

Summary
Epidermal cell migration is critical for restoration of tissue structure and function after damage [1]. However, the mechanisms by which differentiated cells neighboring the wound sense the wound and assume a motile phenotype remain unclear. Here we show that Pvr, a receptor tyrosine kinase (RTK) related to platelet derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) receptors, and one of its ligands, Pvf1, are required for epidermal wound closure. Morphological comparison of wound-edge cells lacking Pvr or the Jun N-terminal kinase (JNK) signaling pathway previously implicated in larval wound closure [2] suggests that Pvr signaling leads wound margin epidermal cells to extend actin-based cell processes into the wound gap while JNK mediates transient dedifferentiation of cells at the wound margin. Genetic epistasis experiments reinforce the conclusion that the JNK and Pvr signaling pathways act in parallel. Tissue-specific knockdown and rescue experiments suggest that epidermally-derived Pvf1 may be sequestered in the blood and that tissue damage exposes blood-borne Pvf1 to Pvr receptors on wound-edge epidermal cells and initiates the extension of cell processes into the wound gap. These results uncover a novel mechanism of sensing tissue damage and suggest that PDGF/VEGF ligands and receptors may have a conserved autocrine role in epidermal wound closure.
Results and Discussion
Pvr is Required for Epidermal Wound Closure
A number of RTK ligands and receptors have been implicated in vertebrate wound closure [3-6]. To identify the signals that initiate epidermal wound closure, we tested homologs of RTK candidate genes that had been implicated in diverse aspects of vertebrate wound closure using a larval Drosophila wound healing assay. We used a larval epidermal Gal4 driver (Figure S1 available online demonstrates tissue specificity of this and other Gal4 drivers used in this study) to drive expression of UAS-RNAi or UAS-dominant-negative versions of RTKs implicated in epidermal [4, 5] or mesenchymal and endothelial responses to wounding [7-9]. Surprisingly, interference with EGF or FGF receptor signaling had no effect on epidermal morphology or wound closure (Figure 1A and data not shown). By contrast, expression of UAS-PvrRNAi, led to a near-total block of wound closure (Figure 1A and 1B) similar to that observed upon inhibition of epidermal JNK signaling (Figure 1A). Simultaneous overexpression of the full-length Pvr cDNA rescued the wound closure defect of UAS-PvrRNAi (Figure 1A) and suggested the wound closure defect was not due to off-target effects, as did the wound closure defect of an RNAi-independent hypomorphic allele of Pvr, Pvrc02859 (Figure 1C).
Figure 1.
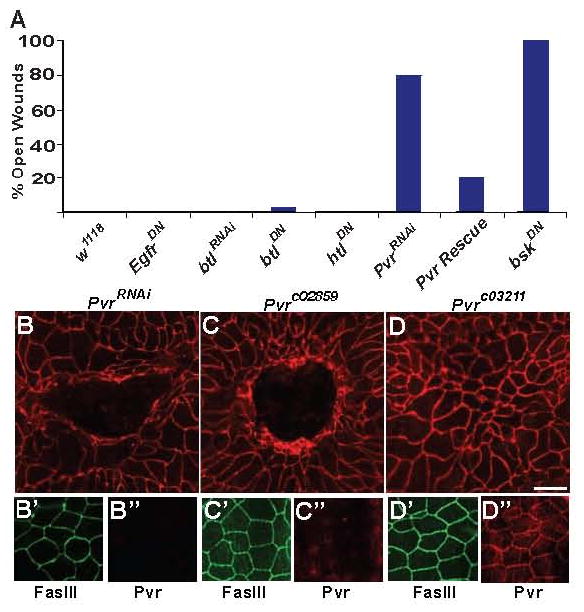
Pvr is an Epidermal Membrane Protein Required for Wound Closure (A) Percentage of larvae of each genotype that showed a defect in epidermal wound closure when knocked down via UAS-RNAi or a UAS-dominant negative transgene. N = 30. (B-D) Dissected epidermal wholemounts immunostained with anti-Fasciclin III (top panels, red) to label membranes and reveal wound architecture 24 hours post-wounding. (B) Larva expressing epidermal UAS-PvrRNAi. An open wound persists. (B′-B″) An unwounded larva of the same genotype expresses epidermal Fasciclin III (B′, green) but not Pvr (b″, blue). (C) Larva homozygous for Pvrc02859. An open wound persists. (C′-C″) An unwounded larva of the same genotype expresses epidermal Fasciclin III (C, green) but not Pvr (C″, blue). (D) Larva homozygous for Pvrc03211. Wound closure is normal. (D′-D″) An unwounded larva of the same genotype expresses epidermal Fasciclin III (D′, green) and Pvr (D″, blue). Scale bar for (B-D) is 100 μm.
Pvr is an Epidermal Membrane Protein Whose Expression is Regulated by Wounding and is Greatly Reduced by Expression of UAS-PvrRNAi
To look at Pvr expression and activation we immunostained dissected larval wholemounts with anti-Pvr [10] or anti-diphospho-ERK antibodies and found that Pvr protein is localized to the plasma membranes outlining epidermal cell-cell contacts, similar to other epidermal membrane markers (Figure S2). Surprisingly, UAS-PvrRNAi expression did not affect wound-induced appearance of dp-ERK staining, a common readout of RTK activation (Figure S3). Importantly, epidermal expression of UAS-PvrRNAi resulted in a complete knockdown of Pvr protein in the epidermal cell membranes with no effect on an independent membrane marker, Fasciclin III (Figure 1B′ and 1B″), or on Pvr expression in other tissues not affected by the larval epidermal Gal4 driver (data not shown). Pvr protein is also absent from epidermal membranes of larvae homozygous for the Pvrc02859 allele (Figure 1C′ and 1C″) but is retained in larvae bearing the silent Pvrc03211 allele that does not affect wound closure (Figure 1D′ and 1D″). To determine the distribution of Pvr protein along the apico-basal axis we used confocal microscopy to view wholemounts of dissected unwounded larvae expressing a Neuroglian-GFP fusion protein (Nrg-GFP) and co-stained for the apical marker DE-Cadherin (Figure S2). Both Nrg-GFP and Pvr were more broadly localized along the apico-basal axis than DE-Cadherin (Figure S2). Pvr protein levels 4 hours post wounding increased along epidermal cell borders proximal to the wound (Figure S2) suggesting that transcriptional upregulation or increased translation may be important for Pvr to promote efficient healing. This increase in Pvr levels was not dependent on JNK signaling as it persisted in larvae expressing a UAS-bskRNAi transgene within the epidermal sheet (Figure S2).
The Pvr and JNK Signaling Pathways Control Distinct Cellular Aspects of Wound Closure
The JNK signaling pathway is also required for larval wound closure[2] and a recent study of thorax closure in Drosophila suggested that Pvr can act upstream of JNK[11]. To understand the specific functions of JNK and Pvr signaling in larval wound closure, we analyzed the morphology of wounded larvae expressing UAS-bskDN and UAS-PvrRNAi by transmission electron microscopy (TEM) and by wholemount staining for the actin cytoskeleton. In control larvae, leading edge cells detach from the overlying cuticle and extend long thin processes that appear to use the debris in the wound gap as a substrate for migration (Figure 2A and 2B). These processes are initially short (Figure 2B) but later reach lengths of over 75 μm (Figure 2A and data not shown). They appear to be actin-based, since anti-GFP staining of control larvae expressing a UAS-actin-GFP transgene[12] within the larval epidermis exhibited numerous actin-based protrusions extending into the wound gap (Figure S4). While extending processes, the epidermal cells cease secreting apical cuticle (Figure 2B). However, once closure is complete they secrete a new cuticle that traps much of the debris in the original wound gap (Figure 2C).
Figure 2.
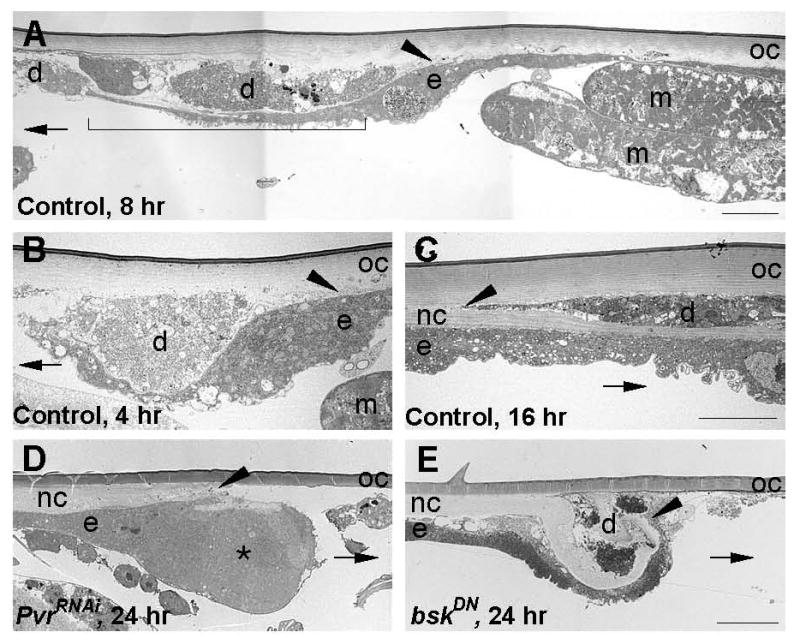
The JNK and Pvr Signaling Pathways Control Distinct Cellular Aspects of Wound Closure (A-E) TEM of transverse sections of epidermal wholemounts of pinch wounded larvae. Control larvae (A-C) contained either w; Pxn-Gal, UAS-nlacZ (A-B) or w;; A58-Gal4, UAS-nlacZ to mark the wound site for sectioning. (A) Control larva, 8 hr post-wounding. Arrowhead, point of detachment from overlying cuticle. Bracket, 50 μm long thin process extending underneath cellular debris and into the wound gap. (B) Control larva, 4 hr post-wounding. Arrowhead, as in a. Note close apposition of process and wound site debris. (C) Control larva, 16 hr post-wounding. A new cuticle traps wound-site debris between itself and the old cuticle. Arrowhead, point of divergence between new and old cuticles. (D) Larva expressing UAS-PvrRNAi in the epidermis. Asterisk, cytoplasmic bulge of leading edge cell. Arrowhead, as in a. (E) Larva expressing UAS-bskDN in the epidermis. Arrowhead, terminus of cell process that continues to secrete cuticle. In all panels: d, wound site debris; e, epidermis; m, muscle; nc, new cuticle; oc, old cuticle; arrow, direction of original wound gap. All bars 10 μm. Bar in C is for B and C and bar in E is for D and E.
In larvae expressing UAS-PvrRNAi, the leading edge cell forms a large cytoplasmic bulge where it detaches from the overlying cuticle (Figure 2D), indicating that it cannot extend a normal thin process into the wound gap. Additionally, labeling of the actin cytoskeleton in UAS-PvrRNAi- and UAS-actin-GFP- expressing larvae showed a marked decrease in wound-margin actin levels and in the density of actin-based protrusions extending from the wound edge (Figure S4). In contrast, larvae expressing UAS-bskDN or UAS-bskRNAi marginal cells extend a thin process that attempts to enter the wound gap, but this process continues to secrete cuticle, often causing it to double back towards the epidermal sheet (Figure 2E and data not shown). At the level of actin visualization, larvae expressing UAS-bskRNAi showed a brighter concentration of actin at the wound edge and did exhibit actin-based protrusions (Figure S4). This closer analysis of the loss-of-function phenotypes of Pvr and bsk indicates that these signaling pathways likely act in parallel to control distinct cellular facets of epidermal wound closure. This conclusion is strengthened by two other experimental approaches. First, genetic reporters of JNK pathway activity[2, 13, 14] are activated normally in the wounded UAS-PvrRNAi-expressing epidermis (Figure S5). Second, gain-of-function alleles of both pathways, also lead to distinct cellular phenotypes when conditionally expressed in the larval epidermis (Figure S6).
Epidermally-Produced Pvf1 Ligand is Necessary for Wound Closure
We next sought to determine the relevant ligand for Pvr during wound closure. Three genes encode secreted PDGF/VEGF-like ligands in the Drosophila genome: Pvf1, Pvf2, and Pvf3[15, 16]. Of these, only Pvf1 has been shown to physically interact with Pvr[16] and to control migration of epithelial cells[10, 16]. More than 90% of heterozygous Pvf1null/+ female larvae healed wounds properly (Figure 3A and 3D) while hemizygous Pvf1null/Y male larvae exhibited a fully penetrant wound closure defect (Figure 3B and 3D). By contrast, larvae homozygous for hypomorphic loss of function alleles of Pvf2 and Pvf3 (Pvf2c06947 and Pvf309531) showed no defects in wound closure (Figure 3D).
Figure 3.
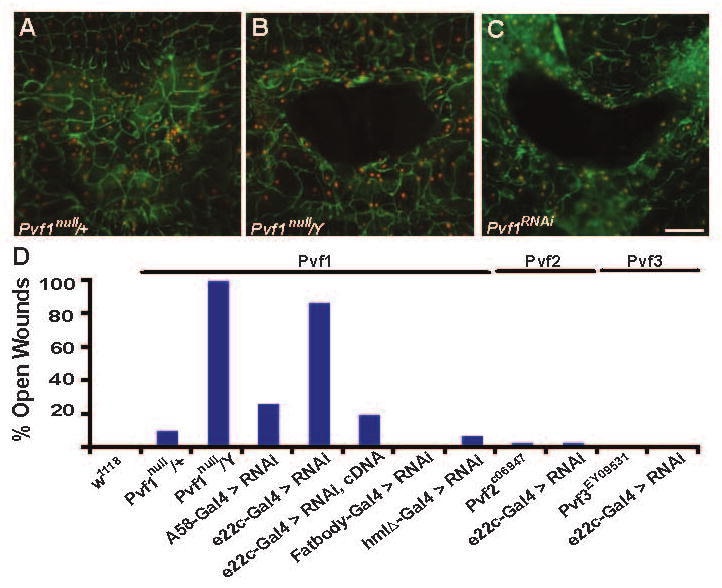
Epidermally-Produced Pvf1 Ligand is Required for Wound Closure (A-C) w, UAS-dsRed2Nuc, A58-Gal4 heterozygous larvae were pinch-wounded, dissected, fixed and immunostained for Fasciclin III (green) 24 hours post-wounding. Red, epidermal nuclei. (A) Female larva heterozygous for Pvf1null. Wound closure is normal. (B) Male larva hemizygous for Pvf1null. An open wound persists. (C) Larva in which A58-Gal4 drives pan-epidermal expression of UAS-Pvf1RNAi. An open wound persists. (D) Percentage of larvae of each genotype that showed a defect in epidermal wound closure. Larvae mutant for Pvf1 or expressing UAS-Pvf1RNAi within the epidermis show defects in wound closure, while larvae mutant for Pvf2 or expressing UAS-Pvf2RNAi or larvae mutant for Pvf3 or expressing UAS-Pvf3RNAi show no defects in wound closure. By contrast, fat body or blood cell expression of UAS-Pvf1RNAi does not perturb wound closure (middle). N = 30. Scale bar in C for A-C, 100 μm.
To test if the epidermis is the cellular source of the Pvf1 protein required for closure, flies bearing a UAS-Pvf1RNAi transgene[17] were crossed to flies bearing a larval epidermal Gal4 driver and progeny larvae were wounded. 23% of larvae expressing the UAS-Pvf1RNAi transgene via the larval epidermal A58-Gal4 driver exhibited open wounds (Figure 3C and 3D). To increase the duration of Pvf1 knockdown, we crossed UAS-Pvf1RNAi flies to flies carrying the slightly weaker e22c-Gal4 driver (Figure S1) which begins driving expression in epidermal cells during embryogenesis [18]. Earlier knockdown increased the percent of open wounds to 87% (Figure 3D) suggesting that it is primarily epidermal Pvf1 produced prior to wounding that acts during wound closure. That Pvf1 might be the only VEGF-like ligand required for wound closure is further supported by the observation that epidermal expression of UAS-Pvf2RNAi or UAS-Pvf3RNAi transgenes via the e22c-Gal4 driver did not perturb wound closure (Figure 3D).
Epidermal Overexpression of Pvf1 Perturbs Wound Closure
Pvf1 acts as a chemoattractant for embryonic blood cells and migrating border cells during Drosophila oogenesis[10, 15, 16]. If Pvf1 functions as a chemoattractant for leading edge cells, we reasoned that uniform epidermal expression of this ligand should interfere with the local concentration gradient formed after wounding and disrupt closure. Consistent with this, we found that only larvae overexpressing Pvf1 (but not larvae overexpressing Pvf2 or Pvf3) in the epidermis exhibited defective wound closure (Figure 4B and 4G) and that epidermal overexpression of Pvf1 in a Pvf1null mutant background could not rescue the null mutant wound closure defect (Figure 4A, 4C, and 4G). Paradoxically, pan-epidermal over expression of activated or wild type Pvr was lethal. However, we also found that lowering the level of Pvf1 over expression resulted in a partial rescue of the Pvf1null mutant wound closure defect (Figure S7) suggesting that interference with wound closure is a function of the level of Pvf1 expression. Taken together, these results argue that Pvf1 normally activates Pvr only at the wound edge and that endogenous Pvf1 produced by the unwounded epidermis is efficiently sequestered from Pvr. These results further suggest that Pvf2 and Pvf3 are not capable of activating Pvr within the epidermal sheet.
Figure 4.
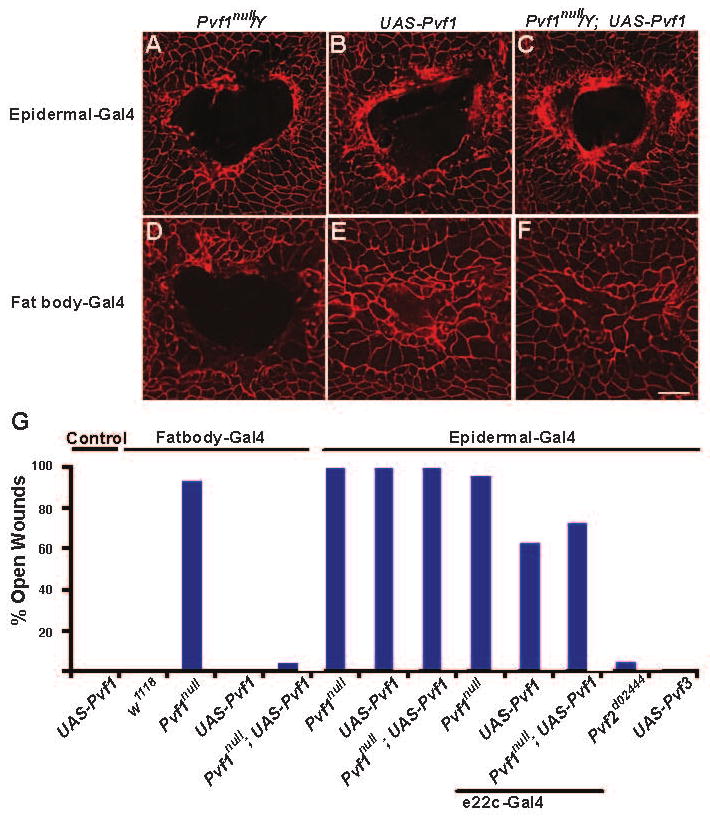
The Proper Spatial Presentation of Pvf1 is Required for Wound Closure (A-F) Dissected epidermal wholemounts immunostained with anti-Fasciclin III (red). (A) Pvf1null/Y male larvae with A58-Gal4. Open wound. (B) Larva in which A58-Gal4 drives pan-epidermal expression of UAS-Pvf1. Open wound. (C) Larva in which A58-Gal4 drives pan-epidermal expression of UAS-Pvf1 in a Pvf1null mutant background. Ectopic Pvf1 expression in the epidermis fails to rescue the wound closure defect of Pvf1null. (D) Pvf1null/Y male larvae with Fat body-Gal4. Open wound. (E) Larva in which Fat body-Gal4 drives expression of UAS-Pvf1. Closed wound. (F) Expression of UAS-Pvf1 via Fat body Gal4 in a Pvf1null mutant background. Ectopic Pvf1 expression rescues the wound closure defect of Pvf1null. (G) Percentage of larvae of each genotype that showed a defect in wound closure. Expression of UAS-Pvf1RNAi via epidermal A58-Gal4 completely blocks wound closure, expression from the weaker e22c-Gal4 driver gives a partial block, and expression from Fatbody-Gal4 rescues the wound closure defect of the Pvf1null mutant. By contrast, epidermal overexpression of Pvf2 (Pvf2d02444) or Pvf3 (UAS-Pvf3) does not block wound closure. N = 30. Scale bar in F for A-F, 100 μm.
Systemic Expression of Pvf1 Rescues the Pvf1null Mutant Wound Closure Defect
Since vertebrate VEGF and PDGF ligands can be released into circulating blood[19] we hypothesized that epidermal cells might secrete Pvf1 across the basal lamina and into the underlying hemolymph (blood) in order to sequester the ligand from epidermal Pvr. We further reasoned that if Pvf1 were a blood protein, then we might be able to rescue the wound closure defect of Pvf1null mutant larvae by expressing Pvf1 in the fat body/oenocytes, tissues that normally produce larval serum proteins[20] and are separated from the epidermis by a basal lamina and the blood filling the open body cavity. Indeed, we found that overexpression of UAS-Pvf1 via a fat body/oenocyte-specific Gal4 driver (Figure S1) did not interfere with normal wound closure (Figure 4E and 4G) but could rescue the wound closure defect of Pvf1null mutant larvae (Figure 4F and 4G). The lack of a wound closure defect upon expressing UAS-Pvf1RNAi via the same fat body/oenocyte driver (Figure 3D), coupled with the nearly fully penetrant wound closure defect observed upon epidermal expression of UAS-Pvf1RNAi (Figure 3D), suggests that the fat body and oenocytes do not normally contribute to the pool of Pvf1 used during normal wound closure. Similar rescue experiments with a blood cell-specific Gal4 driver, hmlΔ-Gal4 (Figure S1), showed that producing Pvf1 from these cells, which circulate within the larval body cavity, did not interfere with wound closure in control larvae but could also rescue the wound closure defect of Pvf1null mutant larvae (Figure S8). As with the fat body, blood cell-specific expression of UAS-Pvf1RNAi does not block epidermal wound closure (Figure 3D). Taken together, these results suggest that although the epidermis is the tissue source of Pvf1 during normal wound closure, ectopically producing this protein in the blood can restore a functional wound closure response in a Pvf1null mutant background.
A Working Model for How the Larval Epidermis Senses and Responds to Tissue Damage
Pvr is required for diverse developmentally-programmed cell migrations, including blood cell migrations during embryogenesis[15, 21], thorax closure during metamorphosis[11], and border cell migration during oogenesis[10, 16]. The latter migration is arguably the most intensively studied, with oocyte-expressed Pvf1 protein proposed to serve as a cellular source of chemoattractant for Pvr-expressing border cells migrating in a cluster from the anterior follicular epithelium. In border cells, Pvr and the EGF receptor (DER) are not required for formation of actin-based protrusions but rather act together to ensure directionality of the migrating cell cluster[22]. Here we implicate Pvr and Pvf1 in a physiologically-induced cell migration and propose a working model for how Drosophila larvae sense damage to the barrier epithelium in the absence of an epidermal source of ligand within the wound gap (Figure 5).
Figure 5.
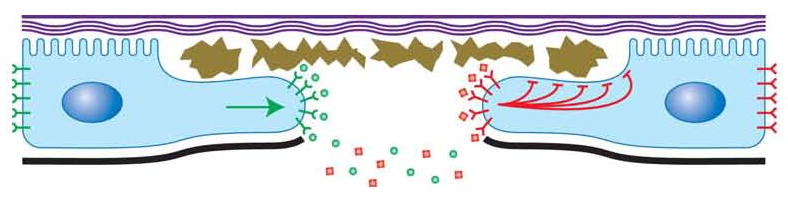
Model of Pvr and JNK Signaling in Larval Epidermal Wound Closure Pvr signaling (green, left side of wound) controls cell process extension into the debris-filled (brown) wound gap. Hemolymph-borne Pvf1 (green circles) originally produced by the epidermis (blue cells) binds to exposed Pvr receptors (green glyphs) at the wound edge to activate signaling. JNK signaling (red, right side of wound) is activated through an as yet undiscovered ligand (red diamonds) and receptor (red glyphs) or mechanical signal. Once JNK is activated it suppresses secretion of and attachment to cuticle (purple) near the wound gap. Thick black line, basal lamina. At a real wound both pathways would be active at all wound margins. See text for details.
The unwounded epidermal sheet expresses both Pvr, which is localized to lateral epidermal cell-cell contacts, and, based on our tissue-specific RNAi experiments, the Pvr ligand, Pvf1, which we propose is segregated from Pvr by secretion across the basal lamina and into the blood filling the open body cavity. When tissue injury creates a gap in the epidermal sheet and underlying basal lamina, we envision a number of events ensuing. First, Pvr receptors on lateral epidermal membranes facing the wound gap would now be exposed to blood-borne Pvf1 ligand and could thus be activated. Subsequent to ligand binding, we infer that the actin polymerization machinery within the cell is locally activated so that the cell can extend processes in the direction of the wound gap. Since the basal lamina underlying the rear of the cell remains intact (data not shown), Pvr receptors would only be activated on the side of the cell facing the wound gap and this could elegantly explain how epidermal cells polarize and extend processes only in this direction, regardless of which side of the wound they occupy. The requirement of Pvr for actin-based protrusion observed here is different from its shared role with DER in border cell migration[22] but this may simply be due to cell type specificity and differences in gene expression or activation of as yet unidentified Pvr downstream signaling components.
Alternative models are conceivable. For example, Pvf1 might be produced in an inactive form by undamaged epidermal cells with wounding somehow locally activating the ligand. However, this possibility would not well explain the data that ectopic overexpression of the ligand in the epidermis can block wound closure and that overexpression of wild-type Pvr in the epidermal sheet is lethal. The latter observation in particular suggests that Pvf1 can activate Pvr even in the absence of wounding. Thus, we favor our current model as it plausibly reconciles the ability of systemic Pvf1 to rescue the wound closure defect of Pvf1null mutants with the fact that the epidermis is the tissue source of functional Pvf1 ligand. We look forward to pursuing the testable predictions raised by our working model and other possible alternatives.
Supplementary Material
Acknowledgments
We thank Andreas Bergmann, Georg Halder, Randy Johnson, and members of the Galko laboratory for comments, Kenn Dunner of the UT-MDACC high-resolution electron microscopy facility for assistance with TEM, Leisa McCord for assistance with graphics, Anthony D'Amelio for assistance with statistics, Hank Adams for assistance with confocal microscopy, Katja Bruckner, Jocelyn McDonald, Mark Krasnow, and Benny Shilo for Pvr/Pvf1 fly stocks and antibodies, the Bloomington Drosophila Stock Center for other fly strains, and the Developmental Studies Hybridoma Bank for antibodies. YWu was supported by NIH predoctoral training grant T32-HD07325-16 and ARB was supported by a cancer research fellowship from the American Legion Auxiliary and American Heart Association grant 0730258N to MJG. The MD Anderson TEM facility was supported by a Cancer Center Core Grant (CA 16672). This work was supported by UT MDACC startup funds, a UT MDACC institutional research grant, and NIH 1 R01GM083031-01 to MJG. The authors declare no competing financial interests.
Footnotes
Supplemental Data: Supplemental Data include seven figures, the Experimental Procedures, and Supplemental Discussion and can be found with this article online at http://www.current-biology.com/supplemental.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martin P. Wound healing--aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 2.Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2004;2:E239. doi: 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, Wisdom RM, Johnson RS. c-Jun Is Essential for Organization of the Epidermal Leading Edge. Developmental Cell. 2003;4:865–877. doi: 10.1016/s1534-5807(03)00159-x. [DOI] [PubMed] [Google Scholar]
- 4.Shirakata Y, Kimura R, Nanba D, Iwamoto R, Tokumaru S, Morimoto C, Yokota K, Nakamura M, Sayama K, Mekada E, et al. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. J Cell Sci. 2005;118:2363–2370. doi: 10.1242/jcs.02346. [DOI] [PubMed] [Google Scholar]
- 5.Werner S, Smola H, Liao X, Longaker MT, Krieg T, Hofschneider PH, Williams LT. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science. 1994;266:819–822. doi: 10.1126/science.7973639. [DOI] [PubMed] [Google Scholar]
- 6.Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177:151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 8.Gao Z, Sasaoka T, Fujimori T, Oya T, Ishii Y, Sabit H, Kawaguchi M, Kurotaki Y, Naito M, Wada T, et al. Deletion of the PDGFR-beta gene affects key fibroblast functions important for wound healing. J Biol Chem. 2005;280:9375–9389. doi: 10.1074/jbc.M413081200. [DOI] [PubMed] [Google Scholar]
- 9.Rossiter H, Barresi C, Pammer J, Rendl M, Haigh J, Wagner EF, Tschachler E. Loss of vascular endothelial growth factor a activity in murine epidermal keratinocytes delays wound healing and inhibits tumor formation. Cancer Res. 2004;64:3508–3516. doi: 10.1158/0008-5472.CAN-03-2581. [DOI] [PubMed] [Google Scholar]
- 10.McDonald JA, Pinheiro EM, Montell DJ. PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development. 2003;130:3469–3478. doi: 10.1242/dev.00574. [DOI] [PubMed] [Google Scholar]
- 11.Ishimaru S, Ueda R, Hinohara Y, Ohtani M, Hanafusa H. PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. Embo J. 2004;23:3984–3994. doi: 10.1038/sj.emboj.7600417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verkhusha VV, Tsukita S, Oda H. Actin dynamics in lamellipodia of migrating border cells in the Drosophila ovary revealed by a GFP-actin fusion protein. FEBS Lett. 1999;445:395–401. doi: 10.1016/s0014-5793(99)00124-6. [DOI] [PubMed] [Google Scholar]
- 13.Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12:557–570. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramet M, Lanot R, Zachary D, Manfruelli P. JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol. 2002;241:145–156. doi: 10.1006/dbio.2001.0502. [DOI] [PubMed] [Google Scholar]
- 15.Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, Krasnow MA. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell. 2002;108:865–876. doi: 10.1016/s0092-8674(02)00676-1. [DOI] [PubMed] [Google Scholar]
- 16.Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- 17.Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence PA, Bodmer R, Vincent JP. Segmental patterning of heart precursors in Drosophila. Development. 1995;121:4303–4308. doi: 10.1242/dev.121.12.4303. [DOI] [PubMed] [Google Scholar]
- 19.Wartiovaara U, Salven P, Mikkola H, Lassila R, Kaukonen J, Joukov V, Orpana A, Ristimaki A, Heikinheimo M, Joensuu H, et al. Peripheral blood platelets express VEGF-C and VEGF which are released during platelet activation. Thromb Haemost. 1998;80:171–175. [PubMed] [Google Scholar]
- 20.Jowett T, Rizki TM, Rizki RM. Regulation of synthesis of larval serum proteins after transplantation of larval fat body into adult Drosophila melanogaster. Dev Biol. 1986;116:23–30. doi: 10.1016/0012-1606(86)90039-4. [DOI] [PubMed] [Google Scholar]
- 21.Wood W, Faria C, Jacinto A. Distinct mechanisms regulate hemocyte chemotaxis during development and wound healing in Drosophila melanogaster. J Cell Biol. 2006;173:405–416. doi: 10.1083/jcb.200508161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad M, Montell DJ. Cellular and molecular mechanisms of border cell migration analyzed using time-lapse live-cell imaging. Dev Cell. 2007;12:997–1005. doi: 10.1016/j.devcel.2007.03.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.