

Abstract
Cytotoxic reactive oxygen species are constantly formed as a byproduct of aerobic respiration, and are thought to contribute to aging and disease. Cells respond to oxidative stress by activating various pathways, whose balance is important for adaptation, or induction of cell death. Our lab recently reported that BiP (GRP78), a proposed negative regulator of the unfolded protein response (UPR), declines during hyperoxia, a model of chronic oxidative stress. Here, we investigate whether exposure to hyperoxia, and consequently loss of BiP, activates the UPR or sensitizes cells to ER stress. Evidence is provided that hyperoxia does not activate the three ER stress receptors IRE1, PERK, and ATF6. Although hyperoxia alone did not activate the UPR, it sensitized cells to tunicamycin induced cell death. Conversely, overexpression of BiP did not block hyperoxia induced ROS production or increased sensitivity to tunicamycin. These findings demonstrate that hyperoxia and loss of BiP alone are insufficient to activate the UPR. However, hyperoxia can sensitize cells to toxicity from unfolded proteins, implying chronic ROS, such as that seen throughout aging could augment the UPR. Moreover, suggesting that therapeutic use of hyperoxia may be detrimental for lung diseases associated with ER stress.
Keywords: hyperoxia, unfolded protein response (UPR), ER stress, BiP(GRP 78)
Introduction
Although oxygen is necessary for aerobic life, persistent exposure causes leakage of enzymatically-derived reactive oxygen species (ROS) [1] that can damage nucleic acids, proteins, and lipids [2]. Exposure to elevated levels of oxygen (hyperoxia) rapidly increases and sustains ROS levels. Because the production of ROS during normal aerobic respiration contributes to aging [3, 4], exposure of cultured cells to hyperoxia has been used as a model of chronic oxidative stress to recapitulate the effects of aging [5, 6]. Additionally, because elevated levels of oxygen are used therapeutically to treat respiratory distress [7, 8], understanding how cells respond to the deleterious effects of oxygen can improve the efficacy of these treatments.
Many signaling pathways have evolved to identify and repair oxidative damage induced by ROS, including those induced by hyperoxia, and to initiate cell death presumably when damage becomes overwhelming [9, 10]. One set of stress response pathways that has been associated with ROS production, and has yet to be investigated during hyperoxia is the ER stress response, or unfolded protein response (UPR) [11, 12]. The UPR is a quality control mechanism that senses unfolded or misfolded proteins, resulting in activation of three ER stress receptors: inositol-requiring protein-1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor-6 (ATF6) [13]. Activation of IRE1 and ATF6 leads to increased transcription of proteins containing an ER stress response element (ERSE) in their promoters, including protein folding chaperones and other proteins that help maintain ER function [14]. Active IRE1 is an endonuclease that cleaves an intron from the X-box binding protein 1 (XBP1) mRNA, producing the active XBP1 transcription factor [15, 16]. Activated ATF6 translocates to the Golgi and is cleaved by proteases, creating a 50kD fragment that promotes transcription of promoters containing ERSE elements [17]. Activation of PERK is marked by its autophosphorylation and subsequent phosphorylation of translation initiation factor eIF2α, resulting in stalled translation. Collectively, these pathways decrease protein load in the ER, upregulate expression of proteins that help manage the existing load, and finally induce cell death if ER homeostasis cannot be restored [13].
Our lab became interested in investigating the UPR in the context of hyperoxia after a recent observation that immunoglobulin binding protein (BiP) decreased in cultured cells exposed to hyperoxia [18]. BiP is an ER resident chaperone that plays two distinct roles in the UPR process. First, BiP is a transcriptional target of the UPR [19]. It is involved in protein folding, translocation into the ER, and targeting of terminally misfolded proteins for degradation by ER associated degradation (ERAD) [20]. Therefore, it is important for reestablishing ER homeostasis in the face of ER stress and preventing the initiation of cell death [21-23]. Second, and more controversial, is the role of BiP as a negative regulator of the UPR. Overexpression of BiP attenuates activation of IRE1 and PERK in response to the ER stress inducer dithiothreitol (DTT) [24]. Further, mutation of the BiP binding domain of ATF6 leads to constitutive localization to the Golgi; the first step in its activation [25]. Also mutation of the BiP binding site in PERK leads to hyperphosphorylation in the absence of ER stress [26]. However, there is strong evidence in yeast suggesting that BiP dissociation from the ER stress receptors alone is not sufficient to activate the UPR. For example, it has been shown that direct binding of unfolded proteins to IRE1 is necessary for UPR activation [27]. Further, genetic studies have shown that deletion of the BiP binding domain of IRE1, does not lead to constitutive UPR activation [28, 29]. Additionally, siRNA knockdown of BiP has been shown in various mammalian cell lines to promote in some as well as have no effect in others on UPR activation [30, 31].
The stimulus for investigating UPR activation in hyperoxia is two fold. First, because other treatments associated with oxidative stress have been shown to activate these signaling pathways [11, 12], the following experiments determine if direct application of pure oxidative stress using hyperoxia can activate the UPR. Second, considering BiP‘s role as a possible negative regulator of the UPR, and as a pro-survival downstream target of the UPR, these studies test if BiP loss during hyperoxia is sufficient to activate the UPR, and if hyperoxia can sensitize cells to classic ER stress.
Materials and Methods
Cell lines and culture
Human lung adenocarcinoma cell lines A549 and H1299 (American Type Culture Collection, Manaassas, VA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, high glucose) with 10% fetal bovine serum, 50 U/ml penicillin, and 50ug/ml streptomycin (Gibco, Carlsbad, CA) in 5% CO2 at 37°C. BiP lentivirus plasmid was created by removing BiP cDNA from pcDNA3/HisGrp78 (gift from Dr. Amy S. Lee, USC [32]) using BamHI and placing it into pFigX, a HIV lentivirus modified vector containing an internal ribosome entry sequence followed by GFP (gift from Dr. Craig Jordan, University of Rochester). High titer lentivirus expressing BiP cDNA with the GFP reporter or the empty GFP reporter were created using the second generation packaging system following established protocols [33, 34]. A549 and H1229 cells stably expressing BiP were created using two rounds of infection with high titer virus. Infection efficiency was shown to be between 96 and 98% using GFP fluorescence measured on an 11 color LSR II (BD Biosciences, San Jose, CA) in the University of Rochester Medical Center (URMC) Flow Core.
Cell and mouse oxygen exposures and cell drug treatment
Cells were exposed to room air plus 5% CO2 (termed room air) or 95% O2 plus 5% CO2 (termed hyperoxia) as previously described [35]. Cells were treated with 0.5ug/ml tunicamycin (TM) (Sigma-Aldrich, St. Louis, MO) or dimethyl sulfoxide (DMSO) (Sigma Aldrich) as vehicle control, and exposed to room air or hyperoxia. For protein half-life studies, cells were exposed to room air or hyperoxia for 36 hours and then treated with 50ug/ml cyclohexamide (Sigma Aldrich) for indicated times.
C57Bl/6J mice (8-12weeks old) were exposed to ambient air or 40-70% humidified 100% FiO2 for 64 hours as previously described [36]. Mice were sacrificed with an intraperitoneal injection of avertin. The University of Rochester‘s animal care and use committee (UCAR) reviewed and approved all studies using mice.
BiP promoter luciferase activity
Cells were transfected with either pGL3basic (Promega, Madison, WI) or pGL3basic containing the BiP −132-+7 promotor region (gift from Dr. K. Mori, Kyoto University) and SV40-Luc for transfection control using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and Opti-MEM (Gibco) according to Invitrogen‘s instructions. After six hours cells were washed twice with Hanks‘ Balanced Salt Solution (HBSS) (Cellgro, Herndon, VA) and DMEM was replaced. Cells were then placed in either room air or hyperoxia for 48 hours or treated with 0.5ug/ml tunicamycin (TM) or dimethyl sulfoxide (DMSO) vehicle control for 24 hours. Cell extracts were harvested and luciferase activity was measured using the Dual-glo Luciferase Assay System (Promega, Madison, WI) as per manufacturer‘s instructions.
Western blot analysis
Cells and whole lungs were homogenized in lysis buffer (50mM Tris (pH 7.4), 150mM NaCl, 2mM EDTA, 25mM sodium fluoride, 25mM sodium B-blycerophosphate, 0.1mM sodium vanadate, 0.1mM phenylmethylsulfonyl fluoride, 0.2% Triton X-100, 0.3% IGEPAL CA-630, 0.1ug/ml pepstatin A, 1.9ug/ml aprotinin and 2ug/ml leupeptin). Protein concentrations were determined using BCA assay (Thermo Scientific, Rockford, IL) and extracts were diluted with 3X Laemmli Buffer. Samples were separated using SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membrane (Pall Life Sciences, Pensacola, FL). All primary antibodies were diluted in 5% milk in Tris buffered saline with 0.1% tween (TBS-T) at the following concentrations: anti-actin (1:10,000, Sigma Aldrich), anti-BiP (1:1000, Cell Signaling, Danvers, MA), anti-BiP (1:1000, Stressgen, Victoria, BC, Canada, for mouse lung) anti-phospho-PERK (1:500, Biolegend, SanDiego, CA), and anti-ATF6 (1:500, Santa Cruz Biotechnology, Santa Cruz, CA). Secondary HRP conjugated antibodies (Southern Biotech, Birmingham, AL) were incubated in 5% milk in TBS-T (1:5000). HRP was detected using the ECL Plus Western Blotting Detection kit (GE Healthcare, UK) and developed using Blue Sensitive film (Laboratory Products Sales, Rochester, NY). Images were scanned and quantified using ImageJ (NIH).
Reverse-transcript and real time PCR
RNA was isolated using Trizol Reagent (Invitrogen). CDNA was made via manufacturer‘s instructions with the iScript cDNA synthesis kit (Biorad, Hercules, CA). Real time PCR was performed on a Stratagene Mx3005p real time machine using SYBR green incorporation. BiP primers were previously published by Taguchi et al [37]. Hypoxanthine guanine phosphoribosyltransferase (HPRT) primers were previously published by van Wijngaarden et al [38]. Fold change was calculated using Stratagene’s MxPro software with BiP normalized to HPRT loading control. XBP1 PCR was performed using primers previously described by Rahmani et al. [39] for 35 cycles using a MyCycler thermocycler (Biorad).
siRNA transfections
Cells were transfected with 50nM of siRNA oligos targeted against BiP (ON-TARGET Plus SmartPool, Dharmacon, Lafayette, CO) and luciferase (Dharmacon) using Lipofectamine 2000 and Opti-MEM according to Invitrogen‘s instructions. Six hours after transfections cells were washed two times with HBSS and DMEM was replaced. Forty eight hours later protein and RNA were isolated.
Cellular fractionation
Cells were harvested in isolation buffer (10mM Hepes buffer (pH 7.2), 1mM EDTA, 320mM sucrose, 25mM sodium fluoride, 25mM sodium B-blycerophosphate, 0.1 mM sodium vanadate, 0.1mM phenylmethylsulfonyl fluoride, 0.1ug/ml pepstatin A, 1.9ug/ml aprotinin and 2ug/ml leupeptin.). The suspension was homogenized with a Dounce homogenizer (20 strokes) and centrifuged at 1000g for 8minutes. The supernatant was removed and pellet was resuspended in isolation buffer and centrifuged at 18000g for 30 minutes. The supernatant was collected for the cytoplasmic rich fraction and the pellet was resuspended in isolation buffer resulting in the membrane rich fraction.
Cell death
Cell death was measured using the Annexin V-PE Apoptosis Detection Kit I (BD PharMingen, San Diego, CA) via manufacturer‘s instructions. Briefly, cells were harvested with mild trypsinization and resuspended in binding buffer. For A549 and H1299 parental cells, samples were stained with Annexin V-PE and 7AAD. To avoid fluorescence overlap between GFP and PE channels, Annexin V-APC was used with the lentivirus cells. Fluorescence was measured using an 11 color LSR II (BD Biosciences, San Jose, CA) in the URMC Flow Core. Ten thousand events were collected per sample. Data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR). Gates were set based on single stain controls. Percent death was measured as cells that stained positive for either or both 7AAD and Annexin V.
Dihydroethidium (DHE) staining
Cells were harvested via trypsinization and washed one time with PBS. Cells were incubated in 5uM DHE (Invitrogen) in phosphate buffered saline (PBS) (Cellgro, Herndon, VA) for 30 minutes at 37°C followed by centrifugation and resuspension in PBS. Fluorescence was detected using the LSRII in the URMC flow core.
Statistical analysis
Values are represented as means ± standard deviations. Significance was determined by ANOVA using Fisher‘s procedure post hoc analysis in Stat View (Adept Scientific, Bethesda, MD). A p-value of less than 0.05 was considered significant.
Results
Loss of BiP protein during hyperoxia does not activate the UPR
Previous studies in A549 and H1299 cells showed BiP levels decline in hyperoxia [18]. To further define this loss, cells were exposed to room air or hyperoxia for 24, 36, 48, and 72 hours and immunoblotted for BiP. In both cell lines hyperoxia significantly decreased BiP protein abundance starting at 36 hours (n=3, p<0.001) (Fig. 1A&B). BiP protein levels did not decrease when cultured for 72 hours in room air (data not shown). To confirm that these changes also occurred in whole lung, BiP expression was evaluated in lungs of adult mice exposed to room air or hyperoxia. As expected, the decreased BiP levels seen in the lung derived cell lines were also observed in protein extracts from whole lungs of mice exposed to hyperoxia as compared to room air. This confirms that decrease of BiP protein during hyperoxia is not unique to immortalized cell lines (Fig. 1C).
Fig. 1.

BiP protein decreases over time in cultured cells and whole lung extract of mice exposed to hyperoxia. (A) A549 and (B) H1299 cells were exposed to room air (RA) or hyperoxia for 24, 36, 48, and 72 hours and immunoblotted for BiP and actin. Band intensities from three independent experiments were quantified using ImageJ and average normalized BiP levels are displayed below each lane as relative to room air control. (C) Mouse lung extracts from 3 sets of mice exposed to room air or 64 hours of oxygen were immunoblotted for BiP and actin. Band intensity of BiP was normalized to actin and displayed as relative to their room air counterpart below each lane.
In order to determine the mechanism through which BiP protein levels decrease in hyperoxia, BiP mRNA levels during hyperoxia were measured by qRT-PCR, revealing no significant change over the entire 72 hour time course (Fig. 2A). PCR products were run on a gel to ensure a single band of appropriate size was obtained with the primers (data not shown). Tunicamycin, a drug that causes ER stress and increases transcription of the ERSE elements in the BiP promoter [13], was used as a positive control for the assay. In these samples BiP mRNA levels increased 37 fold, ensuring the efficacy of the assay (data not shown). Since hyperoxia did not affect Bip mRNA abundance, BiP protein half life was measured after 36 hours of hyperoxia, when BiP protein levels were significantly decreased. Cells were exposed to either room air or hyperoxia and then treated with cyclohexamide to inhibit translation. Subsequently, the level of remaining BiP protein was monitored at one hour intervals and compared between room air and hyperoxia. The average half life was calculated as the time when BiP protein reached half maximal levels. The half life was 1.95 hours in room air and 1.97 hours in hyperoxia, demonstrating that BiP protein half life does not decrease after exposure to hyperoxia (Fig. 2B and C). This experiment was repeated after 48 hours of hyperoxia with similar results (data not shown). The lack of change in BiP mRNA levels and protein stability when total protein is significantly decreased in hyperoxia suggests that hyperoxia inhibits the translation of BiP mRNA.
Fig. 2.

BiP mRNA levels and protein stability do not change in hyperoxia. (A) A549 cells were exposed to room air (RA) or hyperoxia for 24, 36, 48, and 72 hours and total RNA was collected. Reverse transcription followed by quantitative real time PCR was performed using primers specific to BiP. (B) Cells were exposed to RA or hyperoxia for 36 hours and then treated with 50ug/ml cyclohexamide to inhibit translation. Total protein was then collected in 1 hour intervals and BiP protein level was monitored using western blot. (C) Images were scanned and quantified using ImageJ software. BiP protein at all times treated with cyclohexamide was normalized to minus cylcohexamide samples from the corresponding exposure (room air or hyperoxia).
Because BiP binding has been proposed to inactivate the ER stress receptors, we investigated whether loss of BiP during hyperoxia activated the UPR. To test UPR activation, a luciferase reporter containing ERSE elements from the BiP promoter was used to measure transcriptional activity of elements controlled by ER stress activated transcription factors (Fig. 3A). Although this construct contains part of the BiP promoter, in this experiment it is used only as a tool to test response to UPR activated transcription factors, different from measuring whole mRNA levels, which can be affected by factors controlling mRNA stability. Between room air and hyperoxia there was a modest 1.7±0.5 and 1.5±0.3 fold induction in the luciferase activity in A549 and H1299 cells, respectively. This increase was statistically significant only in the A549 cells. Similarly to figure 2, tunicamycin was used as a positive control for the assay. In A549 and H1299 cells, tunicamycin induced reporter activity (12.7±1.8 and 8±1.4 fold, respectively) (n=6, p<0.001) (Fig. 3B). When comparing the fold induction between the tunicamycin and hyperoxia treated samples, the data suggest that the classic UPR is not robustly activated during hyperoxia. However, the relatively slight activation of the luciferase reporter construct in A549 cells exposed to hyperoxia may be because tunicamycin elicits a supra-physiological response, or because hyperoxia activates only a subset of the ER stress receptors.
Fig. 3.

Hyperoxia modestly stimulates an ER stress reporter in A549 but not H1299 cells (A) Schematic of the ER stress response element containing portion of the BiP promotor driving transcription of firefly luciferase (BiP(−132-+7)-FLuc). (B) A549 and H1299 cells were transfected with BiP(−132-+7)-FLuc and SV40-RLuc as transfection control. Cells were then placed in room air (RA) or hyperoxia for 48 hours, or DMSO or 0.5ug/ml tunicamycin (TM) for 24 hours. The luciferase activity of cell extracts was measured and data expressed as a ratio of firefly/renilla luciferase activity. The oxygen treated samples were normalized to RA and the TM treated samples normalized to DMSO. Data graphically presented as relative fold change (n=6, *p<0.001).
In order to determine if a subset of the ER stress receptors were responsible for the slight activation of the ER stress reporter construct seen in A549 cells, activation of the three individual ER stress receptors (IRE1, PERK, and ATF6) was investigated. To test for specific activation of IRE1, the ratio of unspliced/spliced (upper/lower band) XBP1 mRNA was monitored using RT-PCR. When A549 cells were exposed to either room air or hyperoxia, the ratio of unspliced/spliced mRNA did not change over a 72 hour period. However, when comparing cells treated with DMSO vehicle control to tunicamycin, the spliced band became more prominent in relation to the unspliced band during tunicamycin treatment (Fig. 4A). PERK activation was assayed directly using an antibody that recognizes the active, phosphorylated form (pPERK). Although pPERK was not detected in cells exposed to room air or hyperoxia, it was abundantly detected in cells treated with tunicamycin (Fig. 4B). Finally, ATF6 activation was monitored by decrease of the full length inactive precursor. When cells were exposed to hyperoxia there was a decrease of full length ATF6 over time; however, when comparing cells exposed to either room air or hyperoxia for 72 hours, there was no difference in the full length ATF6 protein (Fig. 4C&D). On the contrary, when comparing samples obtained from cells treated with DMSO or tunicmycin for four hours, the average level of full length ATF6 in tunicamycin treated samples was half that of the level in DMSO treated samples (n=3, p<0.004). Therefore, we conclude that the loss of full length ATF6 in cells exposed to hyperoxia is mediated by culturing cells for prolonged periods of time rather than a specific effect of hyperoxia. The collective data demonstrate that although BiP protein levels decrease in hyperoxia, the ER stress receptors (IRE1, PERK, ATF6) remain inactive.
Fig. 4.

Loss of BiP in hyperoxia does not activate the classic ER stress response. A549 cells were exposed to room air or hyperoxia for 24, 36, 48, and 72 hours for XBP1, PERK, and ATF6 assays. For XBP1 and PERK assays cells were treated with 0.5ug/ml tunicamycin (TM) or DMSO vehicle control for 24 hours. For the ATF6 assay cells were treated with 0.5ug/ml tunicamycin (TM) or DMSO vehicle control for 4 hours. (A) RNA was collected and RT-PCR for XBP1 was performed (u=unspliced XBP1, s=spliced XBP1). Whole cell lysates were immunoblotted for (B) phospho-PERK and (C) full length ATF6. (D) ATF6 protein levels were quantified, normalized to actin, and graphed. Data in panels A-C represent similar results from three independent experiments.
Hyperoxia does not inhibit activation of the ER stress receptors
To confirm loss of Bip during hyperoxia is insufficient to activate the UPR, we tested the possibility that hyperoxic conditions inhibit execution of the UPR pathways. This hypothesis was tested in two ways. First, siRNA knockdown of BiP under room air conditions was used to determine if BiP decrease under normoxic conditions could activate the UPR. A549 cells were transfected with siRNA oligonucleotides targeted against BiP or luciferase as a negative control. Protein lysates and RNA were collected 48 hours later. SiRNA oligonucleotides against BiP reduced the protein abundance to 30% ± 16.7% of control (n=3, p<0.002) (Fig. 5A), which is a similar decrease to that seen after 48 hours of exposure to hyperoxia. However, siRNA knockdown of BiP did not change the ratio of unspliced/spliced XBP1 compared to the samples treated with oligonucleotides against luciferase (Fig. 5B). They also did not cause PERK autophosphorylation (Fig. 5C). Likewise, knockdown of BiP did not activate ATF6 (Fig 5D). In contrast, all receptors were activated in the presence of tunicamycin (Fig. 5B-D). These data demonstrate that loss of BiP is not sufficient to activate the ER stress response under normoxic conditions.
Fig. 5.

Knockdown of BiP using siRNA does not activate the ER stress response. A549 cells were transfected with BiP or Luciferase (Luc) control siRNA and protein and RNA were collected 48 hours later. (A) Lysates were immunoblotted for BiP, confirming decreased protein during siRNA treatment. (B) RNA was collected and RT-PCR for XBP1 splicing was performed (u=unspliced XBP1, s=spliced XBP1). (C) Whole cell lysates were immunoblotted for phospho-PERK, and (E) full length ATF6. Data represent similar results from three independent experiments.
Second, to directly test if hyperoxia inhibited activation of the ER stress receptors, cells were exposed to hyperoxia for 48 hours and then treated with tunicamycin for 8 hours. XBP1 splicing and PERK phosphorylation occurred in tunicamycin treated cells that were pre-exposed to either room air or hyperoxia, suggesting that hyperoxia does not inhibit activation of the ER stress response pathways (Fig. 6A&B). Finally, because BiP is a transcriptional target of the UPR, tunicamycin induced BiP levels were measured after pre-treatement with hyperoxia. BiP protein induction upon tunicamycin treatment was attenuated after hyperoxia compared to room air (Fig. 6B). This suggests that although the UPR is activated, BiP protein is unable to accumulate to the same degree under hyperoxic conditions.
Fig. 6.
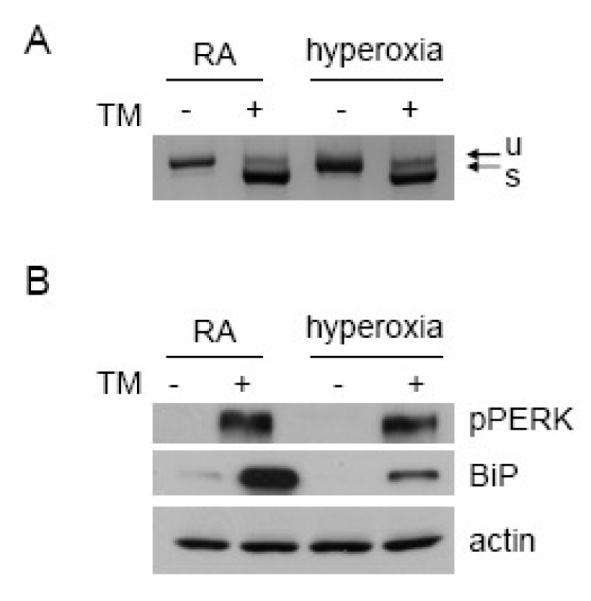
Hyperoxia does not inhibit the ER stress response. A549 cells were exposed to room air (RA) or hyperoxia for 48 hours followed by 0.5ug/ml tunicamycin for 8 hours. (A) RNA was collected and RT-PCR for XBP1 was performed (u= unspliced XBP1, s=spliced XBP1). (B) Whole cell lysates were immunoblotted for phospho-PERK and BiP. Data represent similar results from three independent experiments.
A final possible explanation for why loss of BiP in hyperoxia is insufficient to activate the UPR is that hyperoxia selectively decreases non ER resident BiP. This is unlikely because hyperoxia largely depletes the pool of total BiP protein, and BiP is mostly ER localized. However, during the course of these studies, cellular fractionation showed a small portion of BiP localized to the cytoplasmic enriched fraction. Therefore, to test the possibility that cytosolic BiP protein decreased preferentially in hyperoxia, cellular fractionation was used. After exposure to hyperoxia and separation of membrane (ER enriched) and cytoplasmic rich fractions, a decrease in BiP levels in both the ER and cytosolic enriched fractions occurred, suggesting that BiP levels are in fact decreased in the ER (Fig. 7).
Fig. 7.

BiP protein decrease occurs in both the ER and cytoplasmic rich fractions. A549 cells were exposed to room air (RA) or hyperoxia for 48 hours. Cytoplasmic and membrane rich fractions were separated and immunoblotted for BiP, GAPDH (cytoplasmic marker), and calnexin (ER marker). Data are representative of three independent experiments.
Hyperoxia sensitizes cells to tunicamycin induced cell death
The outcome of UPR activation is thought to be dictated by a balance between pro-survival and pro-death factors. The BiP protein is believed to force this balance toward pro-survival [13]. Because hyperoxia suppressed the induction of BiP in cells treated with tunicamycin, the effect of hyperoxic conditions on cell death induced by tunicamycin was investigated. During the course of the studies it became apparent that both A549 and H1299 cell lines were resistant to tunicamycin induced cell death when using 0.5ug/ml for four days (Fig. 8A&B). Note, this is the same dose used throughout the course of these studies that increased BiP protein levels in both cell lines and activated the ER stress reporter. In contrast to tunicamycin, there was a significant amount of cell death in both cell lines when exposed to hyperoxia alone. Additionally, when exposed to hyperoxia and tunicamycin, the percent of dead A549 cells increased from 52.7% ± 1.9 in hyperoxia alone to 69.0% ± 1.8 with the double treatment (n=3, p=0.0003). The percent of dead H1299 cells increased from 29.6% ± 3.2 in hyperoxia alone to 45.1% ± 1.0 with both treatments (n=3, p=0.001) (Fig. 8A&B). Finally, decreased BiP induction during the four day exposure to hyperoxia plus tunicamycin compared to room air plus tunicamycin was confirmed by western blot (Fig. 8C). These data show that hyperoxic conditions sensitize cells to ER stress induced death pathways.
Fig. 8.

Hyperoxia sensitizes cells to tunicamycin induced cell death. A549 and H1299 cells were exposed to room air plus DMSO vehicle control (RA) or tunicamycin, and hyperoxia plus DMSO vehicle control or tunicamycin for four days. Cell death was assessed using Annexin V / 7AAD staining. (A) Representative histograms are shown for each treatment. (B) Cells that stained positive for either or both Annexin V and 7AAD were counted as dead. Cell death was graphed for each treatment (n=3, *p<0.0001, #p=0.0003). Data are representative of three different experiments with similar results. (C) Whole cell lysates from each treatment were collected and immunoblotted for BiP.
BiP overexpression does not attenuate cell death induced by hyperoxia plus tunicamycin
Because BiP has been shown to be a pro-survival protein in the face of ER stress, it is reasonable to suspect that the increased sensitivity to tunicamycin in hyperoxia exposed cells could be a result of decreased BiP protein abundance. In order to determine if decreased BiP induction is the cause of hyperoxia induced susceptibility to tunicamycin, A549 and H1299 cells constitutively overexpressing BiP were created using lentivirus infection. A high infection efficiency (96-98%) was confirmed by assessing per cell GFP expression from an internal ribosome entry sequence (Fig. 9A). Subcellular fractionation studies confirmed endogenous BiP and His tagged exogenous BiP both localized in ER-enriched fractions. Additionally, immunocytochemical staining showed punctuate, perinuclear staining in both control and BiP overexpressing cells, which is consistent with ER localization (data not shown). A549 and H1299 cells infected with either BiP overexpressing lentivirus or empty lentivirus expressing only GFP (termed GFP) were exposed to hyperoxia or hyperoxia plus 0.5ug/ml tunicamycin. Samples were immunoblotted for BiP to confirm overexpression in the BiP infected cells (Fig. 9B). Cells infected with lentivirus had a higher basal level of cell death than parental cells. However, in both H1299 and A549 cell lines the increase in cell death between the cells treated with oxygen alone and oxygen plus tunicamycin was reproduced in the cells infected with the control GFP virus, allowing the use of this model to study the effects of BiP overexpression on this difference. In both A549 and H1299 cells overexpression of BiP was unable to reverse the increased cell death between hyperoxia and hyperoxia plus tunicamycin treated cells (Fig. 9C). Because ROS, and particularly superoxide, is a known cause of cell death in hyperoxia [40, 41], BiP overexpressing cells were used to test if restoring BiP levels in hyperoxia altered ROS levels. Consistent with BiP’s inability to rescue cell death, overexpression of BiP also did not attenuate superoxide production during hyperoxia in either cell line (Fig. 10). This data suggests that the increased cell death during exposure to hyperoxia plus tunicamycin is independent of BiP protein repression caused by hyperoxia.
Fig. 9.

Overexpression of BiP does not attenuate cell death from hyperoxia plus tunicaymcin. (A) A549 and H1299 cells were infected with lentivirus encoding for BiP or an empty lentivirus control (termed GFP), creating stably induced cell lines. (A) Infection efficiency was monitored by assessing GFP expression on a per cell basis using flow cytometry. (B) Cells were exposed to hyperoxia or hyperoxia plus 0.5ug/ml tunicamycin. Whole cell lysate was colleceted and immunoblotted for BiP. (C) Cell death was assessed using Annexin V / 7AAD staining. Cells that stained positive for either or both Annexin V or 7AAD were counted as dead. Cell death was graphed for each treatment (n=3, *p<0.0001, #p<0.003). Data are representative of three different experiments with similar results.
Fig. 10.

BiP overexpression does not effect ROS production. A549 and H1299 parental, GFP infected (GFP) and BiP overexpressing cells were exposed to 48 hours of hyperoxia and stained with 5uM dihydroethidium (DHE). Panel (A) shows representative histograms of GFP infected and BiP overexpressing cells in room air and hyperoxia. The mean intensity of unstained cells was subtracted from stained cells to account for background changes in fluorescence during hyperoxia. (B&C) The mean intensities of hyperoxia treated cells are expressed as fold change over room air within each cell line (n=3, *p<0.001).
Discussion
The proposed model of BiP binding as the regulator of UPR activation is somewhat controversial. The model is based on the fact that under resting conditions BiP is bound to the luminal domain of all three ER stress receptors, and that UPR activation correlates with BiP dissociation [24-26, 42]. Here we show that BiP decrease in hyperoxia does not activate the UPR in A549 and H1299 lung derived cell lines. Consistent with a lack of UPR activation in cell lines, no difference in spliced XBP1 mRNA from whole lungs of mice exposed to room air or hyperoxia for 72 hours were detected, and all attempts to blot for pPERK in whole lungs of mice exposed to room air or hyperoxia with a mouse specific antibody were unsuccessful (data not shown). Therefore, the lack of UPR activation in cell lines during hyperoxia is recapitulated in mice exposed to hyperoxia. This suggests that while dissociation of BiP from the ER stress receptors may contribute to regulation of the UPR at the cell line and whole lung level, it is not the only factor involved. Our findings in mammalian cells support the recent evidence in yeast showing BiP by itself is not sufficient to activate the UPR [28, 29].
It remains possible that the BiP protein is so abundant that the relatively low level of BiP present after 72 hours of exposure to hyperoxia is still sufficient to bind all of the ER stress receptors. However, if this were the case, a large amount of unfolded proteins would be needed to sequester enough BiP from the receptors to activate the UPR, making it an insensitive switch. These findings imply that although BiP is an important regulator of ER homeostasis, absolute levels of the protein can change without immediate consequences to UPR signaling. Interestingly, Xu et al. show that another ER resident chaperone, ERp57, decreases during hyperoxia [43], suggesting that although hyperoxia does not activate the UPR, it does effect multiple members of the ER protein folding family. However, protein disulfide isomerase (PDI), another ER resident chaperone, does not change during hyperoxia (data not shown), suggesting expression of individual members of the UPR are differentially controlled by chronic ROS created during hyperoxia exposure.
Previous studies suggest that ROS can activate the UPR. For instance, TNF-alpha induced the activation of the UPR in a reactive oxygen species (ROS) dependent manner [11]. These observations suggest that the ROS generated during hyperoxia [1] could activate the UPR pathways. On the contrary, the current study using hyperoxia as a way to induce ROS does not cause ER stress activation, suggesting that the UPR signaling pathways do not contribute to the pathology of hyperoxia. One possible explanation for these seemingly conflicting results is that the amount or type of ROS accrued during hyperoxia is less and/or different than that during TNF-alpha stimulation. Another possibility is that ROS alone are insufficient to activate the ER stress response, and a compounding stimulus is needed. Hyperoxia creates ROS through over activation of the cells‘ own enzymes, and is therefore a unique way to directly create chronic ROS independently of a signaling cascade. Although ROS inhibitors were able to attenuate the ER stress induced by TNF-alpha, inhibition of other effects of TNF-alpha signaling may have also decreased the UPR; suggesting that ROS work in conjunction with other factors missing from the hyperoxic environment to activate the UPR.
Although BiP loss alone in hyperoxia is insufficient to activate the UPR, our data show that hyperoxia exacerbates tunicamyin induced cell death. Because the cells are resistant to death induced by the dose of tunicamycin used, the increase in cell death between oxygen alone and tunicamycin plus oxygen is not simply an additive effect of two death stimuli; suggesting that hyperoxia sensitizes cells to ER stress induced cell death. It is logical to hypothesize that the inability to fully increase the prosurvival molecule, BiP, during hyperoxic conditions could attribute to the increased cell death associated with tunicamycin plus hyperoxia. However, we found that overexpression of BiP protein does not attenuate cell death in hyperoxia plus tunicamycin. Our data suggest increased susceptibility of hyperoxia treated cells to ER stress induced cell death is independent of BiP protein levels.
Although the mechanism through which hyperoxia confers increased sensitivity to UPR induced cell death remains unclear, our observations have interesting implications in relation to lung diseases associated with ER stress. Recently ER stress has been associated with both a familial form of idiopathic pulmonary fibrosis (IPF) caused by an accumulation of unfolded mutant proSP-C and herpesvirus associated IPF. Lung samples from both types of IPF patients showed an increase in multiple UPR markers, including BiP [44]. In the case of the inherited proSP-C mutations, the age of onset for the disease varies within individual families [45-47]. It has been hypothesized that the variance of onset could be due to environmental factors, such as viral infections or differences in modifier genes [48]. Because hyperoxia is used as a model of chronic oxidative stress, our data would suggest that as people with inherited mutations in proSP-C age and accumulate ROS they may become more susceptible to the damaging effects of UPR activation. This implies that while accumulation of mutant unfolded proSP-C may cause a constitutive UPR activation, variance of onset could result from different rates of ROS accumulation, tipping the balance toward epithelial cell death in the lung and subsequent fibrotic repair. Consistent with the hypothesis that environmental factors can trigger the onset of IPF, it has been reported that stable expression of mutant proSP-C is insufficient to kill cells, but can sensitize cells to respiratory syncytial virus (RSV) induced cell death [49]. It is probable that the therapeutic use of high oxygen would act similarly as a “second hit” for patients with proSP-C mutations. Our findings suggest that accumulation of ROS induced by oxygen therapy could compound the negative effects of the ER stress associated with these diseases.
Although BiP protein levels are decreased in hyperoxia, the mRNA levels and protein stability are not changed. This suggests that the translation rate of the protein is changing. BiP translation is known to be uniquely regulated by an internal ribosome entry segment (IRES) that can be either stimulated or repressed under certain conditions [50]. The rate of translation of BiP’s IRES was measured using a dual luciferase assay, showing no difference between room air and hyperoxia (data not shown). Interestingly, hyperoxia has been shown to decrease the general rate of translation [51]. Our data show that in lung derived cells the half life of BiP is short (~2 hours). Therefore, the decrease in protein level could simply be explained by a general decrease in translation, leading to depletion of short half life proteins. In fact, decrease in translation rates of short half life prosurvival molecules affecting their levels has been shown in other systems [52]. Our data suggest that the decrease in translation in hyperoxia may impact specific important proteins, and therefore may become an exciting area in the hyperoxia field.
In conclusion, these studies demonstrate that hyperoxia induced loss of BiP is insufficient to activate the ER stress response. Additionally, BiP protein loss does not contribute to the increased susceptibility that hyperoxia confers on UPR induced cell death. Our data support recent studies that question the simple model of BiP dissociation as an all important switch to activate the UPR [27, 29-31, 53, 54]. Finally, these studies highlight possible complications of using oxygen therapy to treat patients with lung diseases associated with ER stress.
Acknowledgements
This work was funded in part by National Institutes of Health (NIH) Grant HL-67392. NIH Training Grant HL-66988 supported J. Gewandter. NIH Center Grant ES-01247 supported the construction of chambers for continuous exposure of cell lines to hyperoxia. The BiP(−137-+7)-FLuc reporter was generously provided by Dr. K. Mori (University of Kyoto). The pcDNA3/HisGrp78 was generously provided by Dr. Amy S. Lee. We thank Dr. Craig Jordan and John Ashton for technical help and reagents contributing to the production of the BiP lentivirus. We thank Dr. Peter Keng for help with flow cytometry. We thank Dr. Damian Krysan for critical reading of the manuscript.
Abbreviations
- ATF6
activating transcription factor-6
- BiP
immunoglobulin binding protein
- ER
endoplasmic reticulum
- ERSE
ER stress response element
- IRE1
inositol-requiring protein-1
- PERK
protein kinase RNA (PKR)-like ER kinase
- RA
room air
- ROS
reactive oxygen species
- TM
tunicamycin
- XBP1
X-box binding protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Joenje H. Genetic toxicology of oxygen. Mutation Research. 1989;219:192–208. doi: 10.1016/0921-8734(89)90001-5. [DOI] [PubMed] [Google Scholar]
- 2.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 3.Harman Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 4.Gruber J, Schaffer S, Halliwell B. The mitochondrial free radical theory of ageing--where do we stand? Front Biosci. 2008;13:6554–6579. doi: 10.2741/3174. [DOI] [PubMed] [Google Scholar]
- 5.Saretzki G, Feng J, von Zglinicki T, Villeponteau B. Similar gene expression pattern in senescent and hyperoxic-treated fibroblasts. J Gerontol A Biol Sci Med Sci. 1998;53:B438–442. doi: 10.1093/gerona/53a.6.b438. [DOI] [PubMed] [Google Scholar]
- 6.Schoonen WG, Wanamarta AH, van der Klei-van Moorsel JM, Jakobs C, Joenje H. Hyperoxia-induced clonogenic killing of HeLa cells associated with respiratory failure and selective inactivation of Krebs cycle enzymes. Mutat Res. 1990;237:173–181. doi: 10.1016/0921-8734(90)90023-k. [DOI] [PubMed] [Google Scholar]
- 7.O’Brodovich HM, Mellins RB. Bronchopulmonary dysplasia. Unresolved neonatal acute lung injury. Am Rev Respir Dis. 1985;132:694–709. doi: 10.1164/arrd.1985.132.3.694. [DOI] [PubMed] [Google Scholar]
- 8.Jindal SK. Oxygen therapy: important considerations. Indian J Chest Dis Allied Sci. 2008;50:97–107. [PubMed] [Google Scholar]
- 9.Zaher TE, Miller EJ, Morrow DM, Javdan M, Mantell LL. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic Biol Med. 2007;42:897–908. doi: 10.1016/j.freeradbiomed.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee PJ, Choi AM. Pathways of cell signaling in hyperoxia. Free Radic Biol Med. 2003;35:341–350. doi: 10.1016/s0891-5849(03)00279-x. [DOI] [PubMed] [Google Scholar]
- 11.Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- 12.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 16.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 17.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vitiello PF, Wu YC, Staversky RJ, O’Reilly MA. p21(Cip1) protects against oxidative stress by suppressing ER-dependent activation of mitochondrial death pathways. Free Radic Biol Med. 2008 doi: 10.1016/j.freeradbiomed.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMillan DR, Gething MJ, Sambrook J. The cellular response to unfolded proteins: intercompartmental signaling. Curr Opin Biotechnol. 1994;5:540–545. doi: 10.1016/0958-1669(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 20.Gething MJ. Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Miller E, van de Water B, Stevens JL. Endoplasmic reticulum stress proteins block oxidant-induced Ca2+ increases and cell death. J Biol Chem. 1998;273:12858–12862. doi: 10.1074/jbc.273.21.12858. [DOI] [PubMed] [Google Scholar]
- 22.Yu Z, Luo H, Fu W, Mattson MP. The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol. 1999;155:302–314. doi: 10.1006/exnr.1998.7002. [DOI] [PubMed] [Google Scholar]
- 23.Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514:122–128. doi: 10.1016/s0014-5793(02)02289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 25.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 26.Ma K, Vattem KM, Wek RC. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J Biol Chem. 2002;277:18728–18735. doi: 10.1074/jbc.M200903200. [DOI] [PubMed] [Google Scholar]
- 27.Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oikawa D, Kimata Y, Kohno K. Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J Cell Sci. 2007;120:1681–1688. doi: 10.1242/jcs.002808. [DOI] [PubMed] [Google Scholar]
- 29.Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J Cell Biol. 2004;167:445–456. doi: 10.1083/jcb.200405153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhai L, Kita K, Wano C, Wu Y, Sugaya S, Suzuki N. Decreased cell survival and DNA repair capacity after UVC irradiation in association with down-regulation of GRP78/BiP in human RSa cells. Exp Cell Res. 2005;305:244–252. doi: 10.1016/j.yexcr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki T, Lu J, Zahed M, Kita K, Suzuki N. Reduction of GRP78 expression with siRNA activates unfolded protein response leading to apoptosis in HeLa cells. Arch Biochem Biophys. 2007;468:1–14. doi: 10.1016/j.abb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–3740. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 33.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 34.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 35.Helt CE, Rancourt RC, Staversky RJ, O’Reilly MA. p53-dependent induction of p21(Cip1/WAF1/Sdi1) protects against oxygen-induced toxicity. Toxicol Sci. 2001;63:214–222. doi: 10.1093/toxsci/63.2.214. [DOI] [PubMed] [Google Scholar]
- 36.O’Reilly MA, Marr SH, Yee M, McGrath-Morrow SA, Lawrence BP. Neonatal hyperoxia enhances the inflammatory response in adult mice infected with influenza A virus. Am J Respir Crit Care Med. 2008;177:1103–1110. doi: 10.1164/rccm.200712-1839OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi J, Fujii A, Fujino Y, Tsujioka Y, Takahashi M, Tsuboi Y, Wada I, Yamada T. Different expression of calreticulin and immunoglobulin binding protein in Alzheimer’s disease brain. Acta Neuropathol. 2000;100:153–160. doi: 10.1007/s004019900165. [DOI] [PubMed] [Google Scholar]
- 38.van Wijngaarden P, Brereton HM, Coster DJ, Williams KA. Stability of housekeeping gene expression in the rat retina during exposure to cyclic hyperoxia. Mol Vis. 2007;13:1508–1515. [PubMed] [Google Scholar]
- 39.Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P, Grant S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27:5499–5513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buccellato LJ, Tso M, Akinci OI, Chandel NS, Budinger GR. Reactive oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem. 2004;279:6753–6760. doi: 10.1074/jbc.M310145200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Shan P, Sasidhar M, Chupp GL, Flavell RA, Choi AM, Lee PJ. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am J Respir Cell Mol Biol. 2003;28:305–315. doi: 10.1165/rcmb.2002-0156OC. [DOI] [PubMed] [Google Scholar]
- 42.Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009 doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 43.Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297:L44–51. doi: 10.1152/ajplung.90626.2008. [DOI] [PubMed] [Google Scholar]
- 44.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 45.Chibbar R, Shih F, Baga M, Torlakovic E, Ramlall K, Skomro R, Cockcroft DW, Lemire EG. Nonspecific interstitial pneumonia and usual interstitial pneumonia with mutation in surfactant protein C in familial pulmonary fibrosis. Mod Pathol. 2004;17:973–980. doi: 10.1038/modpathol.3800149. [DOI] [PubMed] [Google Scholar]
- 46.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, Roberts R, Haines J, Stahlman M, Loyd JE. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 47.Cameron HS, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, Deutsch G, Nogee LM. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr. 2005;146:370–375. doi: 10.1016/j.jpeds.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 48.Grutters JC, du Bois RM. Genetics of fibrosing lung diseases. Eur Respir J. 2005;25:915–927. doi: 10.1183/09031936.05.00133404. [DOI] [PubMed] [Google Scholar]
- 49.Bridges JP, Xu Y, Na CL, Wong HR, Weaver TE. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol. 2006;172:395–407. doi: 10.1083/jcb.200508016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macejak DG, Sarnow P. Internal initiation of translation mediated by the 5′ leader of a cellular mRNA. Nature. 1991;353:90–94. doi: 10.1038/353090a0. [DOI] [PubMed] [Google Scholar]
- 51.Shenberger JS, Myers JL, Zimmer SG, Powell RJ, Barchowsky A. Hyperoxia alters the expression and phosphorylation of multiple factors regulating translation initiation. Am J Physiol Lung Cell Mol Physiol. 2005;288:L442–449. doi: 10.1152/ajplung.00127.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fritsch RM, Schneider G, Saur D, Scheibel M, Schmid RM. Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem. 2007;282:22551–22562. doi: 10.1074/jbc.M702673200. [DOI] [PubMed] [Google Scholar]
- 53.Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27:1027–1043. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oikawa D, Kimata Y, Takeuchi M, Kohno K. An essential dimer-forming subregion of the endoplasmic reticulum stress sensor Ire1. Biochem J. 2005;391:135–142. doi: 10.1042/BJ20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]