanson K, Sinnamon RH, Kirkpatrick RB, Kerrigan J, Lewis T, Arnone M, King AJ, Lai Z, Copeland RA, Tummino PJ. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc. Natl. Acad. Sci. U. S. A. 2006;103(20):7625–7630. doi: 10.1073/pnas.0602650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moser C, Lang SA, Kainz S, Gaumann A, Fichtner-Feigl S, Koehl GE, Schlitt HJ, Geissler EK, Stoeltzing O. Blocking heat shock protein-90 inhibits the invasive properties and hepatic growth of human colon cancer cells and improves the efficacy of oxaliplatin in p53-deficient colon cancer tumors in vivo. Mol. Cancer Ther. 2007;6(11):2868–2878. doi: 10.1158/1535-7163.MCT-07-0410. [DOI] [PubMed] [Google Scholar]
- 59.Zou L, Jaramillo M, Whaley D, Wells A, Panchapakesa V, Das T, Roy P. Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br. J. Cancer. 2007;97(10):1361–1371. doi: 10.1038/sj.bjc.6604038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.