

Abstract
The Q-cycle mechanism of the cytochrome bc1 complex maximizes energy conversion during electron transport from ubiquinol to cytochrome c (or alternate physiological acceptors). Yet important steps in the Q-cycle are still hotly debated including: bifurcated electron transport; the high yield and specificity of the Q-cycle despite possible short circuits and bypass reactions; and the rarity of observable intermediates in the oxidation of quinol. Mounting evidence shows that some bypass reactions producing superoxide during oxidation of quinol at the Qo site diverge from the Q-cycle rather late in the bifurcated reaction and provide an additional means of studying initial reactions of the Q-cycle. Bypass reactions offer more scope for controlling and manipulating reaction conditions, e.g., redox potential, because they effectively isolate or decouple the Q-cycle initial reactions from later steps, avoiding many complications and interactions. We examine the dependence of oxidation rate on substrate redox potential in the yeast cytochrome bc1 complex and find that the rate limitation occurs at the level of direct one-electron oxidation of quinol to semiquinone by the Rieske protein. Oxidation of semiquinone and reduction of cyt b or O2 are subsequent, distinct steps. These experimental results are incompatible with models in which electron transfer to the Rieske protein is not a distinct step preceding electron transfer to cytochrome b, and with conformational gating models that produce superoxide by different rate limiting reactions from the normal Q-cycle.
The cytochrome (cyt1) bc1 (also known as Complex III or ubiquinol:cyt c oxidoreductase) and related complexes are essential energy transduction components in the respiratory and photosynthetic electron transport chains of a wide range of organisms (1–4). Its physiological role is to pump protons across the inner mitochondrial membrane to drive synthesis of ATP, but its detailed enzyme mechanism and possible roles in oxidative stress are still disputed. Our focus is on the mitochondrial cyt bc1 complex from Saccharomyces cerevisiae whose dimeric functional core is illustrated in cartoon form in the left hand side of Fig. 1. Each monomer consists of three essential catalytic subunits (2, 5–8): cyt b; the Rieske iron-sulfur protein (ISP); and cyt c1. The redox-active cofactors form two distinct redox chains leading away from the quinol oxidase (Qo) site on the positive side (p-side) of the membrane (5). A ‘low-potential chain’ extends across the membrane from the Qo site, through the cyt b subunit with its two b-type hemes (cyt bL and cyt bH), to the quinone reductase (Qi) site on the negatively-charged side (n-side) of the membrane. The ‘high-potential chain’ consists of the Rieske FeS cluster of the ISP, and the c-type heme of cyt c1. This chain, on the p-side of the membrane, relays electrons from the Qo site to a soluble cyt c.
Figure 1. The uninhibited Q-cycle and superoxide production under partially inhibited conditions.
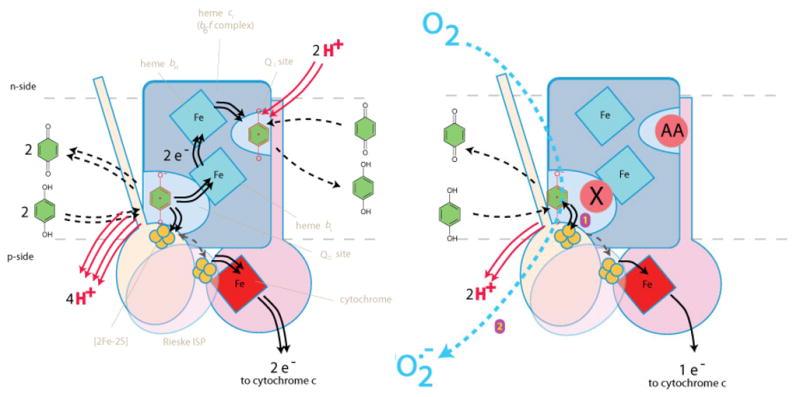
Left) The Q-cycle, showing how two turnovers at the Qo site sends two electrons through the high-potential chain to cyt c, and two electrons through the low-potential chain to reduce a quinone bound at the Qi site. These reactions release four protons from QH2 at the Qo site (red arrows), and the take up two protons by Q at the Qi site (red arrows), resulting in a net 2H+/e− translocation stoichiometry. Right) Blockage of the low-potential chain prevents transfer of the second QH2 electron following the initial transfer to the high-potential chain. The reactive SQ intermediate accumulates in Qo and can reduce oxygen to form superoxide (blue arrow), or participate in other non-productive ‘bypass reactions’. Both panels show pivoting of the Rieske ISP, which partially gates reactions of the SQ to prevent double reduction of the high-potential chain.
The cyt bc1 complex catalyzes a Q-cycle, illustrated in Fig. 1 Left, as first proposed by Mitchell and modified extensively (9–12) as a result of the work of many researchers. In most models of the Q-cycle (1, 13), QH2—originating from complex II in S. cerevisiae—binds in the Qo site and is oxidized in the so-called ‘bifurcated’ reaction, releasing two protons on the positively charged, p-side, of the membrane. The first QH2 electron enters the high-potential chain at the Rieske FeS cluster. A pivoting motion of the Rieske ISP (shown as two superimposed conformations in Fig. 1 Left) carries its FeS cluster close to cyt c1, allowing reduction of the cyt c1, which then relays the electron to a soluble cyt c. Eventually the electrons arrive at complex IV where they reduce O2 to water.
In this model of the Q-cycle, the one-electron oxidation of QH2 at Qo forms a transient semiquinone (SQ) intermediate, termed SQo (14, 15). Instead of reducing the thermodynamically-favored high-potential chain a second time, SQo reduces cyt bL in the low-potential chain. Electrons sent into the low-potential chain travel through cyt bH and cyt bL, and eventually reduce a Q bound at the Qi site. After two turnovers of the Qo site, two electrons pass through the low potential chain, reducing Q to QH2 at the Qi site, with uptake of two protons from the n-side of the membrane. Overall, the Q-cycle oxidizes two QH2 at Qo, with the release of four H+ on the p-side; and reduces one Q at Qi with uptake of two H+ from the n-side. The net result is a highly efficient proton pump that helps produce the transmembrane proton motive force (pmf) that drives ATP synthase. This complicated Q-cycle gives a stoichiometry of 2 H+ translocated across the membrane per e− passed ultimately to cyt c in the steady state, increasing the ATP synthesized per O2 reduced (the P/O ratio) over that of a linear electron flow mechanism in which both QH2 electrons enter the high-potential chain (16, 17).
It is argued that the bifurcated electron flow also serves to ‘quench’ the SQo (18–27) and prevent its reaction with O2 to generate superoxide. Superoxide production by the Qo site (and other reactions that ‘bypass’ the Q-cycle) is readily observed by blocking the low-potential chain in a number of ways, including: high pmf (28–30); high membrane potential (30); mutation of cyt b (17, 31); inhibition of the Qi site, e.g. by antimycin A (‘AA’ in Fig 1 Right); or inhibition of the Qo site niche near heme bL (proximal niche), e.g. by myxothiazole or methoxyacrylate (MOA) stilbene (‘X’ in Fig. 1 Right) (21, 22). Superoxide production at the Qo site is hotly debated as a mitochondrial source of superoxide and other reactive oxygen species in processes such as cell signaling, aging, carcinogenesis and metabolic diseases.
A number of reactions ‘bypass’ the normal Q-cycle, reducing the efficiency of proton pumping and diverting electron flow, at least temporarily (20). The different bypass reactions are not mutually exclusive and might occur in combinations depending on conditions. We focus in this paper on the bypass reaction that produces superoxide during oxidation of quinol at the Qo site because of its very intimate relation to the initial steps in the Q-cycle. Recently, superoxide production has recently been described through a very different bypass reaction that involves reduction of quinone at the Qo site (32, 33), but it will not be considered here.
Forquer et al. (34) recently showed that the rate limiting step for superoxide production in AA-inhibited mitochondrial cyt bc1 complex is the same as for the Q-cycle—involving reduction of the Rieske FeS cluster. Thus, the Q-cycle and superoxide production share intermediates along the same, or similar, reaction pathway(s). EPR of freeze-quenched bacterial cyt bc1 complex suggests that this common intermediate is SQo (14, 15). These recent results allow earlier conclusions about the order of reactions in the Q-cycle the bacterial Rhodobacter sphaeroides cyt bc1 complex (35) to be extended to the mitochondrial cyt bc1 complex.
Little superoxide is generated by cyt bc1 under normal physiological conditions. Several classes of models attempt to explain how the Qo site steers the reaction along the Q-cycle and away from superoxide production (14, 15, 34, 35). The experimental evidence is increasingly at odds with models in which the oxidation of QH2 to SQ is radically different for superoxide production than for the Q-cycle, particularly the ‘double concerted’ electron transfer mechanisms (13, 25, 27, 36). Models that invoke conformational gating (6, 37–41) of the Qo site to steer electron flow are severely constrained because the same activating reaction occurs during quinol oxidation both when the putative gate functions ‘normally’ and when it leaks, to produce superoxide (34).
The Q-cycle and the bypass reaction pathways must diverge at some point. On this point conflicting results have been obtained depending on reaction conditions. In contrast to Forquer et al. (34), Covian and Trumpower (42) concluded that there are different rate limiting steps for the Q-cycle and the bypass reactions from different activation energies under pre-steady state, non-substrate-saturated conditions. The relevance of these results to the reactions of the cyt bc1 complex is limited until the new rate limiting steps are identified and until it is determined which bypass reactions occur under these conditions. Consequently, we will restrict our discussion to substrate-saturated steady-state conditions unless explicitly stated.
Important questions still remain concerning the sequence of reactions and the redox partner of the ISP in the rate limiting step. Does quinol transfer an electron directly to the FeS cluster thereby generating SQo or is there an intervening step? What factors control the rate of superoxide production with other quinols as substrate? Do interactions of the enzyme with particular sidegroups of the substrate guide the reaction and determine the products? To resolve these questions we examine superoxide production in the yeast mitochondrial cyt bc1 complex with a series of seven ubiquinol analogs designed to vary redox potentials and sidegroups on the quinol ring. One of the challenges to such work has been the limited ability to make consistent redox measurements on quinone species with the long, hydrophobic tails needed for activity. The ability to calculate these properties (43) now makes such a study feasible. We report here on the substrate properties that control the substrate saturated rate of superoxide production; we identify the rate limiting reaction; and we determine the sequence of the bifurcated electron transfer reactions.
Experimental Procedures
Chemical Syntheses
Starting materials were purchased from Sigma-Aldrich, USA and used without further purification, except as indicated. Substrates were purified by silica gel chromatography (0.035–0.07 mm particle size, 6 nm pore size, Acros, Geel, Belgium). Syntheses for most of these compounds, or close derivatives, have been described (44–47). Full details are provided as Supporting Information. Product identity was confirmed using 1H and 13C NMR spectra recorded at the Washington State University Center for NMR Spectroscopy on a Varian Mercury 300 MHz NMR spectrometer. Mass spectra were recorded on a Finnegan LCQ mass spectrometer using an ESI ionization source in negative ion mode.
Purification of the cyt bc1 complex
Cyt bc1 complex from S. cerevisiae was isolated from store-bought bakers’ yeast using the protocol of Ljungdahl et al. (48) with modifications (22). The concentration of the cyt bc1 complex was determined by ferricyanide-ascorbate-dithionite absorbance difference spectra using published values of the extinction coefficients (48).
Steady-State Enzyme Turnover Measurements
Steady-state turnover rates were measured from the initial rate of cyt c reduction (19, 22). The stigmatellin-sensitive portion of the rate is reported, so that the activity can be attributed to the Qo site. Q-cycle bypass is measured in the presence of 10 μM of the Qi site inhibitor, AA; and the bypass rate is half the rate of cyt c reduction because each bypass turnover eventually transfers two electrons to cyt c. The rate of superoxide production was measured from the SOD-sensitive fraction of the bypass rate, recognizing that the SOD-dependent decrease in cyt c reduction equals the superoxide production. The initial rates follow Michaelis-Menton kinetics. The reported Vmax are based on measurements at substrate concentrations several times larger than the measured Km, Table 1.
Table 1.
Kinetic parameters for steady state turnover of AA-inhibited cyt bc1 complex. fSOP is the fraction of Q-cycle bypass reactions resulting in superoxide production derived from the superoxide dismutase sensitivity of cyt c reduction in the AA-inhibited complex. Product inhibition constants Ki for a mixed inhibition model are for uninhibited steady –state turnover driven by decyl-ubiquinol. Km and Ki are reported in units of μM. Rates of quinol oxidation are given in s−1 from total bypass reactions (Vmax(bypass)) and from superoxide-producing bypass reaction (Vmax(SO)). In two cases, Vmax(SO) could not be measured but is estimated as a range: 0.5–0.75 Vmax(bypass) covering the range of measured fSOP. Rates noted in italics were measured by stopped flow. The shift in redox potential for the Q/QH2 couple based on measured values of Km and the competitive Ki are reported in V.
| Substrate | Km | Vmax(bypass) | fSOP | Vmax(SO) | Ki(C) | Ki (NC) | ΔEm |
|---|---|---|---|---|---|---|---|
| (1) | 23 | 2.4 | 0.68 | 1.6 | 2.1 | - | −0.060 |
| (2) | 25 | 2.2 | - | 1.1–1.7 | 11.8 | 5.8 | −0.019 |
| (3) | 5 | 0.6 | 0.66 | 0.4 | - | - | - |
| (4) | >150 | 7.5 | 0.71 | 5.3 | - | - | - |
| (5) | 9 | 27 | 0.55 | 15.1 | 4.4 | 7.1 | −0.018 |
| (6) | 35 | 80 | - | 40–60 | - | - | - |
| (7) | 30 | 199 | 0.51 | 101 | 26.2 | 36.8 | −0.003 |
The AA-inhibited complex is a reproducible model of other elicitors of superoxide production, such as mutation of certain Qo and Qi site residues (17, 31, 49, 50), high pmf (28, 30) or high membrane potential (30), in which electron transfer through the low-potential chain is slowed. The use of AA also prevents reactions of substrate at the Qi site from influencing the measurements (see below) and eliminates the sample variability and instability noted with the use of lipid vesicles to control pmf or membrane potential (30). Yeast submitochondrial particles (data not shown), yielded similar results, ruling out artifacts from detergent solubilization or delipidization of the cyt bc1 complex. Use of purified complex precludes interference from other enzymes found in crude preparations or from endogenous substrates. Forquer, et al. (34) found that purified complexes also avoided the kinetic artifacts (e.g., nonlinear or curved responses) seen in native membranes (35) where QH2 concentrations were near Km.
The fast initial rates of some bypass reactions were verified by stopped-flow techniques, measuring the rate of cyt c reduction, as described above (see Table 1). The substrate concentrations used in stopped flow experiments were well in excess of the steady state Km values, resulting in rates that were independent of substrate concentration. Competition assays between non-native Q-species and the native substrate analog (3) were used to estimate the Ki of the non-native Q species and were analyzed using a mixed competitive inhibition model (51) with simultaneous fitting of substrate titration curves over a range of quinone concentrations, holding the uninhibited Km and Vmax values constant. Shifts in the Q/QH2 potential from differential Q/QH2 binding were estimated from the ratio of competitive Ki/Km values. In some cases decyl-ubiquinol was used in place of (3) (differing by one carbon in the tail) with identical results.
Thermodynamic properties of QH2 substrates
The estimation of free energy changes, ΔG, for reactions involving SQ in the Qo site require accurate one- and two-electron redox potentials and pKa values for all the Q/SQ/QH2 species bound to the protein at Qo. Measurement of many of the required quantities has not been experimentally possible. However, for the series of closely-related p-benzoquinone derivatives used here, the redox potentials and pKa values in the Qo site should be offset from the corresponding aqueous solution values by a similar amount if there are no strong specific interactions affecting some molecules in the series much more than the others. The ΔG for a reaction in the protein should be shifted by a similar amount. Thus, the change in free energy of reaction, ΔΔG, for different substrate analogs in this series should be similar whether estimated using the inaccessible values in the protein or the values in aqueous solution.
The use of aqueous properties has its own challenges. Aqueous redox potentials from cyclic voltammetric measurements are not reliable for many lipophilic quinone species (43, 52, 53). Moreover, the reduction potential for Q to Q•− (or the oxidation of QH2 to QH•or Q•−) is nearly impossible to measure reliably by experiment in aqueous solution due to the disproportionation of the semiquinone species in the solutions. Fortunately, there is sufficient experimental data to support the calculation of aqueous redox potentials and pKas in this series of substrate analogs. We use procedures which predict reasonably accurate aqueous redox potentials across a series of homologous water-soluble quinone species (43), similar to ‘benchmarking’ approaches used successfully in other cases to predict accurate redox potentials and pKa values (54, 55). The computed values are summarized in Table 2 with full details in the Supporting Information.
Table 2.
Calculated aqueous redox potentials and pKa values for SQ species, estimated Ks values, and SQ/QH2 potentials for protonated and deprotonated SQ species. KS is defined as the the equilibrium constant for comproportionation of Q + QH2 to form 2 QH•. All potentials and equilibrium constants are adjusted to the experimental conditions of pH 8.0. Redox potentials are given in V versus the standard hydrogen electrode.
| Substrate | E(Q/QH2) | E(Q/Q•−) | pKa SQ | log KS | E(QH•/QH2) | E(Q•−/QH2) |
|---|---|---|---|---|---|---|
| (1) | 0.178 | 0.087 | 4.70 | −9.66 | 0.463 | 0.269 |
| (2) | 0.112 | −0.088 | 5.33 | −12.09 | 0.469 | 0.312 |
| (3) | 0.122 | −0.078 | 5.35 | −12.05 | 0.478 | 0.322 |
| (4) | −0.018 | −0.156 | 4.86 | −10.93 | 0.305 | 0.12 |
| (5) | −0.003 | −0.072 | 5.19 | −7.94 | 0.231 | 0.066 |
| (6) | −0.067 | −0.148 | 4.79 | −9.14 | 0.203 | 0.014 |
| (7) | −0.108 | −0.052 | 5.10 | −3.89 | 0.007 | −0.164 |
Results
Kinetics of Superoxide Production in AA-inhibited Complexes
The apparent Michaelis constant (Km) and the maximum velocity of superoxide production (Vmax) were determined for substrates (1)-(7), Scheme 1 and Table 1. The Km values reflect the partition coefficient for each substrate analog into the detergent or membrane and the apparent binding constant at the Qo site. The Km for all substrate analogs lie in the range of 5–35 μM except for (4), which has solubility problems and a large apparent Km ~150 μM.
Scheme 1.

Synthetic QH2 substrates. -R denotes the n-undecyl group, -C11H23.
For several substrates, the AA-inhibited turnover assays slowed markedly as the product Q accumulated. Such behavior has been observed for other non-native substrates (19, 45) and indicates product inhibition, that is, the oxidized substrate binds more tightly to Qo than the substrate itself. Product inhibition implies that the Q/QH2 redox potential shifts to more negative values when bound at Qo. To measure those shifts, binding constants for the oxidized substrates at Qo were estimated from competition assays on uninhibited, steady-state turnover driven by decyl-ubiquinol. The data was analyzed with mixed competition kinetics with competitive inhibition considered to be binding of the quinol at the Qo site and the non-competitive inhibition as binding of the Q at a site distinct from the Qo binding pocket, probably the Qi site. The Ki values were significant only for four substrates and the calculated shift in the redox potential Em(Q/QH2) based on the Km and competitive Ki values range from −0.003 to −0.060 V, less than the uncertainty in calculated redox potential and much less than the range of potentials for (1)-(7).
Superoxide Production Rates Are Related to the Potentials for SQ Formation
The Vmax for superoxide production varies by more than two orders of magnitude for substrates (1)-(7) and depends strongly on the relative redox potential for the one-electron oxidation of (1)-(7), Figure 2. Remarkably, the present data plotted as a function of the shift in oxidation potential of QH2 relative to UQH2 forms a smooth extension of the Forquer, et al. (34) data plotted as a function of the reduction potential shifts of the Rieske ISP (as varied by site-directed mutagenesis in S. cerevisiae). The reaction rate varies consistently with changes in the redox potential of the QH2/ISP couple whether the redox potential of the substrate or of the ISP is changed. This combined data shows that the rate limiting step for superoxide generation depends on the redox potential for the one-electron reduction of the Rieske ISP by substrate.
Figure 2.

The Vmax for superoxide production, vSOP,, in μmoles of superoxide per μmole of cyt bc1 s−1 measured with saturating amounts of substrate analog and corresponding to Vmax. Rates are plotted as a function of the shift, ΔΔG, in oxidation potential for substrate analogs (Upper for QH2/Q−•; Lower for QH2/QH•) or reduction potential for the Rieske ISP. (●) vSOP for substrate analogs from Table 1, the analog is indicated by number to the left of the symbol; (○) vSOP for Rieske ISP mutants from Forquer, et al.(34) measured using the Amplex Red assay; (*) vSOP for substrate analogs UQ3 and RQ3 from Cape, et al.(19); (◇) Vmax for total bypass in μmoles of substrate analog oxidized per μmole of cyt bc1 s−1 for (2) and (6) with the typical range for the vSOP indicated directly below by ‘error’ bars.
The relation is equally strong whether we consider the redox potential for the QH•/QH2 or Q•−/QH2 couples, Figure 2. The semiquinones of (1)-(7) all have similar pKa values, Table 1, which forces the potentials for oxidation of QH2 to QH• or Q•− to shift by similar amounts. Notably, there is no correlation between the rate of superoxide production and the potential for the second electron transfer, Figure 3, indicating that the oxidation of SQo is not rate limiting for the production of superoxide.
Figure 3.

The Vmax for superoxide production, vSOP, in μmoles of superoxide per μmole of cyt bc1 s−1 measured with saturating amounts of substrate analog. Rates are plotted as a function of the shift in oxidation potential for substrate analogs for QH2/Q. Symbols are the same as in Figure 2A.
Discussion
Specific Molecular Interactions Do Not Have Large Effects on Superoxide Production
Alterations to the QH2 substrate of the Qo site can cause the cyt bc1 complex to produce superoxide at rates exceeding that of normal Q-cycle turnover (~100 s−1), Figure 2 and previous work (19). Altering the substrate could elicit rapid superoxide production by altering specific interactions between the binding site and the substrate that are crucial for steering or gating the reaction into products; or by changing the driving force for formation or subsequent reaction of SQo. The current work was designed to distinguish between these possibilities by probing superoxide production with a homologous series of synthetic substrate analogs.
The substrate analogs were selected, in part, to vary the sidegroups on the benzoquinone ring available to participate in specific interactions with the Qo site. Aside from the obvious effects of ring substituents on redox potentials (see below), there is no discernable relation between superoxide production and the position or type of ring substituent in (1)-(7). The quinol –OH groups and the hydrocarbon tail are certainly necessary for activity; yet, groups at other positions can change the Vmax by two orders of magnitude! The Vmax for superoxide production in AA-inhibited complex is little affected by which hydrophobic tail is attached, with undecylubiquinol, (3), decylubiquinol (34), and ubiquinol-3 (19) giving essentially equal rates. The position and the identity of individual chloro-, methyl-, methoxy-, or amino- sidegroups also are not decisive in determining the Vmax for superoxide production (except through their overall influence on the redox potential, see below). The rate of electron transfer from a substrate analog in this series bound at the Qo site is not controlled by the physical structure of the substrate analog.
Superoxide Production Rates are Controlled by the Driving Force for SQo Formation
The substrate analogs were also selected to provide large variations in both Q/QH2 redox potential and semiquinone stability constant, Ks, Table 2. The large scatter in Ks means that the potential or driving force for the first electron transfer does not track the potential for the second electron transfer (from SQo to O2) and makes it possible to distinguish between the first and second electron transfer reactions. The Vmax for superoxide production increases as the quinol becomes a better one-electron reducing agent and as the Rieske ISP becomes easier to reduce (34), Figure 2. That is, the rate of the reaction depends on the driving force for the one-electron oxidation of the substrate by the Rieske ISP but not on the driving force for the oxidation of SQo. Thus, the rate limiting step with saturating substrate concentrations in superoxide production is the direct, one-electron oxidation of QH2 by the ISP to produce SQo and reduced ISP. Because the activation energy with the natural substrate (34) for this step is the same as for the normal Q-cycle, the first electron transfer step in the bifurcated oxidation of quinol in the cyt bc1 complex must also be the direct, one-electron oxidation of QH2 by the ISP. Covian and Trumpower (42) recently reported that the first electron transfer step is no longer the rate limiting step at less than saturating substrate concentrations. They concluded that the rate limitation occurs at a different step for normal turnover than for the bypass reactions that occur under their conditions. That report did not determine where the rate limitation moved in the complicated, multi-step reactions pathway of the cyt bc1 complex, or whether the bypass reactions under those new conditions results in superoxide production.
Superoxide is Produced by SQ
Figures 2 and 3 clearly show that the rate limiting step for superoxide production is the first electron transfer. But what is the immediate source of the electron to convert O2 into superoxide--the reduced Rieske ISP; the reduced cyt bL; or SQo? It is not likely the Rieske ISP. In contrast to the FeS cluster in Complex I, the reduced Rieske ISP is rather stable in air-saturated solutions either as part of the cyt bc1 complex or as the purified protein owing to its unusually high redox potential (~+0.275 V at pH = 7.0) (56). Its ability to reduce oxygen is incapable of supporting the measured superoxide production rates. Furthermore, measurements of bypass reactions in the cyt bc1 complex find that at least one electron per quinol makes it through the high-potential chain (21, 22), safely passing through the Rieske ISP. An electron that enters the high-potential chain at the Rieske ISP apparently is not easily diverted to superoxide.
The electron to convert O2 to superoxide therefore must come from SQo but its path to O2 is not obvious. Electron transfer from SQo through heme bL or the low-potential chain to produce superoxide is one possibility. The proximal niche inhibitors myxothiazol or MOA stilbene (21, 22) prevent electrons from entering the low-potential chain, yet elicit substantial amounts of such superoxide from the cyt bc1 complex. Muller, et al. (21) showed that Vmax for superoxide production in the presence of proximal niche inhibitors is roughly half that of AA-inhibited complexes. This provides a clear demonstration that reduced cyt bL is not an essential precursor to superoxide because proximal niche inhibitors prevent electrons from entering the low-potential chain and reducing cyt bL. Moreover, when electrons can enter the low-potential chain, as with AA inhibition, the steady state accumulation of electrons on cyt bL ought to cause superoxide production at rates proportional to the amount of reduced cyt bL. Presently available data do not support this prediction (19); very reducing substrates such as rhodoquinol generate similar steady state levels of reduced cyt b as ubiquinol even though superoxide production rates differ by ~2 orders of magnitude. Thus, the low-potential chain does not appear to be able to pass electrons efficiently from SQo to superoxide.
SQo is the only remaining plausible source of the electron for superoxide production and SQo must therefore be considered the last known predecessor of superoxide. However, there is little direct data to indicate whether SQo reacts in the Qo site to produce superoxide or after escape from the Qo site.
Recent studies of superoxide production during ‘semi-reverse’ electron flow (32, 33) conclude that under extreme conditions, electron flow in the low-potential chain can be reversed, with electrons flowing from the Qi site to the Qo site, to reduce quinone in the Qo site to SQo and then producing superoxide. That route to superoxide production supports our results that SQo is the last known precursor to superoxide.
Superoxide Production Shares Intermediates with the Q-Cycle
Superoxide production rates at saturating substrate concentrations are related to shifts in redox potential of both the Rieske ISP and the quinol substrate, Figure 2. The relationship can be summarized following Crofts, et al., p. 1023 (23), based on Marcus Theory as:
This equation has been experimentally tested by variation of the ISP redox potential for normal Q-cycle turnover (23, 34) and for superoxide production (23). We now find that it holds when the redox potential of the substrate is varied. Thus, the free energy change for the one-electron transfer from substrate to the ISP predicts the rate of superoxide production. This indicates that the rate-limiting step for superoxide production is the one-electron transfer reaction to produce SQo (either as the neutral radical or as the radical anion) and reduced ISP. The activation energies at saturating substrate concentrations are the same for superoxide production and for oxidation of quinol in the Q-cycle (34), indicating the same rate limiting step in both processes. The paths then diverge at SQo or soon thereafter as proposed earlier (23, 34).
The initial electron transfer is thought to occur in a hydrogen-bonded complex between quinol and a histidine ligand of the FeS cluster (23, 32, 34, 57). In such a ‘contact ion pair’, back electron transfer should be rapid. A distinct SQo would appear only after the contact ion pair moves apart and back electron transfer slows dramatically, to kinetically ‘trap’ SQo so that it reacts with O2 or proceeds along the Q-cycle. The motion of the ISP headgroup to carry its FeS cluster to cyt c1 is the obvious way for the contact ion pair to dissociate and to terminate the electron transfer step. This view predicts that blocking movement of the ISP would prevent formation of SQo and superoxide. Indeed, Borek, et al. (32) reported no superoxide production in the double alanine insertion mutant of the ISP, where ISP movement is blocked (56). This observation was explained by a gating mechanism in which “the structural changes that occur in the Qo site upon movement of FeS” “act to transiently ‘open’ the Qo site for the reaction with oxygen” (32). The ‘opening’ of this gate is perhaps best viewed in a general sense in which a fully reactive SQo is not formed until the ISP has moved away from Qo.
Conclusion
Mounting evidence shows that some of the bypass reactions, particularly those producing superoxide during oxidation of quinol at the Qo site, diverge from the Q-cycle rather late in the bifurcated reaction. Consequently bypass reactions provide an additional means of studying the initial reactions of the Q-cycle as demonstrated here. There is more scope for controlling and manipulating reaction conditions, e.g., varying redox potential, because the bypass reactions effectively isolate or decouple the initial reactions in the Q-cycle from later steps, avoiding many complications and interactions. We used that approach here to examine the dependence of oxidation rate on the redox potential and structure of the substrate. We find that the direct one-electron oxidation of QH2 by the Rieske ISP produces a semiquinone during oxidation of quinol at the Qo site in the Q-cycle and in superoxide production. The oxidation of the semiquinone and the reduction of cyt b or O2 are subsequent steps, distinct from the rate limiting step. The production of superoxide whether through the oxidation of quinol in the Qo site studied here or through the reduction of quinone in the Qo site by reversed electron flow (32, 33) involves the generation of SQo which is the last known precursor to superoxide. At lower substrate concentrations, other reactions become rate limiting and detailed measurements of carefully designed mutants and substrate analogs or of kinetic isotope effects can make substantial contributions to determining the entire reaction network.
These experimental results reported here are incompatible: 1) with models for cyt bc1 in which electron transfer to the high-potential chain is not a distinct step preceding electron transfer to the low-potential chain, and 2) with conformational gating models that would produce superoxide by a very different set of reactions from the normal Q-cycle.
Supplementary Material
Acknowledgments
The authors acknowledge many useful discussions, in particular with Wolfgang Nitschke, Fevzi Daldal, Christopher Moser, P. Leslie Dutton, Antony R. Crofts, and Isaac Forquer. The authors also thank Daniel Vasseo and Michael Jourdes for their technical assistance.
Footnotes
Abbreviations: cyt, cytochrome; ISP, iron sulfur protein; Qo site, quinol oxidase site; Qi site, quinone reductase site; FeS, two iron-two sulfur cluster in the ISP; SQ, semiquinone; SQo, semiquinone in Qo; pmf, Proton motive force; AA, Antimycin A; MOA, methoxyacrylate; EPR, electron paramagnetic resonance; NMR, nuclear magnetic resonance; ESI, electrospray ionization; SOD, superoxide dismutase; Km, Michaelis constant; Vmax, maximum rate of reaction; Ki, inhibition constant.
This work was supported by a grant from the National Institutes of Health GM061904.
Supporting Information Available
The syntheses of the substrate analogs used in this study and the details of the estimation of redox potentials are described in detail as Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Crofts AR. The cytochrome bc1 complex: Function in the context of structure. Annual Review of Physiology. 2004;66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- 2.Berry EA, Guergova-Kuras M, Huang LS, Crofts AR. Structure and function of cytochrome bc complexes. Annu Rev Biochem. 2000;69:1005–1075. doi: 10.1146/annurev.biochem.69.1.1005. [DOI] [PubMed] [Google Scholar]
- 3.Schutz M, Brugna M, Lebrun E, Baymann F, Huber R, Stetter KO, Hauska G, Toci R, Lemesle-Meunier D, Tron P, Schmidt C, Nitschke W. Early evolution of cytochrome bc complexes. J Mol Biol. 2000;300:663–675. doi: 10.1006/jmbi.2000.3915. [DOI] [PubMed] [Google Scholar]
- 4.Nitschke W, Kramer DM, Riedel A, Liebl U. From naphtho-to benzoquinones — (R)evolutionary reorganizations of electron transfer chains. In: Mathis P, editor. Photosynthesis: From Light to Biosphere. Kluwer Academic Publishers; Dordrecht: 1995. pp. 945–948. [Google Scholar]
- 5.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 6.Crofts AR, Berry EA, Kuras R, Guergova-Kuras M, Hong S, Ugulava NB. Structures of the bc1 complex reveal dynamic aspects of mechanism. In: Garab G, editor. Photosynthesis: Mechanisms and Effects. Kluwer Academic Publ; Dordrecht: 1999. pp. 1481–1486. [Google Scholar]
- 7.Hunte C, Palsdottir H, Trumpower BL. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Letters. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 8.Esser L, Quinn B, Li YF, Zhang MQ, Elberry M, Yu L, Yu CA, Xia D. Crystallographic Studies of Quinol Oxidation Site Inhibitors: a Modified Classification of Inhibitors for the Cytochrome bc1 Complex. Journal of Molecular Biology. 2004;341:281–302. doi: 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell P. The protonmotive Q cycle: A general formulation. FEBS Letters. 1975;59:137–139. doi: 10.1016/0014-5793(75)80359-0. [DOI] [PubMed] [Google Scholar]
- 10.Crofts AR, Shinkarev VP, Kolling DRJ, Hong SJ. The Modified Q-Cycle Explains the Apparent Mismatch Between the Kinetics of Reduction of Cytochromes c1 and bH in the bc1 Complex. Journal of Biological Chemistry. 2003;278:36191–36201. doi: 10.1074/jbc.M305461200. [DOI] [PubMed] [Google Scholar]
- 11.Crofts AR, Wang Z. How rapid are the internal reactions of the ubiquinol: cytochrome c2 oxidoreductase? Photosynthesis Research. 1989;22:69–87. doi: 10.1007/BF00114768. [DOI] [PubMed] [Google Scholar]
- 12.Crofts AR, Wraight CA. The Electrochemical Domain of Photosynthesis. Biochim Biophys Acta. 1983;726:149–185. [Google Scholar]
- 13.Berry EA, Huang LS. Observations Concerning the Quinol Oxidation Site of the Cytochrome bc1 Complex. Febs Letters. 2003;555:13–20. doi: 10.1016/s0014-5793(03)01099-8. [DOI] [PubMed] [Google Scholar]
- 14.Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1, complex: Importance for the Q-cycle and superoxide production. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7887–7892. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang HB, Osyczka A, Dutton PL, Moser CC. Exposing the complex III Qo semiquinone radical. Biochimica Et Biophysica Acta-Bioenergetics. 2007;1767:883–887. doi: 10.1016/j.bbabio.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schultz BE, Chan SI. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annual Review of Biophysics and Biomolecular Structure. 2001;30:23–65. doi: 10.1146/annurev.biophys.30.1.23. [DOI] [PubMed] [Google Scholar]
- 17.Wenz T, Hellwig P, MacMillan F, Meunier B, Hunte C. Probing the role of E272 in quinol oxidation of mitochondrial complex III. Biochemistry. 2006;45:9042–9052. doi: 10.1021/bi060280g. [DOI] [PubMed] [Google Scholar]
- 18.Cape JL, Bowman MK, Kramer DM. Understanding the Cytochrome bc Complexes by What They Don’t Do. The Q-Cycle at 30. Trends in Plant Science. 2006;11:46–55. doi: 10.1016/j.tplants.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Cape JL, Strahan JR, Lenaeus MJ, Yuknis BA, Trieu TL, Shepherd JN, Bowman MK, Kramer DM. The Respiratory Substrate Rhodoquinol Induces Q-cycle Bypass Reactions in the Yeast Cytochrome bc1 Complex: Mechanistic and Physiological Implications. Journal of Biological Chemistry. 2005;280:34654–34660. doi: 10.1074/jbc.M507616200. [DOI] [PubMed] [Google Scholar]
- 20.Kramer DM, Roberts AG, Muller FL, Cape J, Bowman MK. Q-cycle Bypass Reactions at the Qo site of the Cytochrome bc1 (and Related) Complexes. Methods in Enzymology. 2004;382:21–45. doi: 10.1016/S0076-6879(04)82002-0. [DOI] [PubMed] [Google Scholar]
- 21.Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry. 2003;42:6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 22.Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry. 2002;41:7866–7874. doi: 10.1021/bi025581e. [DOI] [PubMed] [Google Scholar]
- 23.Crofts AR, Lhee S, Crofts SB, Cheng J, Rose S. Proton pumping in the bc1 complex: A new gating mechanism that prevents short circuits. Biochimica Et Biophysica Acta-Bioenergetics. 2006;1757:1019–1034. doi: 10.1016/j.bbabio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Mulkidjanian AY. Ubiquinol oxidation in the cytochrome bc1 complex: Reaction mechanism and prevention of short-circuiting. Biochimica Et Biophysica Acta-Bioenergetics. 2005;1709:5–34. doi: 10.1016/j.bbabio.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Osyczka A, Moser CC, Dutton PL. Fixing the Q cycle. Trends in Biochemical Sciences. 2005;30:176–182. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Rich PR. The quinone chemistry of BC complexes. Biochim Biophys Acta-Bioenergetics. 2004;1658(Supplement S):32–32. doi: 10.1016/j.bbabio.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 27.Osyczka A, Moser CC, Daldal F, Dutton PL. Reversible Redox Energy Coupling in Electron Transfer Chains. Nature. 2004;427:607–612. doi: 10.1038/nature02242. [DOI] [PubMed] [Google Scholar]
- 28.Shinkarev VP, Crofts AR, Wraight CA. The electric field generated by photosynthetic reaction center induces rapid reversed electron transfer in the bc1 complex. Biochemistry. 2001;40:12584–12590. doi: 10.1021/bi011334j. [DOI] [PubMed] [Google Scholar]
- 29.Tahara EB, Navarete FDT, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radical Biology and Medicine. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Rottenberg H, Covian R, Trumpower BL. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem. 2009:M109.017376. doi: 10.1074/jbc.M109.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisher N, Castleden CK, Bourges I, Brasseur G, Dujardin G, Meunier B. Human Disease-Related Mutations in Cytochrome b Studied in Yeast. Journal of Biological Chemistry. 2004;279:12951–12958. doi: 10.1074/jbc.M313866200. [DOI] [PubMed] [Google Scholar]
- 32.Borek A, Sarewicz M, Osyczka A. Movement of the Iron-Sulfur Head Domain of Cytochrome bc1 Transiently Opens the Catalytic Qo Site for Reaction with Oxygen. Biochemistry. 2008;47:12365–12370. doi: 10.1021/bi801207f. [DOI] [PubMed] [Google Scholar]
- 33.Drose S, Brandt U. The Mechanism of Mitochondrial Superoxide Production by the Cytochrome bc1 Complex. J Biol Chem. 2008;283:21649–21654. doi: 10.1074/jbc.M803236200. [DOI] [PubMed] [Google Scholar]
- 34.Forquer I, Covian R, Bowman MK, Trumpower BL, Kramer DM. Similar transition states mediate the Q-cycle and superoxide production by the cytochrome bc1 complex. Journal of Biological Chemistry. 2006;281:38459–38465. doi: 10.1074/jbc.M605119200. [DOI] [PubMed] [Google Scholar]
- 35.Hong SJ, Ugulava N, Guergova-Kuras M, Crofts AR. The Energy Landscape for Ubihydroquinone Oxidation at the Qo Site of the bc1 Complex in Rhodobacter Sphaeroides. Journal of Biological Chemistry. 1999;274:33931–33944. doi: 10.1074/jbc.274.48.33931. [DOI] [PubMed] [Google Scholar]
- 36.Trumpower BL. A Concerted, Alternating Sites Mechanism of Ubiquinol Oxidation by the Dimeric Cytochrome bc1 Complex. Biochimica Et Biophysica Acta-Bioenergetics. 2002;1555:166–173. doi: 10.1016/s0005-2728(02)00273-6. [DOI] [PubMed] [Google Scholar]
- 37.Lange C, Hunte C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2800–2805. doi: 10.1073/pnas.052704699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darrouzet E, Valkova-Valchanova M, Moser CC, Dutton PL, Daldal F. Uncovering the [2Fe2S] Domain Movement in Cytochrome bc1 and its Implications for Energy Conversion. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4567–4572. doi: 10.1073/pnas.97.9.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darrouzet E, Valkova-Valchanova M, Ohnishi T, Daldal F. Structure and function of the bacterial bc1 complex: Domain movement, subunit interactions, and emerging rationale engineering attempts. Journal of Bioenergetics and Biomembranes. 1999;31:275–288. doi: 10.1023/a:1005428014548. [DOI] [PubMed] [Google Scholar]
- 40.Crofts AR. The bc1 Complex: What is There Left to Argue About? In: Wikstrom M, editor. Biophysical and Structural Aspects of Bioenergetics. Royal Society of Chemistry; Cambridge: 2006. pp. 123–155. [Google Scholar]
- 41.Iwata M, Bjorkman J, Iwata S. Conformational change of the Rieske [2Fe-2S] protein in cytochrome bc1 complex. J Bioenerg Biomembr. 1999;31:169–175. doi: 10.1023/a:1005407410005. [DOI] [PubMed] [Google Scholar]
- 42.Covian R, Trumpower BL. The Rate-limiting Step in the Cytochrome bc1 Complex (Ubiquinol-Cytochrome c Oxidoreductase) Is Not Changed by Inhibition of Cytochrome b-dependent Deprotonation: Implications for the Mechanism of ubiquinol Oxidation at Center P of the bc1 Complex. J Biol Chem. 2009;284:14359–14367. doi: 10.1074/jbc.M109.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cape JL, Bowman MK, Kramer DM. Computation of the redox and protonation properties of quinones: Towards the prediction of redox cycling natural products. Phytochemistry. 2006;67:1781–1788. doi: 10.1016/j.phytochem.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 44.Folkers K, Catlin JC, Daves GD. Coenzyme Q. 124. New substituted 2,3-dimethoxy-1,4-benzoquinones as inhibitors of coenzyme Q systems. Journal of Medicinal Chemistry. 1971;14:45–48. doi: 10.1021/jm00283a012. [DOI] [PubMed] [Google Scholar]
- 45.Gu LQ, Yu L, Yu CA. Effect Of Substituents Of The Benzoquinone Ring On Electron-Transfer Activities Of Ubiquinone Derivatives. Biochimica Et Biophysica Acta. 1990;1015:482–492. doi: 10.1016/0005-2728(90)90082-f. [DOI] [PubMed] [Google Scholar]
- 46.Keegstra EMD, Huisman BH, Paardekooper EM, Hoogesteger FJ, Zwikker JW, Jenneskens LW, Kooijman H, Schouten A, Veldman N, Spek AL. 2,3,5,6-tetraalkoxy-1,4-benzoquinones and structurally related tetraalkoxy benzene derivatives: Synthesis, properties and solid state packing motifs. Journal of the Chemical Society-Perkin Transactions. 1996;2:229–240. [Google Scholar]
- 47.Gu LQ, Yu L, Yu CA. A ubiquinone derivative that inhibits mitochondrial cytochrome bc1 complex but not chloroplast cytochrome b6f complex activity. J Biol Chem. 1989;264:4506–4512. [PubMed] [Google Scholar]
- 48.Ljungdahl PO, Pennoyer JD, Robertson DE, Trumpower BL. Purification of highly active cytochrome bc1 complexes from phylogenetically diverse species by a single chromatographic procedure. Biochimica et Biophysica Acta (BBA) -Bioenergetics. 1987;891:227–241. doi: 10.1016/0005-2728(87)90218-0. [DOI] [PubMed] [Google Scholar]
- 49.Fisher N, Bourges I, Hill P, Brasseur G, Meunier B. Disruption of the interaction between the Rieske iron-sulfur protein and cytochrome b in the yeast bc1 complex owing to a human disease-associated mutation within cytochrome b. European Journal of Biochemistry. 2004;271:1292–1298. doi: 10.1111/j.1432-1033.2004.04036.x. [DOI] [PubMed] [Google Scholar]
- 50.Wenz T, Covian R, Hellwig P, MacMillan F, Meunier B, Trumpower BL, Hunte C. Mutational analysis of cytochrome b at the ubiquinol oxidation site of yeast complex III. Journal of Biological Chemistry. 2007;282:3977–3988. doi: 10.1074/jbc.M606482200. [DOI] [PubMed] [Google Scholar]
- 51.Fresht A. Enzyme Structure and Mechanism. W. H. Freeman; San Francisco: 1977 . [Google Scholar]
- 52.Marchal D, Boireau W, Laval JM, Moiroux J, Bourdillon C. An electrochemical approach of the redox behavior of water insoluble ubiquinones or plastoquinones incorporated in supported phospholipid layers. Biophysical Journal. 1997;72:2679–2687. doi: 10.1016/S0006-3495(97)78911-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michalkiewicz S. Cathodic reduction of coenzyme Q10 on glassy carbon electrode in acetic acid-acetonitrile solutions. Bioelectrochemistry. 2007;70:495–500. doi: 10.1016/j.bioelechem.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 54.Baik MH, Friesner RA. Computing redox potentials in solution: Density functional theory as a tool for rational design of redox agents. Journal of Physical Chemistry A. 2002;106:7407–7412. [Google Scholar]
- 55.Fu Y, Liu L, Wang YM, Li JN, Yu TQ, Guo QX. Quantum-chemical predictions of redox potentials of organic anions in dimethyl sulfoxide and reevaluation of bond dissociation enthalpies measured by the electrochemical methods. Journal of Physical Chemistry A. 2006;110:5874–5886. doi: 10.1021/jp055682x. [DOI] [PubMed] [Google Scholar]
- 56.Cooley JW, Roberts AG, Bowman MK, Kramer DM, Daldal F. The Raised Midpoint Potential of the [2Fe2S] Cluster of Cytochrome bc1 Is Mediated by Both the Qo Site Occupants and the Head Domain Position of the Fe-S Protein Subunit. Biochemistry. 2004;43:2217–2227. doi: 10.1021/bi035938u. [DOI] [PubMed] [Google Scholar]
- 57.Cape JL, Bowman MK, Kramer DM. Reaction Intermediates of Quinol Oxidation in a Photoactivatable System That Mimics Electron Transfer in the Cytochrome bc1 Complex. Journal of the American Chemical Society. 2005;127:4208–4215. doi: 10.1021/ja043955g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.