

Abstract
Prochlorococcus, an abundant phototroph in the oceans, are infected by members of three families of viruses: myo-, podo- and siphoviruses. Genomes of myo- and podoviruses isolated on Prochlorococcus contain DNA replication machinery and virion structural genes homologous to those from coliphages T4 and T7 respectively. They also contain a suite of genes of cyanobacterial origin, most notably photosynthesis genes, which are expressed during infection and appear integral to the evolutionary trajectory of both host and phage. Here we present the first genome of a cyanobacterial siphovirus, P-SS2, which was isolated from Atlantic slope waters using a Prochlorococcus host (MIT9313). The P-SS2 genome is larger than, and considerably divergent from, previously sequenced siphoviruses. It appears most closely related to lambdoid siphoviruses, with which it shares 13 functional homologues. The ∼108 kb P-SS2 genome encodes 131 predicted proteins and notably lacks photosynthesis genes which have consistently been found in other marine cyanophage, but does contain 14 other cyanobacterial homologues. While only six structural proteins were identified from the genome sequence, 35 proteins were detected experimentally; these mapped onto capsid and tail structural modules in the genome. P-SS2 is potentially capable of integration into its host as inferred from bioinformatically identified genetic machinery int, bet, exo and a 53 bp attachment site. The host attachment site appears to be a genomic island that is tied to insertion sequence (IS) activity that could facilitate mobility of a gene involved in the nitrogen-stress response. The homologous region and a secondary IS-element hot-spot in Synechococcus RS9917 are further evidence of IS-mediated genome evolution coincident with a probable relic prophage integration event. This siphovirus genome provides a glimpse into the biology of a deep-photic zone phage as well as the ocean cyanobacterial prophage and IS element ‘mobilome’.
Introduction
Phages (viruses that infect prokaryotes) represent the largest source of uncharacterized genetic diversity in the biosphere (Pedulla et al., 2003). One particular group of these phages, the ocean cyanophages, has been relatively well studied because of the global abundance of their cyanobacterial hosts (Partensky et al., 1999; Waterbury et al., 1979; 1986), for which a number of genome sequences are available (Rocap et al., 2003; Palenik et al., 2003; 2006; Dufresne et al., 2003; 2008; Coleman et al., 2006; Kettler et al., 2007). The abundance of ocean cyanophages often covaries with cyanobacterial abundance in the wild (Waterbury and Valois, 1993; Suttle and Chan, 1994; Lu et al., 2001; Marston and Sallee, 2003; Sullivan et al., 2003; Muhling et al., 2005). Though estimating the quantitative impact of cyanophages on mortality of their cyanobacterial hosts is challenging due to the current need to compare strain-specific cyanophage titres to total cyanobacterial counts, cyanophages are thought to be responsible for a small, but significant fraction of cell mortality (Waterbury and Valois, 1993; Suttle and Chan, 1994; Fuhrman, 2000).
Three morphologies of viruses – myo-, podo- and siphoviruses – are known to infect ocean cyanobacteria. The myovirus and podovirus cyanophage families have been relatively well characterized by morphology, host range and genomics (Waterbury and Valois, 1993; Chen and Lu, 2002; Sullivan et al., 2003; 2005; Mann et al., 2005), and almost universally contain homologues to their host's photosynthetic machinery, including the core reaction centre genes of the photosystem (Mann et al., 2003; Millard et al., 2004; Lindell et al., 2004, Zeidner et al., 2005; Sullivan et al., 2006). The core photosynthesis reaction centre gene, psbA, has been shown to be expressed during infection for a podovirus (Lindell et al., 2005; 2007) and a myovirus (Clokie et al., 2006), and is hypothesized to play a role in cyanophage fitness (Lindell et al., 2007; Bragg and Chisholm, 2008; Hellweger, 2009). Not only do these genes appear to be important for the cyanophage, but sequence analysis has shown that subsections of the phage copy can be traced back to their host genome (Sullivan et al., 2006). Thus, ocean cyanophages appear to influence the evolution of cyanobacterial genomes via horizontal gene transfer events, even at the level of the core reaction centres (Zeidner et al., 2005; Sullivan et al., 2006).
Cyanophage studies to date have focused on lytic phages, which infect the host, use its machinery to replicate and burst the cell, releasing phage progeny. In contrast, temperate phages infect their hosts and may temporarily insert their DNA into the host genome as a prophage, which is replicated with the host genome as part of the cell cycle. Expression of prophage genes often fundamentally changes the host's physiology – a process known as lysogenic conversion (Calendar, 1988). For example, pathogen-associated toxin genes are commonly encoded by prophages (Miao and Miller, 1999; Boyd et al., 2001; Wagner and Waldor, 2002), which act as a mechanism for horizontally transferring such toxins between microbial species (Banks et al., 2002). More than 70% of sequenced bacterial genomes contain prophages (Canchaya et al., 2003; Casjens, 2003). They often represent the primary constituent of strain-to-strain variability (Simpson et al., 2000; Baba et al., 2002; Beres et al., 2002; Smoot et al., 2002), and their genes are among the most highly expressed genes in genome-wide expression studies (Smoot et al., 2001; Whiteley et al., 2001).
Curiously, the genomes of currently available freshwater and marine cyanobacterial genomes lack identifiable prophage (Canchaya et al., 2003; Casjens, 2003; Dufresne et al., 2003; 2008; Coleman et al., 2006; Kettler et al., 2007), in spite of two lines of indirect evidence that suggest prophages exist in cyanobacteria. First, strain-to-strain variability in marine cyanobacterial genomes is often clustered in genomic islands with signatures of phage and mobile element activity even including phage-like integrase genes (Palenik et al., 2003; Coleman et al., 2006; Kettler et al., 2007; DuFresne et al., 2008). Second, addition of inducing agents to natural seawater communities have yielded increases in culturable Synechococcus cyanophage thought to be induced prophage (McDaniel et al., 2002; Ortmann et al., 2002).
Here, we characterize the genome and proteome of an ocean siphovirus that was isolated from 83 m deep Atlantic Ocean slope waters using Prochlorococcus MIT9313 as a host strain. The data are analysed on their own, and in an evolutionary context using comparative genomics of Prochlorococcus and Synechococcus genomes. To this end, we uncover basic biology of the siphovirus genome, identify a possible integration site in its host, and explore the evolutionary link between insertion sequence (IS) activity and prophage integration in the Prochlorococcus and related Synechococcus host genomes.
Results and discussion
The architecture of the P-SS2 particle and its genome
P-SS2 has the morphology of a siphovirus, with a ∼75 nm diameter elongated (∼140 nm long) capsid and a ∼325 nm flexible, non-contractile tail (Fig. 1A). This is the largest siphovirus for which a complete genome has been sequenced (Table 1), and the size of its genome is also large: at 107 595 bp (Fig. 1B; Table 1) it is surpassed only by the 122 kb coliphage T5 genome (Table 1). Of the 131 predicted open reading frames (ORFs) in the P-SS2 genome, only 38 have recognizable homologues (Table 2). This is proportionally fewer than in other siphoviruses, where often half the predicted proteins have recognizable homologues (Brussow and Desiere, 2001; Proux et al., 2002; Pedulla et al., 2003). It is, however, proportionally similar to the alpha-proteobacterial marine siphovirus phi-JL001 where only 17 of 91 ORFs had homologous proteins in the database (Lohr et al., 2005), and consistent with the idea that marine siphoviruses encode proteins that are under-represented in the database. Of the 38 ORFs in the P-SS2 genome that have homologues (Table 2), 24 have ascribed functions, eight are hypotheticals predominately from cyanobacteria or their phages, and six are ORFan proteins with a single database match.
Fig. 1.

The morphology (A) and genome/structural proteome (B) of Prochlorococcus siphovirus P-SS2. A. Electron micrograph of uranyl acetate negative-stained, purified P-SS2 viral particle. B. The open reading frames (ORFs) are indicated on either the positive (above grey line) or negative (below grey line) DNA strand. Bioinformatically determined promoters and terminators are indicated, as is a putative host integration site (see Fig. 2). Structural proteins detected using mass-spectrometry are indicated by the diagonal lines in the corresponding ORFs, with structural modules indicated by the red lines and the text underneath the genome. For further detail, the number of virion structural peptides detected per ORF is provided in Table 2. The genome sequence is deposited in GenBank under accession #GQ334450.
Table 1.
Genome-wide characteristics of marine siphoviruses P-SS2 (this study) and phi-JL001 (Lohr et al., 2005) relative to other recognized phage groups within the Siphoviridae. Siphoviruses are all non-enveloped and contain double-stranded DNA genomes, non-contractile, flexible tails, and are distinguished by different combinations of alleles of structural and DNA replication proteins
| Genome features |
Particle features |
||||
|---|---|---|---|---|---|
| Phage genusa | Size (kb)b | # ORFs | %G+C | Capsid diameter (nm) | Tail (nm) – L × W |
| Marine, non-classified siphoviruses | |||||
| cyanophage P-SS2 | 108 | 131 | 52.3 | 75 | 325 × 12 |
| Alpha-protebacteria φJL001 | 63 | 91 | 62 | 75 | 125 × N.D. |
| Lambda-likesb | |||||
| Enterobacteria phage λ | 48.5 | 92 | 49 | 60 | 150 × 8 |
| Enterobacteria phage HK022 | 40.8 | 57 | 49 | 51 | 106 × N.D. |
| Enverobacteria phage HK97 | 39.7 | 62 | 49 | 54 | 179 × N.D. |
| T1-likes | |||||
| Enterobacteria phage T1 | 48.8 | 78 | 45 | 60 | 150 × 8 |
| Enterobacteria phage TLS | 49.9 | 87 | 42 | 50 | N.D. |
| Enterobacteria phage RTP | 46.2 | 75 | 44 | 60 | 160 × N.D. |
| L5-likes | |||||
| Mycobacterium phage L5 | 52.3 | 88 | 62 | 60 | 135 × 8 |
| Mycobacterium phage D29 | 49.1 | 84 | 63 | N.D. | N.D. |
| Mycobacterium phage Bxb1 | 50.6 | 86 | 63 | 60 | 135 × N.D. |
| φC31-likes | |||||
| Streptomyces phage φC31 | 41.5 | 54 | 63 | 53 | 100 × 5 |
| Streptomyces phage φBT1 | 41.8 | 56 | 62 | N.D. | N.D. |
| N15-likes | |||||
| Enterobacteria phage N15 | 46.4 | 60 | 51 | 60 | 140 × 8 |
| T5-likes | |||||
| Enterobacteria phage T5 | 121.7 | 195 | 39 | 80 | 180 × 9 |
| c2-likes | |||||
| Lactococcus phage bIL67 | 22.2 | 37 | 35 | 41 | 98 × 9 |
| Lactococcus phage c2 | 22.2 | 41 | 36 | N.D. | N.D. |
| ψM1-likes | |||||
| Methanobacterium phage ψM1 | 26.1 | 31 | 46 | 55 | 210 × 10 |
Genus as recognized by the International Committee on the Taxonomy of Viruses (van Regenmortel et al., 2000) and recently described Sfi21-like siphovirus families (Proux et al., 2002). There are 19 sequenced genomes currently recognized as part of the lambda supergroup. Here we present a representative genome from each major group.
Genome sizes are from the classified siphovirus genomes from the NCBI TaxBrowser database.
Table 2.
Summary table of P-SS2 predicted proteins that contained relevant annotation information as determined from (a) significant blastp hits (e-value < e−3) against the GenBank non-redundant database, (b) experimental proteomics on the virus particle, or (c) detection in viral metagenomes.
| P-SS2 ORF # | Strand | LeftEnd | RightEnd | Size (aa) | Gene | Putative function | e-value | Average peptides detected |
|---|---|---|---|---|---|---|---|---|
| 001 | + | 1 | 528 | 176 | terS | Terminase – small subunit | e−8 | 0 |
| 002 | + | 525 | 2018 | 498 | terL | Terminase – large subunit | e−63 | 0.5 |
| 003 | + | 2091 | 2732 | 214 | Type III rpoS | Cyanobacterial type III RNAP sigma factor | e−13 | 0 |
| 005 | + | 3417 | 3608 | 64 | Unknown protein in metagenomes | No hits | 0 | |
| 009 | + | 6004 | 6192 | 63 | Structural protein | No hits | 1.5 | |
| 010 | + | 6423 | 6713 | 97 | Thioredoxin | Thioredoxin | e−3 | 0 |
| 011 | + | 6853 | 9423 | 857 | nrd | Cyanobacterial class II ribonucleotide reductase | e = 0 | 0 |
| 014 | + | 10792 | 11481 | 230 | Hypothetical protein | e−5 | 0 | |
| 020 | + | 13316 | 13579 | 88 | Unknown structural protein | No hits | 2.5 | |
| 025 | + | 15118 | 16674 | 519 | Structural prophage protein | e−46 | 33 | |
| 028 | + | 17092 | 17280 | 63 | Conserved T4-like protein in metagenomes | e−5 | 0 | |
| 030 | + | 17648 | 22147 | 1500 | Major capsid protein | e−18 | 96.5 | |
| 031 | – | 22144 | 22347 | 68 | Unknown structural protein, also in metagenomes | No hits | 1.5 | |
| 032 | + | 22350 | 22535 | 62 | Unknown structural protein | No hits | 1 | |
| 033 | + | 22567 | 22767 | 67 | Unknown structural protein | No hits | 2.5 | |
| 036 | + | 23802 | 24836 | 345 | cobO | Cyanobacterial cobO | e−98 | 0 |
| 038 | + | 25557 | 25730 | 58 | Conserved marine cyanobacterial protein | e−12 | 0 | |
| 045 | + | 27442 | 27702 | 87 | Syn5_026 | Cyanopodophage Syn5 ORFan protein (gp26) | e−13 | 0 |
| 049 | + | 28259 | 28486 | 76 | Conserved marine Synechococcus protein | e−4 | 0 | |
| 053 | + | 29861 | 30058 | 66 | 9313_1008 | Prochlorococcus eMIT9313 ORFan protein | e−3 | 0 |
| 058 | + | 31797 | 32363 | 189 | Kinase | Possible phage kinase | e−5 | 0.5 |
| 061 | + | 32988 | 33524 | 179 | Unknown structural protein | No hits | 16 | |
| 062 | + | 33526 | 33993 | 156 | Unknown structural protein | No hits | 3 | |
| 063 | + | 33993 | 34499 | 169 | Unknown structural protein | No hits | 5 | |
| 066 | + | 36202 | 36831 | 210 | Unknown structural protein | No hits | 2.5 | |
| 067 | + | 36831 | 37625 | 265 | Fibre | Cyanophage T4-like fibre | e−7 | 9.5 |
| 068 | + | 37635 | 42854 | 1740 | Fibre | Unknown structural protein, tail fibre | e = 0.015 | 6 |
| 069 | + | 42886 | 43926 | 347 | Unknown structural protein | No hits | 13 | |
| 071 | + | 44422 | 44988 | 189 | Unknown structural protein | No hits | 1 | |
| 072 | + | 44988 | 46352 | 455 | Fibre | Lambdoid phage tail fibre | e−11 | 3 |
| 073 | + | 46354 | 51234 | 1627 | Fibre | Tail fibre with low %G+C | e−35 | 5 |
| 074 | + | 51512 | 52852 | 447 | Capsid decoration protein | Lambdoid tail collar/fibre decoration protein (gpH) | e−45 | 2 |
| 076 | + | 53164 | 53838 | 225 | Unknown structural protein | No hits | 9.5 | |
| 077 | + | 54104 | 59761 | 1886 | Tail tape measure | Lambdoid tail tape measure protein | e−36 | 102 |
| 078 | + | 59795 | 60211 | 139 | Unknown structural protein | No hits | 3 | |
| 079 | + | 60216 | 63218 | 1001 | Unknown structural protein | No hits | 43 | |
| 080 | + | 63255 | 63593 | 113 | Cyanophage T4-like hypotheticals | e−5 | 9 | |
| 081 | + | 63603 | 64040 | 146 | Unknown structural protein | No hits | 7.5 | |
| 082 | + | 64040 | 64306 | 89 | M2_082 | Cyanophage P-SSM2 ORFan protein (gp082) | e−6 | 3.5 |
| 083 | + | 64460 | 64657 | 66 | Unknown structural protein | No hits | 2 | |
| 084 | + | 64656 | 72495 | 2613 | Unknown structural protein, also in metagenomes | No hits | 43 | |
| 085 | + | 72530 | 73087 | 186 | Unknown protein in metagenomes | No hits | 0 | |
| 086 | + | 73129 | 78666 | 1846 | Structural protein similar to marine siphophage JL001 ORFan protein (gp88) | e−4 | 45 | |
| 087 | + | 78666 | 80936 | 757 | Structural protein similar to cyanophage MaTMM01 ORFan protein (gp105) | e−10 | 11 | |
| 088 | + | 80960 | 82057 | 366 | Unknown structural protein | No hits | 17.5 | |
| 089 | + | 82057 | 82920 | 288 | Unknown structural protein | No hits | 6 | |
| 090 | + | 82920 | 83348 | 143 | Unknown structural protein | No hits | 4 | |
| 091 | + | 83401 | 84141 | 247 | Host specificity | Lambdoid host specificity protein (gpJ) | e−4 | 14 |
| 092 | + | 84331 | 85251 | 307 | Structural cyanobacterial prophage protein | e−65 | 43.5 | |
| 093 | + | 85317 | 85643 | 109 | Unknown structural protein | No hits | 12.5 | |
| 095 | + | 86065 | 86304 | 80 | Unknown protein in metagenomes | No hits | 0 | |
| 097 | – | 86560 | 87231 | 224 | hyp_Syn | SynRS9917 ORFan protein | e−23 | 0 |
| 098 | + | 87302 | 88528 | 409 | Lysozyme | Lysozyme | e−11 | 0 |
| 101 | – | 89241 | 90614 | 458 | int | Site-specific integrase (int) | e−12 | 0 |
| 102 | – | 91145 | 92104 | 320 | bet | Recombination protein (bet) | e−15 | 0 |
| 103 | – | 92216 | 92956 | 247 | Conserved cyanobacterial protein | e−5 | 0 | |
| 108 | + | 94460 | 96085 | 542 | Helicase | DNA helicase | e−8 | 0 |
| 109 | + | 96089 | 97663 | 525 | Primase | Cyanobacterial DNA primase | e−58 | 0.5 |
| 111 | + | 98065 | 98655 | 197 | dcd | Cyanobacterial dCTP deaminase (dcd) | e−19 | 0 |
| 113 | + | 98987 | 99841 | 285 | Type II rpoS | Type II RNAP sigma factor (rpoS) | e−17 | 0 |
| 114 | + | 99889 | 100242 | 118 | ssb | Cyanobacterial single-stranded DNA binding protein (ssb) | e−22 | 0 |
| 123 | + | 103253 | 104206 | 318 | exo | 5′-3′ exonuclease recombination protein (exo) | e−11 | 0 |
| 126 | + | 105166 | 106119 | 318 | thy1 | Cyanobacterial thymidylate synthase | e−57 | 0 |
For each protein, the genome locus information is paired with our annotations, as well as the top e-value and the average number of peptides detected from three biological replicate proteomic analyses (see text and methods).
Twenty-two of the P-SS2 ORFs appear phage-related, with 13 of these most similar to proteins of the lambdoid siphoviruses and nine most similar to other viral types (Fig. 1B). Six have sequence homology to lambdoid structural proteins (tail fibre, tail collar gpH, tail tape measure, host specificity gpJ), recombination (bet), and lysis (lysozyme) proteins. Another six include ‘cyanobacterial’ analogues of lambdoid proteins (dCTP deaminase, single-stranded DNA binding protein, integrase, thymidylate synthase, and the small and large subunits of terminase) and the last encodes a ‘non-cyanobacterial’ (exonuclease, exo) lambdoid analogue. The remaining nine ORFs with phage-related homologues are most similar to unclassified prophage proteins (ORFs 007, 014, 025, 030, 092), T4-like myovirus proteins (ORFs 028, 067, 080), and a T7-like podovirus protein (ORF 045).
Gene expression in cyanobacteria is commonly regulated at the level of transcription, so we investigated the potential transcriptional regulatory machinery available to siphovirus P-SS2 (Fig. 1B). A search for classic sigma-70 promoter sequences and rho-independent terminators revealed 19 and 22 respectively (details in Experimental procedures, genome locations in Table S1 and Fig. 1B, weblogo promoter consensus sequence in Fig. S1). Notable among these were transcriptionally autonomous genes (termed ‘morons’ by Hendrix et al., 1999) that are thought to be the basis for mosaicism among siphoviruses; examples here include the Prochlorococcus MIT9313 ORFan (P-SS2 ORF 053) and Syn5 cyanopodophage ORFan (P-SS2 ORF 045). As well, reverse promoters that were identified often provided support that predicted opposite strand ORFs may indeed be functional (e.g. P-SS2 ORFs 016–020). Sequence analysis also identified two sigma factors that are likely used by P-SS2 to modulate host RNAP activity during infection. The first, a group 2 sigma factor (ORF 113), most probably recognizes the canonical sigma-70 promoter sequences identified above, while the second, a group 3 sigma factor (ORF 003), likely recognizes sequences specific for a particular regulon (Lonetto et al., 1992). Such functionally specific group 3 sigma factors are uncommon among the sequenced marine Prochlorococcus genomes to date; found only in ProMIT9303 and the original P-SS2 host strain, ProMIT9313. However, the P-SS2 group 3 sigma factor appears significantly diverged from that of its host (Fig. S2), so if the phage or host version was acquired from the other entity then it has greatly diverged or one of the two acquired the sigma factor from outside cyanobacteria and their phages.
While the bulk of P-SS2's phage-related proteins are most similar to those from lambdoid phages, P-SS2 is a distantly related lambdoid phage at best. First, the sequence similarity of the aforementioned six ‘lambdoid’ proteins is quite poor. Second, a phylogeny of the large terminase protein (diagnostic for DNA packaging characteristics; Casjens et al., 2005), suggests that the P-SS2 TerL and the homologues from remnants of marine Synechococcus prophage integration events (see Discussion below) comprise a novel terminase class quite divergent from known phage terminases (Fig. 2). Third, most of the protein components that comprise the P-SS2 virus particle are unrecognizable; only six structural proteins could be assigned by sequence, as elaborated upon below.
Fig. 2.
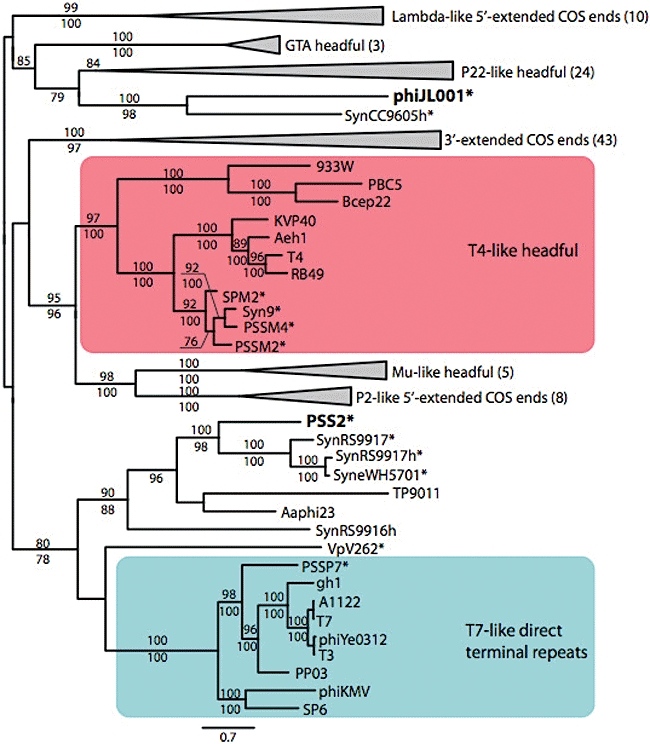
Phylogenetic relationships of the large terminase protein across diverse phage types. This protein is diagnostic of phage DNA packaging mechanisms (Casjens et al., 2005) and was here used to initially characterize the P-SS2 large terminase protein relative to known phage terminases. The asterisk (*) denotes marine phage and cyanobacterial host genomes. Notably, the terminase from the other marine siphovirus whose genome is sequenced (phi-JL001) clusters separately from known terminases, while that from cyanophage P-SS2 clusters with terminases from marine cyanobacterial host genomes (likely remnant prophages, see text). The tree shown is a maximum likelihood tree constructed from 1513 positions (significantly divergent protein and gapped alignment) as described in Experimental procedures. Numbers above and below branches represent bootstrap values over 75 from maximum likelihood and distance analyses respectively. Numbers in parentheses with taxa labels represent number of taxa in collapsed nodes.
Structural proteins
To expand our understanding of the genes encoding the P-SS2 structure, we identified the structural proteins in purified virus particles experimentally using mass spectrometry (see Experimental procedures). We detected 35 structural proteins (Table 2, hashed lines in ORFs in Fig. 1B), including all six that were identified by sequence. As is common in phage genomes (Brussow and Desiere, 2001; Proux et al., 2002; Casjens, 2003), these structural genes were clustered on the genome into ‘modules’ (red lines below genome in Fig. 1B). The largest cluster, consisting of 28 structural genes, included homologues to tail fibre structural genes (ORFs 067, 072, 074, 077, 091). Notably, one putative tail fibre gene (ORF 073) is predicted to encode a 1627-amino-acid protein and has a significantly low %G+C content (Fig. S3). This anomalous %G+C, and the fact that the ORF is most similar to a non-siphovirus tail fibre gene from the myovirus P-SSM2 genome, suggests possible horizontal transfer into P-SS2 from another phage class. If true, such tail fibre switching might have significant implications for host-range among cyanophages.
Another five proteins detected in the virus particle were mapped to a genome cluster that contained a capsid protein homologue (ORF 030) and a highly conserved marine prophage protein that was among the most abundant proteins in the proteomics analysis (ORF 025; averaged 33 detected peptides across biological replicates). This region likely defines proteins involved in capsid formation. The last two structural proteins detected in the virus particle are small proteins in the 5′-end of the P-SS2 genome (ORFs 009, 020) with unknown function.
Cyanobacterial and marine features of the P-SS2 genome
The P-SS2 genome is 108 kb whereas, with one exception, most of the other siphoviruses sequenced to date have genomes on the order of 20–50 kb (Table 1). What comprises this extra DNA? Unlike the majority of cultured marine myovirus and podovirus (Mann et al., 2003; Millard et al., 2004; Lindell et al., 2004; 2005; 2007; Sullivan et al., 2006), P-SS2 does not encode cyanobacterial photosynthesis genes. Because cyanomyoviruses were commonly isolated from similar deep-photic zone depths that also contain psbA (12 are documented in Sullivan et al., 2006), we posit that the lack of such photosynthesis genes in siphovirus P-SS2 is more likely to be due to the hypothesized temperate phage lifestyle of this virus.
However, P-SS2 does encode 14 genes with homology to genes from ocean cyanobacteria. Six of these proteins are also phage-encoded in the lambda/Escherichia coli system, and likely, as described above for cyanobacterial lambdoid analogues in the genome section have important DNA synthesis and packaging functions. The remaining eight, which have host but not phage parallels in the lambda/E. coli system, include a cyanobacterial DNA primase (paired with a phage-encoded non-cyanobacterial DNA helicase), ribonucleotide reductase (RNR), cobalamin synthesis gene cobO, three conserved marine cyanobacterial hypothetical proteins, and two cyanobacterial ORFan genes. The last six of these genes have not been seen previously in any phage genome, and their functional roles and importance to phage fitness remain unclear. In contrast, primase, helicase and RNR-encoding genes are common in phage genomes. While not found in lambda, primase and helicase genes are often present in other siphovirus genomes, including the divergent ocean siphovirus phi-JL001, suggesting that these genes encode critical protein functions not required in lambda. Further, while RNR-encoding genes are uncommon among siphoviruses (found in 12 of 107 siphoviruses at http://rnrdb.molbio.su.se), two lines of evidence suggest their importance in marine ecosystems. First, the other marine siphovirus, phi-JL001, contains a RNR-encoding gene (Lohr et al., 2005). Second, they are also found in non-siphovirus marine phage: all marine T4-like and T7-like phage sequenced to date contain them (Rohwer et al., 2000; Chen and Lu, 2002; Mann et al., 2005; Sullivan et al., 2005; Pope et al., 2007; Weigele et al., 2007) even though they are absent in non-marine T7-like phages. We hypothesize that the prevalence of RNR-encoding genes in marine phage of all types reflects the importance of scavenging nucleic acids for DNA synthesis during phage infection in the often nitrogen- and phosphorous-limited oceanic environments.
Host genomic islands and a putative P-SS2 integration site
The P-SS2 genome contains three of the four genes that are considered hallmark lysogeny genes in lambda: int, exo and bet (Table 2, Fig. 1B). If capable of integration as a prophage, then two more protein functions must be present as well. First, the function of the fourth hallmark lysogeny gene, excisionase, would need be filled by a host-encoded version as has been observed for other phages and plasmids that use host-encoded site-specific recombinases (Barre and Sherratt, 2002; Huber and Waldor, 2002). Second, a repressor is critical to remain integrated as a prophage to prevent expression of lytic genes that would induce the prophage out of its host genome. While P-SS2 lacks an identifiable repressor, these are small proteins that are highly divergent and often not recognizable even in known functional prophages (e.g. marine Silicibacter prophages; Chen et al., 2006). In addition to int, exo and bet, the P-SS2 genome contains a 53 bp intragenic non-protein-coding sequence between int and bet that exactly matches an intergenic, non-coding region of host ProMIT9313 and includes 36 bp that exactly match a nearby tRNA-Met (Fig. 3). No other matches outside of the 36 bp coding region of the tRNA-Met were found in GenBank, the Global Ocean Survey microbial metagenomes, or the microbial genomes at Microbes Online. Prophages commonly integrate into conserved host genome sites such as tRNA (Campbell, 2003) and tmRNA (Williams, 2002). Thus this exceedingly rare 53 bp match to the non-coding host genome sequence which should be prone to amelioration by neutral mutation and that maintains partial identity to a tRNA in the host genome may represent the phage (attP) and host (attB) site-specific attachment sites. Near the putative integration site in the host genome are signatures of mobile genetic element activity including eight transposase genes, five pseudo-tRNA-Met genes and five copies of 36 bp of the 53 bp exact match (Fig. 3). However, these mobile genetic elements are likely relics of long-ago transposition events because the transposase genes are variously degraded and lack identifiable inverted-repeat ‘ends’ (see Experimental procedures).
Fig. 3.
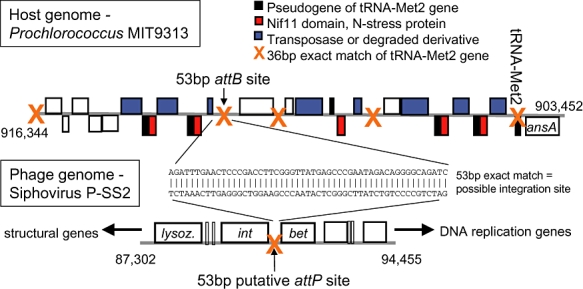
Schematic representation of genome regions surrounding the putative phage (P-SS2) and host (Prochlorococcus MIT9313, GenBank ID: NC_005071) integration sites. This site consists of a 53 bp exact match between the phage sequence downstream of its integrase gene at position 90,836–90,888, and the non-coding sequence in the host genome at position 912,261–912,313. This general region of the host genome is a genomic island, and thus hypervariable (see text). Numbers at the genome ends represent the nucleotide position in the respective genomes.
While future work is required to experimentally prove that this 53 bp match is the integration site for P-SS2 in its host, we chose to examine the available marine Prochlorococcus and Synechococcus genomes in this tRNA-Met + ansA region. Across 21 genomes, this homologous region revealed a complex evolutionary story (Fig. 4A). In each examined host genome, this region is highly conserved right up to the tRNA-Met gene from the ansA gene side of this tRNA (blue bar in Fig. 4A). Among some genomes, synteny continues among subgroups of these strains (ProI, ProII, ProIII, Synechococcus labels in Fig. 4A). In contrast, a number of genomes have hypervariable ‘genomic island’ regions (sensuColeman et al., 2006) at this tRNA breakpoint (red squares in Fig. 4A). Some of these ‘island’ regions are small, as in the Prochlorococcus strains MED4 and MIT9515, and contain numerous hli genes, which appear to have been horizontally transferred to these genomes by phages (Lindell et al., 2004). Others are more extensively hypervariable, as in ProMIT9313 (described above, detailed in Fig. 3) and SynRS9917 (detailed in Fig. 4B). The SynRS9917 island region is 41 kb in size and lacks hli genes but contains six transposases and two P-SS2-like genes – lysozyme and ORF97 (Fig. 4B). Further, ∼27 kb of highly syntenic genome away is a second genomic island of ∼42 kb, bounded by a tRNA-Ser (Fig. 4B). This island contains another transposase, assorted genes related to prophage induction including maintenance proteins, an antirepressor and a possible repressor, as well as four P-SS2-like genes – large terminase, integrase, RNAP sigma factor, ORF25 structural gene, lysozyme. This is most certainly a relic prophage that shared some similarity with P-SS2.
Fig. 4.
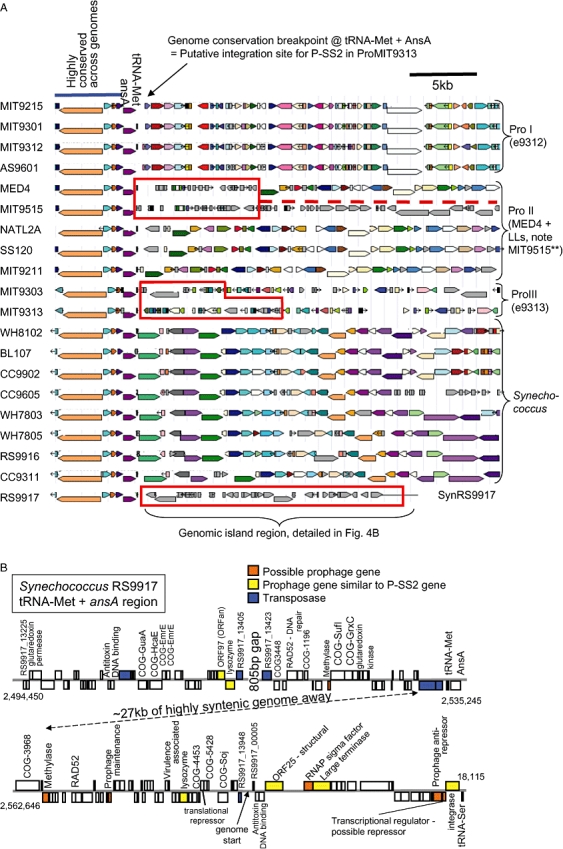
Genome arrangement at the tRNA-Met +ansA locus across (A) Prochlorococcus and Synechococcus genomes, and (B) detailed for Synechococcus RS9917. A. Comparative genomics of marine Prochlorococcus and Synechococcus at the tRNA-Met +ansA locus identified as the putative P-SS2 integration site in ProMIT9313. Across the marine cyanobacteria, this region is highly syntenic with four basic genome patterns observed – denoted as ProI, ProII, ProIII, and Synechococcus in the figure. However, some strains lack synteny and have hypervariable or ‘genomic islands’ regions, indicated by the red boxes in the figure. MED4 has a small ∼8 kb island with phage high-light inducible genes (this is equivalent to ISL2, Coleman et al., 2006), while MIT9515 has a slightly larger and similar island to MED4s then a region that is a large genome rearrangement (red dashed line) that is syntenic to another region of the MED4 genome (647,805–687,505). The eMIT9313 variability in this region is detailed in Fig. 3. B. The tRNA-Met + AnsA region in Synechococcus RS9917 that is homologous to the putative attB integration site in ProMIT9313 from Fig. 3. This ∼41 kb ‘island’ region contains four transposases, an antitoxin gene, and two PSS2-like genes – lysozyme and structural protein ORF97. Genomic synteny to all the other marine cyanobacteria then continues for ∼27 kb until reaching a second ∼42 kb ‘island’ that is bounded on the other side by tRNA-Ser, and contains a transposase, as well as numerous prophage-related genes including a possible repressor, antirepressor, prophage maintenance protein, RNAP sigma factor, and four PSS2-like genes – large terminase, integrase, ORF25 structural gene, lysozyme. The COG categories refer to those at Microbes Online.
In contrast to the ProMIT9313 transposases described above, the bulk of the SynRS9917 transposase genes appear as intact composite IS element with identifiable ‘ends’ (Fig. 5). The presence of two RAD52 family proteins (Fig. 4B) suggests the need for double-stranded DNA repair as if this region were under heightened ‘attack’ from IS elements. Notably, a second hot-spot of IS elements occurs at pyrE in the SynRS9917 genome. This region contains another tRNA-Met and RAD52 family protein, and is syntenic across all host genomes examined except for a 65 kb genomic island in SynRS9917 (Fig. 6A). This SynRS9917 island contains 13 phage-related genes (seven of which are similar to P-SS2 genes, Fig. 6B) and, while clearly incomplete, represents the most intact marine cyanobacterial prophage observed to date. Notably, among the 21 Prochlorococcus and Synechococcus host strains available for testing at the time, P-SS2 infected only its original host used for isolation, ProMIT9313 (note: SynRS9917 was not available for testing, Sullivan et al., 2003).
Fig. 5.
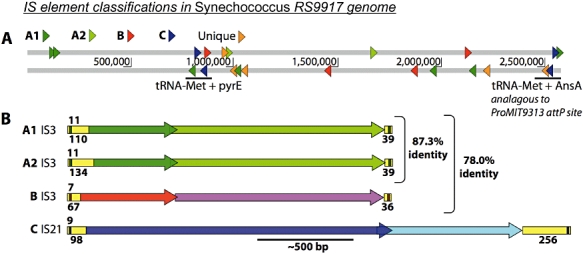
Characterization of insertion sequence (IS) elements in Synechococcus RS9917. The 22 transposase genes and surrounding regions (the IS element) revealed four groups of multicopy IS elements, and five unique or degraded IS elements in the SynRS9917 genome. Using the ACLAME database, we classified these IS elements as follows. The two multicopy groups ‘A1’ and ‘A2’ are IS3-like mobile elements and have identical inverted repeats, identical lengths and > 87% sequence identity. IS group ‘B’, is also IS3-like element but has a shorter inverted repeat. IS group ‘C’ is longer and most similar to IS21-like elements. A. Location and orientation of IS elements, represented by coloured arrows, in relation to the proposed P-SS2-like phage integration sites (regions represented by black bars). B. Diagrams of IS elements including size of inverted repeat (size in bp indicated above the 5′-end of the yellow box), position and orientation of ORFs, and size of flanking non-coding regions (shown in yellow, with size in bp indicated below the yellow box).
Fig. 6.
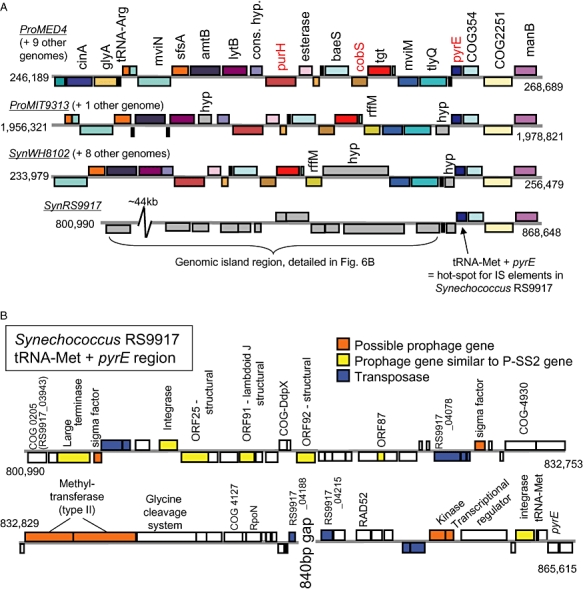
Genomic arrangement of the tRNA-Met +pyrE site in Synechococcus RS9917 identified as a secondary hot-spot for insertion sequence elements. A. Schematic of the highly syntenic tRNA-Met +pyrE region from representative Prochlorococcus and Synechococcus genomes. Minor insertions in ProMIT9303 and ProMIT9313 (rffM insertion, small hypothetical ORFs) and the marine Synechococcus (rffM+ large hypothetical ORF) are the only deviations from complete synteny in this region, except for the genomic island detailed for Synechococcus RS9917 (see Fig. 6B). Gene names are listed for the top genome only, and homologues across the genomes are similarly coloured. Red gene names have been previously observed in myovirus cyanophage genomes (Sullivan et al., 2005). Nine other genomes are similar to the Prochlorococcus MED4 arrangement, one other for the MIT9313 arrangement, and eight other for the Synechococcus WH8102 arrangement (details in Table S2). B. In contrast to the genome conservation observed in other Prochlorococcus and Synechococcus genomes, SynRS9917 contains a ∼65 kb genomic island region that contains nine transposases, seven P-SS2-like genes and seven phage-like genes. This is the most intact prophage in any marine Prochlorococcus or Synechococcus genome, but it is still significantly degraded.
Insertion sequence-mediated genome evolution in ProMIT9313
While the tRNA-Met + AnsA genomic island in ProMIT9313 described above contains variously degraded IS elements, this region may not simply be a ‘graveyard’ of degraded genes and pseudogenes. All five transposases are bordered by paralogous genes with sequence similarity to a nif11-domain (Fig. 3) that suggests a nitrogen stress-related function based upon annotation alone. Indeed, nitrogen stress, and not phosphate stress, alters the gene expression of four of five of these nif11-domain-containing genes (Martiny et al., 2006; Tolonen et al., 2006). Across diverse bacterial genomes, IS elements often alter expression of neighbouring genes using outward facing promoters (Mahillon and Chandler, 1998). Insertion sequence elements are also capable of transporting ‘cargo’ genes around genomes (Poirel et al., 2005; Toleman et al., 2006; Bartosik et al., 2008), often with greater transposition efficiency than wild-type elements (Bartosik et al., 2008) and lead to fitness gains under experimental evolutionary conditions (Schneider and Lenski, 2004) and in the generation of reduced symbiont genomes (Moran and Plague, 2004; Plague et al., 2008). Thus we hypothesize that these ProMIT9313 IS elements may both move around (as ‘cargo’) and regulate (via outward-facing promoters) these proximal nitrogen stress genes thereby contributing to host niche differentiation.
Further, this region is coincident with the putative P-SS2 siphovirus integration site, suggesting two layers of genome evolution. Insertion sequence elements may mediate intragenomic innovation by bringing genes to this region as an evolutionary ‘sandpit’ where selection challenges new combinations of alleles and genes as appears to be the case for the nif11-domain genes above. Then, if indeed P-SS2 is capable of integration at this site, the phage could obtain such IS-mediated genetic innovation through aberrant excision and distribute it to other host genomes (intergenomic innovation) via new infections. If this were occurring, then one might expect to occasionally observe IS elements from such evolutionary action in phage genomes. Indeed, in spite of very few environmental phage genomes available, IS transposase genes have been observed in marine phages (e.g. vibriophage VHML; Oakey et al., 2002; Chibani-Chennoffi et al., 2004) and freshwater cyanophages (Ma-LMM01 has 3 transposases; Yoshida et al., 2008). Further, IS elements are known to facilitate the spread and expression of genes enabling antibacterial resistance and degradation of toxic compounds (Berg and Howe, 1989; Bushman, 2002; Nojiri et al., 2004). Perhaps here nitrogen stress response is similarly tied to IS-mediated evolution in marine cyanobacteria, which are often N-limited.
The acquisition of ‘host genes’ into the cyanophage genome pool
While ‘host genes’ or ‘auxiliary metabolic genes’ (AMGs) are commonly observed in cyanophage genomes (reviewed in Breitbart et al., 2007), it remains unclear how they are obtained by cyanophages. Notably, a second IS-element hot-spot in SynRS9917 is again located at a tRNA-Met which in 21 other Prochlorococcus and Synechococcus genomes is proximal to pyrE, cobS and purH (Fig. 6A) – three genes that are found in lytic myovirus cyanophage genomes (Syn9, P-SSM2 and P-SSM4; Sullivan et al., 2005, Weigele et al., 2007). In contrast, this region in the SynRS9917 genome has significant evidence of past prophage-integration activity including 13 phage-related genes, seven of which share sequence similarity with P-SS2 genes (Fig. 6B). While this prophage is clearly a relic (nine transposases, missing many genes), could such a prophage have introduced these three ‘host genes’ or AMGs into the phage genome pool? Induced prophages can improperly excise from the host genome and mispackage up to 10% of the host genome proximal to the integration site in place of part or all of the phage genome (Calendar, 1988). Thus the remnant SynRS9917 prophage(s) may have initially obtained such AMGs proximal to this site-specific integration site. These genes could then have been disseminated to super-infecting lytic cyanophages through recombination. Such prophage-to-lytic-phage recombination events are thought to be among the most probable means of spreading new genetic material through the phage genome pool as has been observed in Streptococcus thermophilus and Lactococcal phages (Brussow and Desiere, 2001), mycobacteriophages (Pedulla et al., 2003), and more generally the siphoviruses (Hendrix et al., 1999).
Prevalence of P-SS2-like siphoviruses in the surface oceans
Given that siphoviruses have rarely been isolated in studies using a diversity of marine cyanobacterial hosts to isolate phage from seawater (Waterbury and Valois, 1993; Suttle and Chan, 1994; Lu et al., 2001; Marston and Sallee, 2003, Sullivan et al., 2003), one wonders whether this was a function of isolation procedures or whether they occur in relatively low abundances in the wild. To begin to address this question, we used the P-SS2 genome to ‘recruit’ homologous fragments from the microbial fraction Global Ocean Survey surface ocean metagenomes (see Experimental procedures). There were only seven GOS reads with a best hit to the P-SS2 genome (Table S3), and the alignment lengths of these hits were short, ranging from 47 to 242 bp. Homologues of the ProMIT9313 genome are also rare in the GOS data set (< 0.35% of the total hits in any given site, data not shown), which is not surprising as these LL-adapted Prochlorococcus cells are not abundant in surface waters (Johnson and Zinser et al., 2006). Thus this limited analysis suggests that siphoviruses similar to the one used in this study are not abundant in surface ocean waters, but may be in undersampled lower euphotic zone waters.
Conclusions
The ocean cyanobacterial siphovirus P-SS2 contains a large genome that is significantly divergent from the siphovirus genomes sequenced to date, so much so that even structural proteins required experimental validation to annotate. This contrasts with the classically lytic phages (e.g. T4-like myoviruses and T7-like podoviruses) which exemplify a cohesive genomic architecture ranging from non-marine coliphages (Miller et al., 2003; Nolan et al., 2006) to marine representatives of roseophages (Rohwer et al., 2000), vibriophages (Miller et al., 2003) and cyanophages (Chen and Lu, 2002; Mann et al., 2005; Sullivan et al., 2005; Pope et al., 2007; Weigele et al., 2007). The siphoviruses, however, are thought to be prone to extensive genetic module ‘swapping’ through intensive recombination (Hendrix et al., 1999; Juhala et al., 2000; Brussow and Desiere, 2001; Proux et al., 2002; Pedulla et al., 2003), thus the divergence observed is not surprising. This intense mosaicism causes siphoviruses to display web-like phylogenies (Brussow and Desiere, 2001) and to represent the most taxonomically challenging phage group (Hendrix et al., 1999; Edwards and Rohwer, 2005; Lawrence et al., 2002; Proux et al., 2002). Beyond the P-SS2 genome, exploration of a putative phage integration site in the host genome revealed extensive genomic islands in the host and IS elements among some ocean Prochlorococcus and Synechococcus genomes at this location. Particularly striking are the IS element hot-spots in SynRS9917 where the comingling components of the cyanobacterial ‘mobilome’ revealed evidence of prophages under IS element attack, as well as a possible mechanism for phage-captured ‘host ‘AMGs central to cyanophage biology (reviewed in Breitbart et al., 2007).
Experimental procedures
Isolation of the phage and preparation for genomic sequencing
The siphovirus P-SS2 was isolated from Atlantic Ocean slope waters (38°10′N, 73°09′W) collected on 17 September 2001 on the R/V Endeavor cruise number 360. The sampled water was from 83 m depth with a salinity 36.6 ppt and temperature 20.8°C. This water was 0.2 μm filtered and stored at 4°C until it was used directly in a plaque assay with Prochlorococcus strain MIT9313 as a host on 12 December 2001. A large, well-resolved plaque was picked from the lawn of host cells on 29 December 2001, plaque purified two more times and stored as a lysate of a P-SS2 clonal stock isolate.
P-SS2 was prepared for sequencing as previously described (Lindell et al., 2004). Briefly, phage particles were concentrated from large volume (2 l) lysates using polyethylene glycol. Concentrated DNA-containing phage particles were purified from other material in phage lysates using a density caesium chloride gradient. Purified phage particles were broken open (SDS/proteinase K), and DNA was extracted (phenol:chloroform) and precipitated (ethanol) yielding small amounts of DNA (< 1 ng). A custom 1–2 kb insert linker-amplified shotgun library was constructed by Lucigen (Middletown, WI, USA) as described previously (Breitbart et al., 2002). Additional larger insert (3–8 kb) clone libraries were constructed from genomic DNA by the Department of Energy (Joint Genome Institute, Walnut Creek, CA, USA) using a similar protocol to provide larger scaffolds during assembly. Inserts were sequenced by the Department of Energy Joint Genome Institute from all clone libraries and used for initial assembly of these phage genomes. The Stanford Human Genome Center Finishing Group (Palo Alto, CA, USA) closed the genomes using primer walking.
Genome annotation
Gene identification and characterization was done as in Sullivan and colleagues (2005). Briefly, protein coding genes were predicted using GeneMark and manual curation. Translated ORFs were compared with known proteins in the non-redundant GenBank and in the KEGG databases using the blastp program. Where blastpe-values were high (> 0.001) or no sequence similarity was observed, ORF annotation was aided by the use of PSI-blast, gene size, domain conservation, and/or synteny (gene order). Identification of tRNA genes was done using tRNAscan-SE. Additionally, rho-independent transcription terminators were identified with TransTermHP (Kingsford et al., 2007) using default parameters. All terminators had a confidence score > 80% with an energy score of < −15 and a tail score of < −6. Bacterial σ70 promoters were predicted using BPROM (Softberry, Mount Kisco, NY, USA) using default parameters. All intergenic promoters with a linear discriminant function > 3.5 were considered candidate promoters. Inverted repeats of IS elements were found using the Palindrome program in the EMBOSS software suite (Rice et al., 2000). Approximately 200 bp upstream and downstream of each putative IS element was fed into Palindrome and the output of many inverted repeats identified were screened manually to identify those that were exact matches and at least 7 bp long. Insertion sequence elements were classified using the ACLAME database blast tool (Leplae et al., 2004). Genome visualizations were done in Artemis (Rutherford et al., 2000), while comparative genomics analyses were greatly aided by the tools available at MicrobesOnline (http://www.microbesonline.org). For figure labels where genes are denoted as ‘prophage’ or ‘cyanobacterial’, these assignments were made using NCBI taxonomy lineages of the top 5 blast hits. In all cases where these are denoted, all five top hits were of one of these two organismal types, cyanobacteria or known temperate phages or integrated prophages, with e-values < 0.001. The resulting genome sequence is deposited under GenBank accession #GQ334450.
Ocean microbial metagenomic analyses
To determine whether P-SS2 occurred in the wild, we queried the Global Ocean Survey (GOS; Rusch et al., 2007) microbial surface ocean water metagenomes. We created a database of all sequenced marine isolates, including Gordon and Betty Moore Foundation Marine Microbial Initiative genomes, NCBI marine isolates, and cyanophage available from the GenBank and CAMERA databases as of November 2008. Environmental metagenomic reads were blasted (blastall -p blastn -e 1e-5 -z 25000000000 -m 7 -a 4 -F ‘m L’ -X 150 -U T) against this database. Best hits to each GOS read were retrieved and filtered by alignment length. Reads with best hits to P-SS2 and ProMIT9313 (GenBank ID: NC_005071) are the focus of this study.
Large terminase (TerL) and group 3 sigma factor protein phylogenies
Protein alignments were generated using the Promals web server (Pei and Grishin, 2007; Pei et al., 2007) using default parameters and manually edited as needed. Amino acid distance trees were constructed using the paup*4.0b10 software. Neighbour joining was used to reconstruct distance trees using minimum evolution as the objective function and uncorrected distances. Amino acid maximum likelihood trees were inferred using the CIPRES web portal RAxML rapid bootstrapping and ML search (Stamatakis, 2006; Stamatakis et al., 2008) assuming the James-Taylor Thornton model of substitution using empirical base frequencies and estimating the proportion of invariable sites from the data.
Virion structural proteomics
Briefly, the samples were incubated in a denaturing solution of 8 M Urea/1% SDS/100 mM ammonium bicarbonate/10 mM DTT pH 8.5 at 37°C for 1 h. Next, the samples were alkylated for 1 h by the addition of iodoacetamide to a final concentration of 40 mM and then quenched with 2 M DTT. Following the addition of 4× LDS loading buffer (Invitrogen), each sample was centrifuged at 14 000 r.p.m. for 5 min at room temperature, and each sample was fractionated on a NuPAGE 10% Bis-Tris 10 lane gel (Invitrogen) for 2.5 h at 125 V, 50 mA and 8 W. Gels were shrunk overnight by the addition of 50% ethanol and 7% acetic acid, and then allowed to swell for 1 h by the addition of deionized water. Gels were stained with SimplyBlue Safe Stain (Invitrogen) for 2–4 h, imaged, and sliced horizontally into fragments of equal size based on the molecular weight markers.
In-gel digestion was performed after destaining and rinsing the gel sections with two washes of 50% ethanol and 7% acetic acid, followed by two alternating washes with 50 mM ammonium bicarbonate and acetonitrile. After removal of the last acetonitrile wash, 100 μL of sequencing grade trypsin (Promega) was added to each gel slice at a concentration of 6.6 ng μl−1 in 50 mM ammonium bicarbonate/10% acetonitrile. The gel slices were allowed to swell for 30 min on ice, after which the tubes were incubated at 37°C for 24 h. Peptides were extracted with one wash of 100 μl of 50 mM ammonium bicarbonate/10% acetonitrile and one wash of 100 μl of 50% acetonitrile/0.1% formic acid. The extracts were pooled and frozen at −80°C, lyophilized to dryness and redissolved in 40 μl of 5% acetonitrile, 0.1% formic acid.
Samples were then loaded into a 96-well plate (AbGene) for mass spectrometry analysis on a Thermo Fisher Scientific LTQ-FT. For each run, 10 μl of each reconstituted sample was injected with a Famos Autosampler, and the separation was performed on a 75 mM × 20 cm column packed with C18 Magic media (Michrom Biosciences) running at 250 nl min−1 provided from a Surveyor MS pump with a flow splitter with a gradient of 5–60% water 0.1% formic acid, acetonitrile 0.1% formic acid over the course of 120 min (150 min total run). Between each set of samples, standards from a mixture of 5 angiotensin peptides (Michrom Biosciences) were run for 2.5 h to ascertain column performance and observe any potential carryover that might have occurred. The LTQ-FT was run in a top five configuration with one MS 200K resolution full scan and five MS/MS scans. Dynamic exclusion was set to 1 with a limit of 180 s with early expiration set to 2 full scans.
Peptide identifications were made using SEQUEST (ThermoFisher Scientific) through the Bioworks Browser 3.3. The data were searched with a 10 ppm window on the MS precursor with 0.5 Dalton on the fragment ions with no enzyme specificity. A reverse database strategy (Elias and Gygi, 2007) was employed with a six frame translation of the genomic sequence reversed and concatenated with the forward sequences supplemented with common contaminates and filtered to obtain a false discovery rate of less than or equal to 1%. Peptides passing the filters were mapped back onto the genome and compared with predicted ORFs.
Acknowledgments
The siphovirus P-SS2 genome was sequenced and assembled by the DOE JGI under the auspices of the Community Sequencing Program. This work was supported in part by grants to S.W.C. from the Gordon and Betty Moore Foundation, NSF, DOE-GTL. We thank Maureen Coleman for generating the draft P-SS2 genome figure, Brian Binder and the crew of the R/V Endeavor for the sampling opportunity, and Sherwood Casjens, Patrick Degnan, Howard Ochman, Luke Thompson, Marcia Osburne, Maureen Coleman, Melissa Duhaime and Li Deng for the original terminase alignments (S.C.) and engaging discussion of IS elements (P.D., H.O.), ribonucleotide reductases (L.T.) and siphovirus biology (M.O., M.C., M.D., L.D.). We thank three anonymous reviewers whose comments also improved the manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Weblogo representation of the consensus promoter sequences predicted across the siphovirus P-SS2 genome. Genomic locations of the predicted promoter sequence locations are presented in Suppl. Table 1 (along with predicted terminators).
Fig. S2. Phylogenetic relationships of group 3 sigma factors among phages and microbes. In contrast to group 1 sigma factors which are universal among microbes, these group 3 sigma factor transcriptional regulatory proteins are uncommon among microbes. This is particularly notable among the marine Prochlorococcus where they are only found in ProMIT9313 and ProMIT9303. Tree details are as in the Fig. 2 legend, and methods, while in-figure table contains taxa names.
Fig. S3. %G+C plot of the siphovirus P-SS2 genome. The black line indicates a sliding base-pair (100 bp) window of %G+C along the genome, while the red line indicates 2.5 times the standard deviation. Notably the major deviation from the genome average (highlighted with the grey box) is where the anomalous tail fibre protein of putative lateral gene transfer origin.
Table S1. Genomic locations of predicted promoters and terminators.
Table S2. Details of genomic regions sharing synteny with representative genomes presented in Fig. 6A.
Table S3. Environmental sequence reads from the Global Ocean Survey (Rusch et al., 2007) that were best hits to the P-SS2 genome.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- Banks DJ, Beres SB, Musser JM. The fundamental contribution of phages to GAS evolution, genome diversification and strain emergence. Trends Microbiol. 2002;10:515–521. doi: 10.1016/s0966-842x(02)02461-7. [DOI] [PubMed] [Google Scholar]
- Barre FX, Sherratt DJ. Xer site-specific recombination: promoting chromosome segregation. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington DC, USA: American Society for Microbiology Press; 2002. pp. 149–161. [Google Scholar]
- Bartosik D, Putyrski M, Dziewit L, Malewska E, Szymanik M, Jagiello E, et al. Transposable modules generated by a single copy of insertion sequence ISPme1 and their influence on structure and evolution of natural plasmids of Paracoccus methylutens DM12. J Bacteriol. 2008;190:3306–3313. doi: 10.1128/JB.01878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, et al. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. 2002;99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DE, Howe MM. Mobile DNA. Washington DC, USA: American Society for Microbiology Press; 1989. [Google Scholar]
- Boyd EF, Davis BM, Hochhut B. Bacteriophage–bacteriophage interactions in the evolution of pathogenic bacteria. Trends Microbiol. 2001;9:137–144. doi: 10.1016/s0966-842x(01)01960-6. [DOI] [PubMed] [Google Scholar]
- Bragg JG, Chisholm SW. Modelling the fitness consequences of a cyanophage-encoded photosynthesis gene. PLoS One. 2008;3:3550. doi: 10.1371/journal.pone.0003550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Thompson LT, Suttle CA, Sullivan MB. Exploring the vast diversity of marine viruses. Oceanography. 2007;20:135–139. [Google Scholar]
- Brussow H, Desiere F. Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol Microbiol. 2001;39:213–222. doi: 10.1046/j.1365-2958.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- Bushman F. Lateral DNA Transfer: Mechanisms and Consequences. Cold Spring Harbor, NY, USA: Cold Spring Harbor University Press; 2002. [Google Scholar]
- Calendar R. The Bacteriophages. New York, USA: Plenum; 1988. [Google Scholar]
- Campbell A. Prophage insertion sites. Res Microbiol. 2003;154:277–282. doi: 10.1016/S0923-2508(03)00071-8. [DOI] [PubMed] [Google Scholar]
- Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- Casjens S, Gilcrease EB, Winn-Stapley DA, Schicklmaier P, Schmieger H, Pedulla ML, et al. The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J Bacteriol. 2005;187:1091–1104. doi: 10.1128/JB.187.3.1091-1104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Lu J. Genomic sequence and evolution of marine cyanophage P60: a new insight on lytic and lysogenic phages. Appl Environ Microbiol. 2002;68:2589–2594. doi: 10.1128/AEM.68.5.2589-2594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wang K, Stewart J, Belas R. Induction of multiple prophages from a marine bacterium: a genomic approach. Appl Environ Microbiol. 2006;72:4995–5001. doi: 10.1128/AEM.00056-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brussow H. Phage–host interaction: an ecological perspective. J Bacteriol. 2004;186:3677–3686. doi: 10.1128/JB.186.12.3677-3686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clokie MRJ, Shan J, Bailey S, Jia Y, Krisch HM. Transcription of a ‘photosynthetic’ T4-type phage during infection of a marine cyanobacterium. Environ Microbiol. 2006;8:827–835. doi: 10.1111/j.1462-2920.2005.00969.x. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, Delong EF, Chisholm SW. Genomic islands and the ecology and evolution of Prochlorococcus. Science. 2006;311:1768–1770. doi: 10.1126/science.1122050. [DOI] [PubMed] [Google Scholar]
- Dufresne A, Salanoubat M, Partensky F, Artiguenave F, Axmann IM, Barbe V, et al. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc Natl Acad Sci USA. 2003;100:10020–10025. doi: 10.1073/pnas.1733211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufresne A, Ostrowski M, Scanlan DJ, Garczarek L, Mazard S, Palenik BP, et al. Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol. 2008;9:R90. doi: 10.1186/gb-2008-9-5-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Rohwer F. Viral metagenomics. Nat Rev Microbiol. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Fuhrman JA. Impact of viruses on bacterial processes. In: Kirchman DL, editor. Microbial Ecology of the Oceans. New York, USA: Wiley-Liss; 2000. pp. 327–350. [Google Scholar]
- Hellweger FL. Carrying photosynthesis genes increases ecological fitness of cyanophage in silico. Environ Microbiol. 2009;11:1386–1394. doi: 10.1111/j.1462-2920.2009.01866.x. [DOI] [PubMed] [Google Scholar]
- Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc Natl Acad Sci USA. 1999;96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KE, Waldor MK. Filamentous phage integration requires the host recombinases XerC and XerD. Nature. 2002;417:656–659. doi: 10.1038/nature00782. [DOI] [PubMed] [Google Scholar]
- Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EM, Chisholm SW. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science. 2006;311:1737–1740. doi: 10.1126/science.1118052. [DOI] [PubMed] [Google Scholar]
- Juhala RJ, Ford ME, Duda RL, Youlton A, Hatfull GF, Hendrix RW. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J Mol Biol. 2000;299:27–51. doi: 10.1006/jmbi.2000.3729. [DOI] [PubMed] [Google Scholar]
- Kettler GC, Martiny AC, Huang K, Zucker J, Coleman ML, Rodrigue S, et al. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 2007;3:e231. doi: 10.1371/journal.pgen.0030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Hatfull GF, Hendrix RW. Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J Bacteriol. 2002;184:4891–4905. doi: 10.1128/JB.184.17.4891-4905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Hebrant A, Wodak SJ, Toussaint A. ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 2004;32:D45–D49. doi: 10.1093/nar/gkh084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell D, Sullivan MB, Johnson ZI, Tolonen AC, Rohwer F, Chisholm SW. Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc Natl Acad Sci USA. 2004;101:11013–11018. doi: 10.1073/pnas.0401526101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell D, Jaffe JD, Johnson ZI, Church GM, Chisholm SW. Photosynthesis genes in marine viruses yield proteins during host infection. Nature. 2005;438:86–89. doi: 10.1038/nature04111. [DOI] [PubMed] [Google Scholar]
- Lindell D, Jaffe JD, Coleman ML, Futschik ME, Axmann IM, Rector T, et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature. 2007;449:83–86. doi: 10.1038/nature06130. [DOI] [PubMed] [Google Scholar]
- Lohr JE, Chen F, Hill RT. Genomic analysis of bacteriophage PhiJL001: insights into its interaction with a sponge-associated alpha-proteobacterium. Appl Environ Microbiol. 2005;71:1598–1609. doi: 10.1128/AEM.71.3.1598-1609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross CA. The sigma-70 family: sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Chen F, Hodson RE. Distribution, isolation, host specificity, and diversity of cyanophages infecting marine Synechococcus spp. in river estuaries. Appl Environ Microbiol. 2001;67:3285–3290. doi: 10.1128/AEM.67.7.3285-3290.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel L, Houchin LA, Williamson SJ, Paul JH. Lysogeny in marine Synechococcus. Nature. 2002;415:496. doi: 10.1038/415496a. [DOI] [PubMed] [Google Scholar]
- Mahillon J, Chandler M. Insertion sequences. Microbiol Mol Biol Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann NH, Cook A, Millard A, Bailey S, Clokie M. Bacterial photosynthesis genes in a virus. Nature. 2003;424:741. doi: 10.1038/424741a. [DOI] [PubMed] [Google Scholar]
- Mann NH, Clokie MR, Millard A, Cook A, Wilson WH, Wheatley PJ, et al. The genome of S-PM2, a ‘photosynthetic’ T4-type bacteriophage that infects marine Synechococcus. J Bacteriol. 2005;187:3188–3200. doi: 10.1128/JB.187.9.3188-3200.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston MF, Sallee JL. Genetic diversity and temporal variation in the cyanophage community infecting marine Synechococcus species in Rhode Island's coastal waters. Appl Environ Microbiol. 2003;69:4639–4647. doi: 10.1128/AEM.69.8.4639-4647.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny AC, Coleman ML, Chisholm SW. Phosphate acquisition genes in Prochlorococcus ecotypes: evidence for genome-wide adaptation. Proc Natl Acad Sci USA. 2006;103:12552–12557. doi: 10.1073/pnas.0601301103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Miller SI. Bacteriophages in the evolution of pathogen–host interactions. Proc Natl Acad Sci USA. 1999;96:9452–9454. doi: 10.1073/pnas.96.17.9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard A, Clokie MR, Shub DA, Mann NH. Genetic organization of the psbAD region in phages infecting marine Synechococcus strains. Proc Natl Acad Sci USA. 2004;101:11007–11012. doi: 10.1073/pnas.0401478101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, et al. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Plague GR. Genomic changes following host restriction in bacteria. Curr Opin Genet Dev. 2004;14:627–633. doi: 10.1016/j.gde.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Muhling M, Fuller NJ, Millard A, Somerfield PJ, Marie D, Wilson WH, et al. Genetic diversity of marine Synechococcus and co-occurring cyanophage communities: evidence for viral control of phytoplankton. Environ Microbiol. 2005;7:499–508. doi: 10.1111/j.1462-2920.2005.00713.x. [DOI] [PubMed] [Google Scholar]
- Nojiri H, Shintani M, Omori T. Divergence of mobile genetic elements involved in the distribution of xenobiotic-catabolic capacity. Appl Microbiol Biotechnol. 2004;64:154–174. doi: 10.1007/s00253-003-1509-y. [DOI] [PubMed] [Google Scholar]
- Nolan JM, Petrov V, Bertrand C, Krisch HM, Karam JD. Genetic diversity among five T4-like bacteriophages. Virol J. 2006;3:30. doi: 10.1186/1743-422X-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakey HJ, Cullen BR, Owens L. The complete nucleotide sequence of the Vibrio harveyi bacteriophage VHML. J Appl Microbiol. 2002;93:1089–1098. doi: 10.1046/j.1365-2672.2002.01776.x. [DOI] [PubMed] [Google Scholar]
- Ortmann AC, Lawrence JE, Suttle CA. Lysogeny and lytic viral production during a bloom of the cyanobacterium Synechococcus spp. Microb Ecol. 2002;43:225–231. doi: 10.1007/s00248-001-1058-9. [DOI] [PubMed] [Google Scholar]
- Palenik B, Brahamsha B, McCarren J, Waterbury J, Allen E, Webb EA, et al. The genome of a motile marine Synechococcus. Nature. 2003;424:1037–1041. doi: 10.1038/nature01943. [DOI] [PubMed] [Google Scholar]
- Palenik B, Ren Q, Dupont CL, Myers GS, Heidelberg JF, Badger JH, et al. Genome sequence of Synechococcus CC9311: insights into adaptation to a coastal environment. Proc Natl Acad Sci USA. 2006;103:13555–13559. doi: 10.1073/pnas.0602963103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partensky F, Hess WR, Vaulot D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol Mol Biol Rev. 1999;63:106–127. doi: 10.1128/mmbr.63.1.106-127.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, et al. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003;113:171–182. doi: 10.1016/s0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- Pei J, Grishin NV. PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics. 2007;23:802–808. doi: 10.1093/bioinformatics/btm017. [DOI] [PubMed] [Google Scholar]
- Pei J, Kim BH, Tang M, Grishin NV. PROMALS web server for accurate multiple protein sequence alignments. Nucleic Acids Res. 2007;35:W649–W652. doi: 10.1093/nar/gkm227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plague GR, Dunbar HE, Tran PL, Moran NA. Extensive proliferation of transposable elements in heritable bacterial symbionts. J Bacteriol. 2008;190:777–779. doi: 10.1128/JB.01082-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirel L, Lartigue MF, Decousser JW, Nordmann P. ISEcp1B-mediated transposition of blaCTX-M in Escherichia coli. Antimicrob Agents Chemother. 2005;49:447–450. doi: 10.1128/AAC.49.1.447-450.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope WHWP, Chang J, Pedulla ML, Ford ME, Houtz JM, Jiang W, et al. Genome sequence, structural proteins, and capsid organization of the cyanophage Syn5: a ‘horned’ bacteriophage of marine Synechococcus. J Mol Biol. 2007;368:966–981. doi: 10.1016/j.jmb.2007.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proux C, van Sinderen D, Suarez J, Garcia P, Ladero V, Fitzgerald GF, et al. The dilemma of phage taxonomy illustrated by comparative genomics of Sfi21-like Siphoviridae in lactic acid bacteria. J Bacteriol. 2002;184:6026–6036. doi: 10.1128/JB.184.21.6026-6036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, et al. Virus Taxonomy: The Classification and Nomenclature of Viruses. San Diego, USA: Academic Press; 2000. [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rocap G, Larimer FW, Lamerdin J, Malfatti S, Chain P, Ahlgren NA, et al. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature. 2003;424:1042–1047. doi: 10.1038/nature01947. [DOI] [PubMed] [Google Scholar]
- Rohwer F, Segall A, Steward G, Seguritan V, Breitbart M, Wolven F, Azam F. The complete genomic sequence of the marine phage Roseophage SIO1 shares homology with nonmarine phages. Limnol Oceanogr. 2000;45:408–418. [Google Scholar]
- Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Schneider D, Lenski RE. Dynamics of insertion sequence elements during experimental evolution of bacteria. Res Microbiol. 2004;155:319–327. doi: 10.1016/j.resmic.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Simpson AJ, Reinach FC, Arruda P, Abreu FA, Acencio M, Alvarenga R, et al. The genome sequence of the plant pathogen Xylella fastidiosa. The Xylella fastidiosa Consortium of the Organization for Nucleotide Sequencing and Analysis. Nature. 2000;406:151–157. doi: 10.1038/35018003. [DOI] [PubMed] [Google Scholar]
- Smoot JC, Barbian KD, Van Gompel JJ, Smoot LM, Chaussee MS, Sylva GL, et al. Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc Natl Acad Sci USA. 2002;99:4668–4673. doi: 10.1073/pnas.062526099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot LM, Smoot JC, Graham MR, Somerville GA, Sturdevant DE, Migliaccio CA, et al. Global differential gene expression in response to growth temperature alteration in group A Streptococcus. Proc Natl Acad Sci USA. 2001;98:10416–10421. doi: 10.1073/pnas.191267598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web-servers. Syst Biology. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Waterbury JB, Chisholm SW. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;424:1047–1051. doi: 10.1038/nature01929. [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Coleman M, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 2005;3:e144. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006;4:e234. doi: 10.1371/journal.pbio.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA, Chan AM. Dynamics and distribution of cyanophages and their effects on marine Synechococcus spp. Appl Environ Microbiol. 1994;60:3167–3174. doi: 10.1128/aem.60.9.3167-3174.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toleman MA, Bennett PM, Walsh TR. ISCR elements: novel gene-capturing systems of the 21st century? Microbiol Mol Biol Rev. 2006;70:296–316. doi: 10.1128/MMBR.00048-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolonen AC, Aach J, Lindell D, Johnson ZI, Rector T, Steen R, et al. Global gene expression of Prochlorococcus ecotypes in response to changes in nitrogen availability. Mol Syst Biol. 2006;2:53. doi: 10.1038/msb4100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PL, Waldor MK. Bacteriophage control of bacterial virulence. Infect Immun. 2002;70:3985–3993. doi: 10.1128/IAI.70.8.3985-3993.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterbury JB, Valois FW. Resistance to co-occurring phages enables marine Synechococcus communities to coexist with cyanophage abundant in seawater. Appl Environ Microbiol. 1993;59:3393–3399. doi: 10.1128/aem.59.10.3393-3399.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterbury JB, Watson SW, Guillard RRL, Brand LE. Widespread occurrence of a unicellular marine planktonic cyanobacterium. Nature. 1979;277:293–294. [Google Scholar]
- Waterbury JB, Watson SW, Valois FW, Franks DG. Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Can Bull Fish Aquat Sci. 1986;214:71–120. [Google Scholar]
- Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF, et al. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ Microbiol. 2007;9:1675–1695. doi: 10.1111/j.1462-2920.2007.01285.x. [DOI] [PubMed] [Google Scholar]
- Whiteley M, Bangera MG, Bumgarner RE, Parsek MR, Teitzel GM, Lory S, Greenberg EP. Gene expression in Pseudomonas aeruginosa biofilms. Nature. 2001;413:860–864. doi: 10.1038/35101627. [DOI] [PubMed] [Google Scholar]
- Williams KP. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 2002;30:866–875. doi: 10.1093/nar/30.4.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Nagasaki K, Takashima Y, Shirai Y, Tomaru Y, Takao Y, et al. Ma-LMM01 infecting toxic Microcystis aeruginosa illuminates diverse cyanophage genome strategies. J Bacteriol. 2008;190:1762–1772. doi: 10.1128/JB.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidner G, Bielawski JP, Shmoish M, Scanlan DJ, Sabehi G, Beja O. Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Environ Microbiol. 2005;7:1505–1513. doi: 10.1111/j.1462-2920.2005.00833.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.