

Abstract
Decarbonization of the transport system and a transition to a new diversified energy system that is scalable and sustainable, requires a widespread implementation of carbon-neutral fuels. In biomimetic supramolecular nanoreactors for solar-to-fuel conversion, water-splitting catalysts can be coupled to photochemical units to form complex electrochemical nanostructures, based on a systems integration approach and guided by magnetic resonance knowledge of the operating principles of biological photosynthesis, to bridge between long-distance energy transfer on the short time scale of fluorescence, ~10−9 s, and short-distance proton-coupled electron transfer and storage on the much longer time scale of catalysis, ~10−3 s. A modular approach allows for the design of nanostructured optimized topologies with a tunneling bridge for the integration of storage with catalysis and optimization of proton chemical potentials, to mimic proton-coupled electron transfer processes in photosystem II and hydrogenase.
Introduction
Although solar energy plays a major role in all scenarios for a secure, sustainable and efficient energy supply, direct solar-to-fuel conversion remains a distant goal due to major scientific and technological challenges. During his career, Lubitz et al. [1] has been one of the pioneers in the scientific underpinning of the principles of photosynthetic energy conversion, with a particular focus on resolving molecular mechanisms of multi-electron catalysis in photosystem II and in hydrogenases. The recent scientific breakthroughs in basic research in photosynthesis have made it possible to learn from Nature how to construct artificial devices that can be put to use for harnessing solar energy for sustainable production of primary energy carriers, like hydrogen from water or carbon-based fuels from CO2. Building bio-inspired photoelectrochemical cells for fuel production from solar energy will require the development and systems design of integrated supramolecular modular systems that combine functionalities for light harvesting, charge separation, proton-coupled electron transfer and multi-electron catalysis (Fig. 1). With a nanostructured device, it is possible to use the physical–chemical principles for efficient photochemical conversion and storage at early stages of natural photosynthesis as a blueprint for the construction of artificial systems that convert solar energy into fuel in a rational higher order supramolecular assembly design by systems integration. The crucial point that is brought forward in this contribution, is that a full merger between catalysis and storage will allow for optimal matching of time, length and energy scales for solar energy to fuel conversion [2].
Fig. 1.
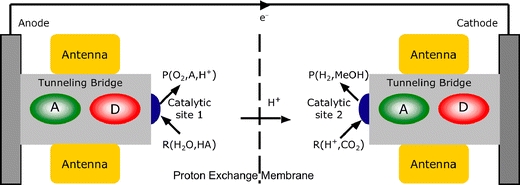
Solar energy-driven photoelectrochemistry for production of hydrogen or methanol with high efficiency (schematic). Systems integration of modules will combine a light-harvesting antenna system and photochemical reaction center, containing a donor (D) and an acceptor (A), to produce a stable photocurrent that drives novel catalysts for multi-electron reactions (R → P). Such “smart matrices” represent build upon knowledge about natural photosynthesis and hydrogenase to extract electrons from water and produce fuel(s) from protons or CO2
Photochemical Energy Storage
In nature, the photosynthetic apparatus is connected to a steady-state network of catalytic conversion reactions in the organism that is continuously dissipating energy and generating entropy. From a biological perspective, photosynthesis is an expensive process in terms of resources required, and organisms generally produce only the photosynthetic capacity that they need to serve their requirements within the limits of the environmental and developmental constraints. At low light intensities, however, the productive storage of energy is an efficient chemical thermodynamics process, which is optimized against depletion by wasteful back reactions that feed into the decay of the absorber with rate g = 1/τ from the excited chlorophyll state into the ground state at the start of the conversion chain [3]. Natural photosynthesis requires coupling of primary conversion into biochemical networks and catalytic cycles that are subject to regulation for protection against excess light, leading to limited photosynthetic capacity. In contrast, for artificial photosynthesis it is necessary to consider the minimal design requirements for the most efficient solar-to-fuel conversion process, operating close to the theoretical limits on solar energy conversion.
To understand how thermodynamic constraints can guide the design of artificial systems for photochemical storage, it is best to start from minimal models that consider the thermodynamic constraints on the photovoltaic or photochemical solar energy conversion by exploring the analogy with efficient conversion in the solar or photovoltaic cell [3–5]. In productive photochemical conversion, the storage or downstream utilization is described as a single step conversion of solar energy into Gibbs free energy. Like the silicon solar cell, the photosynthetic solar cell is in its simplest form a molecular absorber that is excited and produces energy conversion by charge separation with rate I into an electron and hole in dynamic equilibrium with the absorber (Fig. 2). The excitation of a molecular Chl absorber in exchange with the field of solar irradiation leads to a difference in chemical potential
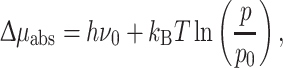 |
1 |
with p and p 0 being the populations of the excited and ground state of the absorber, respectively [4]. In the dark, the excited and ground state are in equilibrium and Δμ abs = 0, leading to p/p 0 = exp(−hv 0/k B T), the Boltzmann distribution. For Chl a in green plants, hν 0 = 1.8 eV. Detailed balance has been used to calculate the upper limit at the absorber stage, which leads to the maximum efficiency η = 1 − 4T/3T S = 93%, for reversible operation, due to entropic losses originating from the different temperatures of the incoming radiation at T S ~ 5800 K and of the heat reservoir at T ~ 300 K [3, 5, 6].
Fig. 2.
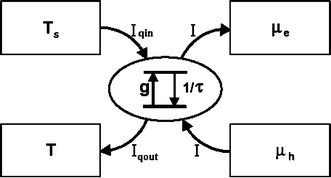
Basic electrochemistry and energetics of the photosynthetic solar cell, after ref. [4]. A chlorophyll molecule is connected to a reservoir at ambient temperature T ~ 300 K, and emitting heat at a rate I qout, is excited due to the heat flow I qin from the solar reservoir at temperature T = 5800 K, with a rate g to an excited state separated by an energy ην 0 from the ground state. The excited chlorophyll state either decays with loss rate 1/τ or produces an electron–hole pair with net charge separation rate I and free energy Δμ = μ e − μ h
A photovoltaic (PV) solar cell produces electricity, while a photosynthetic reaction center produces a photochemical steady state with a voltage over the membrane and charge separation in dynamic equilibrium with the absorber. Recently, the solar energy conversion has been considered in a general setting, including both the photochemical and photovoltaic mechanisms [4]. The equation
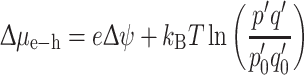 |
2 |
represents the difference in chemical potential or free energy produced by light-induced charge separation and contains an electronic term eΔψ. In addition, a photochemical term 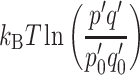 measures the concentration p′q′ of the charge separated state, i.e., the probabilities of electron and hole occupation of the acceptor and donor states, in the light relative to the concentration p
0′q
0′ in the dark. This term is proportional to the temperature. Equation (2) illustrates how the electrochemical potential Δμ
e−h can comprise a light-induced electrostatic potential or charge separation eΔψ, a photochemical
measures the concentration p′q′ of the charge separated state, i.e., the probabilities of electron and hole occupation of the acceptor and donor states, in the light relative to the concentration p
0′q
0′ in the dark. This term is proportional to the temperature. Equation (2) illustrates how the electrochemical potential Δμ
e−h can comprise a light-induced electrostatic potential or charge separation eΔψ, a photochemical 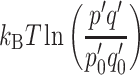 , or both, as in photosynthesis [4].
, or both, as in photosynthesis [4].
The net production rate of the charge separation and energy storage in a photosynthetic process can then be described by the solar cell equation
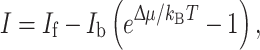 |
3 |
with I f and I b being the forward and backward reaction rates, respectively [6, 7]. The difference I represents the energy extraction. For PV, this is electrical energy, and for artificial photosynthesis, I is the net flow of energy into proton-coupled electron transfer and temporary storage, followed by the multi-electron chemical reaction that oxidizes water or produces fuel.
The second principle of thermodynamics, stating that entropy cannot be destroyed, limits the efficiency of solar energy conversion to 0.93 for reversible operation [6]. Extraction of energy implies a non-reversible process with a theoretical limit less than 93% for the reversible process [3]. Processing the incident solar energy current is thus not possible without lowering the efficiency, and Eq. (2) can be generalized according to
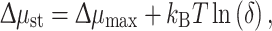 |
4 |
with a quantum yield
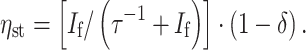 |
5 |
Here the first factor is due to the kinetics of the steady state, while δ measures the thermodynamic activity of the trap [3]. Hence, in order to favor the forward reaction over the backward process, the energy of the combined products is lower than for the excited state by at least k B Tln(δ). The maximum power is generated for δ ~ k B T/μmax, a compromise between a high conversion efficiency and a high storage efficiency.
Since multi-electron catalysis for water oxidation or hydrogen production requires storage for downstream accumulation of electronic charges at longer time scales than the initial light absorption process, a catalytic site in an artificial photosynthesis device will be in Boltzmann equilibrium with the initial excited state throughout the entire process, both during the accumulation of charge and during the actual catalytic reaction. This makes that the processes of storage and catalysis have to go hand in hand. Catalytic processes operate on the millisecond time scale, and the accumulation of several electrons or holes on this time scale is rate limiting. Thus, Δμ st inside the catalyst should be sufficiently low to trap electrons or holes for allowing multi-electron catalysis to proceed against back reactions. For efficient solar-to-fuel conversion, the overall rate of energy extraction has to be close to the net rate of excitation of the molecular absorber at the start of the conversion chain. When the absorber operates at an energy hν 0 with an excited state lifetime τ, the theoretical limit to the electrochemical potential Δμ st that can be established inside the catalyst in a reversible process in a multi-electron catalytic chain will depend on the required storage time t for accumulation prior to the actual catalytic process, according to [2, 8]
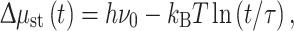 |
6 |
and will decrease in order to establish a net energy flow I through the catalyst for the chemical reaction, in line with Eqs. (3)–(5).
An Integrated Modular Approach for Solar-to-Fuel Conversion
The fundamental limits on solar energy conversion in photosynthesis have been described many times in the literature. The crucial point about Eq. (6) is, however, that it links the time scale of the chemical process to the electrochemical potential that is established inside the catalyst, and to the free energy difference hν 0 − Δμ st(t) that is minimally required to achieve the desired steady state for thermodynamic reasons. It puts a fundamental restraint on how to match length, energy and time scales, and allows for the design of nanostructured artificial solar-to-fuel conversion topologies based on the knowledge about length, time and energy scales from natural photosynthesis that have been discussed extensively recently [9].
In Fig. 1, catalysis is integrated with storage to allow for a multi-electron process. When operating a multi-electron catalyst in artificial photosynthesis, part of the initial free energy, stored in the solar energy assimilation hν 0, will be necessary to produce a free energy difference hν 0 − Δμ st(t). This free energy difference is inevitable, and must be put to use in promoting confinement of electrons or holes inside catalytic sites, in line with Eq. (6). Viewed in this way, the term k B Tln(t/τ) leads to a nanostructure design concept that is fundamentally different from alternatives, that work from delocalized electrons at moderately functionalized extended semiconductor interfaces or try to combine the different required functions of storage and catalysis into essentially one or two complex molecules that perform all the functions of an anode or a cathode.
In particular, the incorporation of insulating tunneling bridges between the catalyst and the electrode are critical parameters in the design. Inside the nanostructure, the eΔψ and the 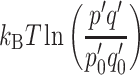 are interchangeable, as described by Eq. (2). During accumulation, energy is to be preserved locally, initially in the form of eΔψ, followed by chemical
coupling
into a
proton
process to temporarily
neutralize the
charge. It may appear counterintuitive at first sight, but the tunneling bridge ensures that the overvoltage for catalysis produces a suitable confinement potential for electrons or holes in the catalytic sites. A tunneling bridge of ~20 Å will allow the accumulation of charges on a time scale necessary for multi-electron catalysis [9]. Similarly, the spatial orientation and separation between the antenna and the donor–acceptor pairs will be adjusted for optimal exciton trapping and charge separation [9].
are interchangeable, as described by Eq. (2). During accumulation, energy is to be preserved locally, initially in the form of eΔψ, followed by chemical
coupling
into a
proton
process to temporarily
neutralize the
charge. It may appear counterintuitive at first sight, but the tunneling bridge ensures that the overvoltage for catalysis produces a suitable confinement potential for electrons or holes in the catalytic sites. A tunneling bridge of ~20 Å will allow the accumulation of charges on a time scale necessary for multi-electron catalysis [9]. Similarly, the spatial orientation and separation between the antenna and the donor–acceptor pairs will be adjusted for optimal exciton trapping and charge separation [9].
While it is essential to have the free energy difference operational in confining electrons and holes, it will be equally essential that the subsequent chemical conversion can proceed downhill, without any restraints. This implies that there should not be a potential barrier between the proton exchange membrane and the catalytic nanostructure, to allow for barrierless proton-coupled electron transfer and smooth charge transfer across the proton exchange membrane. Previous attempts to perform multi-electron catalysis on an electrode often work from catalysts embedded in proton exchange membranes optimized for proton conduction, such as NAFION, with a pKa ~ −6. In this case, the chemical potential for protons at the catalytic site is very high. While such an approach may work well for hydrogen production at a photocathode, for water oxidation at a photoanode it will give rise to a free energy difference that competes with the tunneling barrier, quenching catalysis. For a photoanode it would be preferred to work from an alkaline environment to produce a low proton chemical potential and to tune the chemical potential of intermediates in the multi-electron pathway for optimal proton-coupled electron transfer and a charged resting state. This is in line with the mechanisms in natural photosynthesis that have been resolved with EPR by Lubitz and others [1], and recently also by NMR and theoretical analyses [10, 11].
Concluding Remarks
Many scientists believe that it is now possible, given sufficient resources, to capitalize on recent scientific progress in the understanding of photosynthesis, to learn from Nature how to harness solar energy for sustainable production of primary energy carriers, like hydrogen from water or carbon-based fuels from CO2, at an affordable cost. Photochemical devices that mimic the photosynthesis of plant leaves and produce renewable fuels from solar energy using water as raw material, require the integration of efficient light harvesting and photocatalysis in nano-structured “smart matrices” based on powerful biochemical and biophysical insights on natural water splitting and hydrogenases to address the complex chemistry of conversion and storage by multi-electron photocatalysis. When vigorously pursued, such systems represent one of the very few major options that mankind has to provide clean energy, provide its security in a humanitarian manner instead of confrontation, and eliminate the ‘overshoot’ of our footprint that is now maintained by liquidating the planet’s ecological resources.
Acknowledgments
This research was financed in part by the Chemical Sciences Section of the Netherlands Organization for Scientific Research (NWO) through a TOP grant. Discussions with Hans van Gorkom on the thermodynamics and energetics of photosynthesis are gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Lubitz W, Reijerse EJ, Messinger J. Energy Environ. Sci. 2008;1:15–31. doi: 10.1039/b808792j. [DOI] [Google Scholar]
- 2.A. Pandit, H.J.M. de Groot, A. Holzwarth, Harnessing Solar Energy for the Production of Clean Fuels (European Science Foundation, Strasbourg, France, 2006)
- 3.Ross RT, Calvin M. Biophys. J. 1967;7:595–614. doi: 10.1016/S0006-3495(67)86609-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markvart T, Landsberg PT. Physica E. 2002;14:71–77. doi: 10.1016/S1386-9477(02)00352-1. [DOI] [Google Scholar]
- 5.Wurfel P. Physica E. 2002;14:18–26. doi: 10.1016/S1386-9477(02)00355-7. [DOI] [Google Scholar]
- 6.Shockley W, Queisser HJ. J. Appl. Phys. 1961;32:510. doi: 10.1063/1.1736034. [DOI] [Google Scholar]
- 7.Archer MD, Bolton JR. J. Phys. Chem. 1990;94:8028–8036. doi: 10.1021/j100384a011. [DOI] [Google Scholar]
- 8.van Gorkom HJ. Bioelectrochem. Bioenerg. 1986;16:77–87. doi: 10.1016/0302-4598(86)80047-2. [DOI] [Google Scholar]
- 9.Noy D, Moser CC, Dutton PL. Biochim. Biophys. Acta. 2006;1757:90–105. doi: 10.1016/j.bbabio.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Rossmeisl J, Dimitrievski K, Siegbahn P, Norskov JK. J. Phys. Chem. C. 2007;111:18821–18823. doi: 10.1021/jp077210j. [DOI] [Google Scholar]
- 11.Alia A, Wawrzyniak PK, Janssen GJ, Buda F, Matysik J, de Groot HJM. J. Am. Chem. Soc. 2009;131:9626–9627. doi: 10.1021/ja9028507. [DOI] [PubMed] [Google Scholar]