

Abstract
Herpes simplex virus-1 primase misincorporates the natural NTPs at frequencies around 1 error per 30 NTPs polymerized, making it one of the least accurate polymerases known. We used a series of nucleotide analogues to further test the hypothesis that primase requires Watson-Crick hydrogen bond formation in order to efficiently polymerize a NTP. Primase could not generate base pairs containing a complete set hydrogen bonds in an altered arrangement (iso-guanine:iso-cytosine), and did not efficiently polymerize dNTPs completely incapable of forming Watson-Crick hydrogen bonds opposite templating bases incapable of forming Watson-Crick hydrogen bonds. Similarly, primase did not incorporate most NTPs containing hydrophobic bases incapable of Watson-Crick hydrogen bonding opposite natural template bases. However, 2-pyridone NTP and 4-methyl-2-pyridone NTP provided striking exceptions to this rule. The effects of removing single Watson-Crick hydrogen bonding groups from either the NTP or templating bases varied from almost no effect to completely blocking polymerization depending both on the parental base pair (G:C vs. A:T/U) and which base pair of the growing primer (second, third or fourth) was examined. Thus, primase does not absolutely need to form Watson-Crick hydrogen bonds in order to efficiently polymerize a NTP. Additionally, we found that herpes primase can misincorporate nucleotides both by misreading the template and by a primer-template slippage mechanism. The mechanistic and biological implications of these results are discussed.
Keywords: Polymerase, fidelity, misincorporation, Watson-Crick, hydrogen bond, kinetics, mechanism
The herpes simplex virus 1 (HSV-1 1) primase-helicase is an essential component of the herpes DNA replication machinery and consists of 3 subunits coded for by three genes - UL5, UL8, and UL52. This heterotrimeric complex possess three main activities: primase, DNA dependent NTPase and 5′-3′ helicase. The helicase tracks along the lagging strand and unwinds the DNA in front of the replication fork, and the NTPase provides the energy needed for unwinding. Primase synthesizes short RNA primers (up to 10 – 13 nucleosides long) on single-stranded DNA that a replicative polymerase extends via dNTP polymerization (1-3). The helicase-primase can initiate primer synthesis at any consecutive template pyrimidine residues but for longer primer synthesis (>3 nucleotides long), the 3′-G-Pyr-Pyr trimer is strictly required (Pyr = pyrmidine). Even on templates containing 3′-G-Pyr-Pyr, however, primase still synthesizes very large quantities of dinucleotides and trinucleotides (1) .
A subassembly containing UL5 and UL52 subunits retains all 3 activities, while the UL8 subunit modulates these activities. UL8 also binds to other herpes replication proteins, and may be involved in the organization of the herpes replisome (1, 2, 4-9). UL52 contains the complete primase active site in terms of phosphodiester bond formation, but cannot initiate primer synthesis in the absence of UL5 (7, 10). Thus, UL5 plays a critical but as yet undefined role during initiation of primer synthesis.
The question of how polymerases choose whether or not to polymerize a (d)NTP remains unresolved, although enzymes from different evolutionary families clearly use different mechanisms. A and B family DNA polymerases do not require the formation of Watson-Crick hydrogen bonds in order to polymerize a dNTP (11, 12). B family enzymes use specific functional groups on the base of the incoming dNTP to encourage correct dNTP incorporation and prevent misincorporation (13-17). Data for the A family are less clear cut, with some strongly supporting shape as a primary determinant and others suggesting shape does not play a critical role (16, 18-27). Human primase, an enzyme that shares a small amount of sequence similarity to herpes primase, may require the formation of Watson-Crick hydrogen bonds in order to polymerize a NTP (28, 29). Likewise, it has been suggested that the Y family DNA polymerases also require the formation of Watson-Crick hydrogen bonds in order to polymerize a dNTP (30, 31).
HSV-1 DNA primase misincorporates the natural NTPs extremely efficiently, on average only 30-fold less efficiently than it correctly incorporates a NTP, and readily generates all types of mismatches - purine:purine, purine:pyrimidine, and pyrimidine:pyrimidine (32). Misincorporation largely, but not necessarily exclusively, occurs via misreading of the templating base as opposed to occurring via template or primer slippage (32). Herpes primase clearly is a low fidelity polymerase, although this infidelity is not a problem biologically since the RNA primer is removed and replaced with DNA.
Previous work examined polymerization of NTP analogues containing modified bases as the first, second or third nucleotide of the primer, and suggested that efficient NTP incorporation requires formation of Watson-Crick hydrogen bonds (33). Loss of either N-1 or N6 from ATP reduced incorporation much more severely than converting ATP to CTP, GTP, or UTP. The natural NTPs can always form Watson-Crick hydrogen bonds with any natural base by using either the major or a minor tautomer (28, 33, 34). Adding a halogen at C-2 of ATP, a modification that only weakens the ability of the resulting base to form Watson-Crick hydrogen bonds (35, 36), likewise had relatively small effects on incorporation.
We examined how HSV-1 primase interacts with a series of NTP analogues to better define the role of Watson-Crick hydrogen bonds and minor groove hydrogen bond acceptors during NTP polymerization. Surprisingly, primase exhibited very unpredictable requirements for polymerizing a NTP. The requirements varied depending upon the base-pair being generated, whether the incoming NTP will become the second, third, or fourth nucleotide of the primer, and if the NTP was a purine or pyrimidine. The enzyme also did not require formation of any Watson-Crick hydrogen bonds between the incoming NTP and templating base with a few select NTPs. Besides showing that herpes primase can be a remarkably selective enzyme, these studies raise the question of whether other polymerases proposed to require Watson-Crick hydrogen bonds between the incoming and templating nucleotides actually do require them.
EXPERIMENTAL PROCEDURES
Materials
All reagents were of the highest quality commercially available. Unlabeled natural NTPs were from Sigma and radiolabeled NTPs were from Perkin Elmer. Protected phosphoramidites of 2′-deoxyribonucleosides containing the bases 5-methyl-2(1H)-pyrimidinone, 3-deazaadenine, and 5-methyl-isocytosine were purchased from Glen Research. C-2′-deoxyribonucleosides (pyridin-3-yl, 4-oxopyridin-3-yl, and 4-aminopyridin-3-yl) were synthesized as described (37). ITP and iso-GTP were purchased from Trilink. 2-Hydroxypyridine, 2-hydroxy-4-methylpyridine, 2,3-diaminopyridine, 2-amino-4-methyl-3-nitropyridine, benzimidazole, and 2,3-diaminotoluene were purchased from Sigma-Aldrich. Purine and protected 1-chlorodeoxyribose were purchased from Berry and Associates. Synthetic DNA oligonucleotides of defined seqeuence were purchased from IDT or Biosearch Technologies. HSV-1 primase (UL5/LU8/UL52 complex) was purified and stored as previously described (1, 33).
Methods
Primase assays
Assays (10 μL) were performed as previously described (1, 33) and typically contained 600 nM HSV-1 primase, 2 μM DNA template, 50 mM tris(hydroxymethyl)aminomethane, HCl salt pH 8.0, 1 mM dithiothreitol, 0.1 mg/mL bovine serum albumin, 10 mM MgCl2, 1% glycerol, 800 μM GTP, and various concentrations of a NTP (analogue). Reactions were incubated at 37°C for 60 min and quenched with 25 μL formamide/0.05% xylene cyanol and bromophenol blue. Products were separated using 20 – 25% polyacrylamide, 8 M urea gels and analyzed using a Typhoon Phosphorimager (Molecular Dynamics). The relative Vmax / KM for polymerization of a NTP (analogue) versus GTP was determined using a partitioning analysis as described previously (38).
Synthesis of nucleotide analogues
1-Deazapurine
1,2-Diaminopyridine (4.36 g, 40 mmol) was dissolved in formic acid (90 %) and refluxed for 48 h. Formic acid was evaporated under reduced pressure and the crude product was chromatographed on silica gel (250 mL) using a gradient of 0–10% MeOH in CH2Cl2. The product was crystallized from a mixture of MeOH/EtOAc. Yield of yellow crystals was 3.43 g (65 %). 1H NMR (400 MHz, CDCl3): 8.45 (s, 1H, H-8), 8.35 (dd, 1H, J1 = 4.7 Hz, J2 = 2.4 Hz), 8.01 (bd, 1H, J = 7.9 Hz), 7.21 (dd, 1H, J1 = 8.3 Hz, J2 = 4.8 Hz, H-1), 3.41 (bs, 1H, NH).
6-Methyl-1-deazapurine
2-Amino-4-methyl-3-nitropyridine (5 g, 32.6 mmol) was dissolved in a mixture of THF (50 mL), isopropyl alcohol (20 mL) and water (5 mL). Pd (1 g, 10 % on charcoal, 0.9 mmol) was added and the mixture was repeatedly evacuated and saturated with H2 at r.t. and laboratory pressure. The mixture was vigorously stirred for 3 h and then the Pd/C was filtered off, organic solvents were removed under reduced pressure. Resulting 2,3-diamino-4-methylpyridine was dissolved (without any further purification) in formic acid (70 mL, 90 % in water) to get 6-methyl-1-deazapurine in the same procedure as for 1-deazapurine. The whole procedure yielded 2.2 g (51 %) of 6-methyl-1-deazapurine. 1H NMR (400 MHz, CDCl3): 8.38 (s, 1H, H-8), 8.20 (d, 1H, J = 4.9 Hz), 7.04 (dd, 1H, J1 = 4.9 Hz, J2 = 0.7 Hz), 3.17 (bs, 1H, NH), 2.54 (s, 3H, Me).
7-Methylbenzimidazole
2,3-diaminotoluene (5 g, 41 mmol) was dissolved in formic acid (100 mL, 90 % in water) and heated to get 7-methylbenzimidazole in the same procedure as for 1-deazapurine. This yielded 4.1 g (76 %) of 7-methylbenzimidazole. 1H NMR (400 MHz, CDCl3): 8.23 (s, 1H, H-2), 8.16 (bs, 1 H, NH), 7.41 (d, 1H, J = 6.8 Hz), 7.09 (dd, 1H, J1 = 7.3 Hz, J2 = 7.3 Hz), 6.98 (dt, 1H, J1 = 7.1 Hz, J2 = 0.9 Hz), 2.51 (s, 3H, Me).
9-β-d-(1-Deazapurin)-2′-deoxy-3′,5′-di-O-(4-toluoyl)-d-ribofuranose
1-Deazapurine (500 mg, 3.78 mmol) was dissolved in MeCN (11 mL) and NaH (5.67 mmol, 60 % in oil, 1.5 eq.) was added. The mixture was stirred 3 h at r.t. and then 1-chloro-3,5-bis(p-toluoyl)-2-deoxy-β-d-ribofuranose (1.5 g, 3.78 mmol) was added. The resulting slurry was stirred overnight, resulting in most of the slurry dissolving. The mixture was then poured into saturated aqueous NH4Cl, extracted with EtOAc, and the product purified by chromatography on silica gel (100 mL) using a gradient from 50–100 % EtOAc in hexanes. The yield of colorless oil was 750 mg (43 %). 1H NMR (400 MHz, CDCl3): 8.38 (dd, 1H, J1 = 4.9 Hz, J2 = 1.5 Hz, H-2), 8.23 (s, 1H, H-8), 8.07 (dd, 1H, J1 = 4.9 Hz, J2 = 1.5 Hz, H-6), 7.98 (bdd, 2H, J1 = 6.7 Hz, J2 = 1.7 Hz, Tol), 7.93 (bdd, 2H, J1 = 6.7 Hz, J2 = 1.7 Hz, Tol), 7.20 – 7.30 (m, 5H, H-1 and 4xH-Tol), 6.70 (dd, 1H, J1 = 8.5 Hz, J2 = 5.8 Hz, H-1′), 5.82 (m, 1H, H-3′), 4.6–4.8 (m, 3H, H-5′a, H-5′b, H-′4), 3.18 (m, 1H, H-2′a), 2.84 (ddd, 1H, Jgem = 12.1 Hz, J2′b,1′ = 5.8 Hz, J2′b, 3′ = 2.0 Hz, H-2′b), 2.44 (s, 3H, CH3-Tol), 2.40 (s, 3H, CH3-Tol).
9-β-d-(1-Deazapurine)-2′-deoxyribofuranose
9-β-d-(1-Deazapurin)-2′-deoxy-3′,5′-di-O-(4-toluoyl)-d-ribofuranose (0.5 g, 1.08 mmol) was deprotected in 30 min using 0.1 M MeONa in MeOH and purified by silica gel chromatography (20 mL) using a gradient of 0–20 % MeOH in CHCl3. This procedure gave 0.2 g (78 %) of product. 1H NMR (400 MHz, CDCl3): 8.61 (s, 1H, H-8), 8.37 (dd, 1H, J1= 4.9 Hz, J2 = 1.5 Hz, H-2), 8.10 (dd, 1H, J1 = 8.1 Hz, J2 = 1.5 Hz, H-6), 7.37 (dd, 1H, J1 = 8.1 Hz, J2 = 4.9 Hz, H-1), 6.57 (dd, 1H, J1 = 8.0 Hz, J2 = 6.0 Hz, H-1′), 4.83 (bs, 3H, OH, H2O), 4.61 (m, 1H, H-3′), 4.09 (dd, 1H, J1 = 6.0 Hz, J2 = 3.2 Hz, H-4′), 3.85 (dd, 1H, J5′a, 5′b = 12.2 Hz, J5′a, 4′ = 3.1 Hz, H-5′a), 3.75 (dd, 1H, J5′b, 5′a = 12.3 Hz, J5′a, 3′ = 3.4 Hz, H-5′a), 2.88 (m, 1H, H-2′a), 2.43 (ddd, 1H, Jgem = 13.5 Hz, J2′b,1′ = 6.1 Hz, J2′b, 3′ = 2.7 Hz, H-2′b).
9-β-d-(6-Methyl-1-deazapurin)-2′-deoxy-3′,5′-di-O-(4-toluoyl)-d-ribofuranose
The nucleoside was prepared from 6-Methyl-1-deazapurine (490 mg, 2.67 mmol) in the same procedure as 1-deazapurine nucleoside. The whole procedure yielded 580 mg (44 %) of colorless oil. 1H NMR (400 MHz, CDCl3): 8.24 (d, 1H, J1 = 4.0 Hz, H-2), 8.18 (s, 1H, H-8), 7.98 (bdd, 2H, J1 = 6.7 Hz, J2 = 1.7 Hz, Tol), 7.93 (bdd, 2H, J1 = 6.7 Hz, J2 = 1.7 Hz, Tol), 7.20 – 7.30 (m, 4H, 4xH-Tol), 7.06 (dd, 1H, J1 = 3.7 Hz, J2 = 0.8 Hz, H-1), 6.67 (dd, 1H, J1 = 8.4 Hz, J2 = 5.7 Hz, H-1′), 5.82 (m, 1H, H-3′), 4.6-4.8 (m, 3H, H-5′a, H-5′b, H-′4), 3.15 (ddd, 1H, Jgem = 14.3 Hz, J2′a,1′ = 8.7 Hz, J2′a, 3′ = 6.3 Hz, H-2′a), 2.82 (ddd, 1H, Jgem = 14.3 Hz, J2′b,1′ = 5.7 Hz, J2′b, 3′ = 1.9 Hz, H-2′b), 2.65, 2.42, 2.38 (3xs, 3H, 3xCH3).
9-β-d-(6-Methyl-1-deazapurine)-2′-deoxyribofuranose
9-β-d-(6-Methyl-1-deazapurin)-2′-deoxy-3′,5′-di-O-(4-toluoyl)-d-ribofuranose (530 mg, 1.1 mmol) was deprotected in the same procedure as 1-deazapurine nucleoside to give 200 mg (76 %) of crystals. 1H NMR (400 MHz, CDCl3): 8.55 (s, 1H, H-8), 8.41 (dd, 1H, J1= 4.7 Hz, J2 = 1.4 Hz, H-2), 7.36 (d, 1H, J1 = 4.1 Hz, H-1), 6.57 (dd, 1H, J1 = 8.2 Hz, J2 = 5.9 Hz, H-1′), 4.80 (bs, 3H, OH, H2O), 4.60 (m, 1H, H-3′), 4.09 (dd, 1H, J1 = 6.0 Hz, J2 = 3.2 Hz, H-4′), 3.85 (dd, 1H, Jgem= 12.2 Hz, J5′a, 4′ = 3.1 Hz, H-5′a), 3.75 (dd, 1H, Jgem= 12.3 Hz, J5′a, 3′ = 3.4 Hz, H-5′a), 2.88 (m, 1H, H-2′a), 2.43 (ddd, 1H, Jgem = 14.5 Hz, J2′b,1′ = 5.8 Hz, J2′b, 3′ = 1.9 Hz, H-2′b).
1-β-d-(2-Oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose
2-Hydroxypyridine (510 mg, 5.34 mmol) was dissolved in MeCN (11 mL) and bis-trimethylsilylacetamide (1.32 mL, 5.34 mmol) was added dropwise. The mixture was stirred for 70 min at r.t., cooled on ice, and a mixture of MeCN (11 mL) and 1-chloro-3,5-bis(p-toluoyl)-2-deoxy-β-d-ribofuranose (1.7 g, 4.35 mmol) was then added. The mixture was stirred overnight while warming up to r.t., and worked up as described above. Silica gel chromatography (200 mL) in 28 % EtOAc/hexane yielded 1.8 g (37 %) of the desired 1-β-d-(2-oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose, along with 0.5 g (10 %) of mixture of α and β anomers and 1.4 g (29 %) of the α anomer of the product. 1H NMR (400 MHz, CDCl3): 7.97 (bdd, 2H, J1 = 6.4 Hz, J2 = 1.7 Hz, Tol), 7.87 (bdd, 2H, J1 = 6.4 Hz, J2 = 1.7 Hz, Tol), 7.71 (dd, 1H, J1 = 7.1 Hz, J2 = 1.9 Hz, 1H-Pyr), 7.31 (ddd, 1H, J1 = J2 = 6.5 Hz, J3 = 2.0 Hz, 1H-Pyr), 7.28 (bd, 2H, J = 6.5 Hz, Tol), 7.22 (bd, 2H, J = 6.5 Hz, Tol), 6.61 (dd, 1H, J1 = 8.1 Hz, J2 = 5.6 Hz, H-1′), 6.54 (bd, 1H, J1 = 9.15 Hz, J2 = 1.3 Hz, J3 = 0.6 Hz 1H-Pyr), 6.12 (dd, J1 = 6.8 Hz, J2 = 1.1 Hz, 1H-Pyr), 5.60 (dt, 1H, J1 = 6.6 Hz, J2 = 2.0 Hz, H-3′), 4.71 (m, 2H, H-5′a, H-5′b), 4.62 (m, 1H, H-4′), 3.00 (ddd, 1H, Jgem = 14.5 Hz, J2′a,1′= 5.7 Hz, J2′a,3′ = 1.9 Hz, H-2′a), 2.43 (s, 3H, CH3-Tol), 2.41 (s, 3H, CH3-Tol), 2.16 (ddd, 1H, Jgem = 14.5 Hz, J2′b,1′ = 7.9 Hz, J2′b,3′ = 6.6 Hz, H-2′b). The NMR spectrum was identical to that reported in the literature (39).
1-β-d-(2-Oxopyridine)- 2′-deoxyribofuranose
The 1-β-d-(2-oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose (525 mg, 1.2 mmol) was deprotected using 0.1 M MeONa in MeOH and purified by silica gel chromatography (20 mL) using a gradient of 0–20 % MeOH in CHCl3. This procedure gave 224 mg (78 %) of product. 1H NMR (400 MHz, DMSO-d6): 7.93 (dd, 1H, J1 = 7.0 Hz, J2 = 1.8 Hz, 1H-Pyr), 7.42 (ddd, 1H, J1 = J2 = 6.5 Hz, J3 = 2.0 Hz, 1H-Pyr), 6.35 (m, 2H, 2H-Pyr), 6.28 (td, J1 = 6.9 Hz, J2 = 1.2 Hz, H-1′), 5.26 (bd, 1H, J = 4.3 Hz, 3-OH), 5.04 (t, 1H, J = 5.2 Hz, 5-OH), 4.22 (m, 1H, H-3′), 3.84 (m, 1H, H-4′), 3.60 (m, 2H, H-5′a, H-5′b), 2.24 (m, 1H, H-2′a), 1.94 (ddd, 1H, Jgem = 13.2 Hz, J2′b,1′ = 6.1 Hz, J2′b,3′= 6.1 Hz, H-2′b).
1-β-d-(4-Methyl-2-oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose
This nucleoside was prepared from 4-methyl-2-hydroxypyridine (583 mg, 5.34 mmol) in the same procedure as 2-oxopyridine nucleoside which yielded 790 mg (32 %) of the desired 1-β-d-(4-methyl-2-oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose (along with 40 % of the mixture of desired product with the α anomer). 1H NMR (400 MHz, CDCl3): 7.97 (bdd, 2H, J1 = 6.7 Hz, J2 = 1.6 Hz, Tol), 7.87 (bdd, 2H, J1 = 6.7 Hz, J2 = 1.6 Hz, Tol), 7.56 (d, 1H, J = 7.2 Hz, 1H-Pyr), 7.28 (dd, 2H, J1 = 8.5 Hz, J2 = 0.6 Hz, Tol), 7.23 (dd, 2H, J1 = 8.5 Hz, J2 = 0.6 Hz, Tol), 6.61 (dd, 1H, J1 = 8.2 Hz, J2 = 5.6 Hz, H-1′), 6.35 (1H, s, H-3-Pyr), 5.96 (dd, J1 = 7.2 Hz, J2 = 1.7 Hz, H-Pyr), 5.60 (dt, 1H, J3′,2′b = 6.6 Hz, J3,2′a = 1.9 Hz, H-3′), 4.70 (m, 2H, H-5′a, H-5′b), 4.60 (m, 1H, H-4′), 2.96 (ddd, 1H, Jgem = 14.4 Hz, J2′a,1′= 5.6 Hz, J2′a,3′ = 1.8 Hz, H-2′a), 2.44 (s, 3H, CH3-Tol), 2.42 (s, 3H, CH3-Tol), 2.24 (ddd, 1H, Jgem = 14.6 Hz, J2′b,1′ = 7.2 Hz, J2′b,3′= 6.6 Hz, H-2′b). The NMR spectrum was identical to that reported in the literature (39).
1-β-d-(4-Methyl-2-oxopyridine)-2′-deoxyribofuranose
The 1-β-d-(2-oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose (730 mg, 1.6 mmol) was deprotected using 0.1 M MeONa in MeOH and purified by silica gel chromatography (20 mL) using a gradient of 0–20 % MeOH in CHCl3. This procedure gave 330 mg (93 %) of product. 1H NMR (400 MHz, DMSO-d6): 7.80 (d, 1H, J1 = 7.1 Hz, 1H-Pyr), 6.34 (t, 1H, J1 = 6.9 Hz, 1H-Pyr), 6.14 (m, 3H, 2H-Pyr, H-1′), 5.25 (d, 1H, J = 4.1 Hz, 3-OH), 5.03 (t, 1H, J = 5.2 Hz, 5-OH), 4.23 (m, 1H, H-3′), 3.84 (m, 1H, H-4′), 3.58 (m, 2H, H-5′a, H-5′b), 2.21 (ddd, 1H, Jgem = 13.2 Hz, J2′a,1′= 5.9 Hz, J2′a,3′ = 3.3 Hz, H-2′a), 2.11 (s, 1H, Me), 1.92 (ddd, 1H, Jgem = 13.2 Hz, J2′b,1′ = 7.1 Hz, J2′b,3′= 6.1 Hz, H-2′b).
General procedure of the preparation of purine ribonucleoside analogs
Glycosylation was performed essentially as previously described (28, 40). Benzimidazole (591 mg, 5 mmol) was dissolved in MeCN (50 mL) and N,O-Bis(trimethylsilyl)acetamide (1.8 mL, 7.5 mmol) was added dropwise. The mixture was heated under reflux for 15 min. After cooling to r.t., 1,2,3,5-Tetra-O-acetyl-β-d-ribofuranose (1.8 g, 5.6 mmol) in MeCN (20 mL) was added followed by 1.2 mL (6.25 mmol) of trimethylsilyl trifluoromethanesulfonate (1.3 mL, 7.2 mmol) and the mixture was heated under reflux another 3 h. After cooling to r.t., the mixture was poured into saturated NaHCO3 and the product was extracted to EtOAc. The organic layer was dryed with MgSO4 and solvents were removed at vacuum. The product was purified by the chromatography on silica gel (100 g) in gradient from EtOAc to 5 % of MeOH in EtOAc to give colorless oil of product in yields of 25 – 60 %. The nucleoside was then deprotected using MeONa in MeOH (0.1M) and purified by the chromatography on silica gel (25 g) in gradient from CH2Cl2 to 30 % MeOH in CH2Cl2.
2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(benzimidazol-1-yl)-β-d-ribofuranose
The yield was51 %, and the 1H NMR spectrum was identical to that reported previoiusly (40).
1′-deoxy-1′-(benzimidazol-1-yl)-β-d-ribofuranose
The yield was 85 % and the 1H NMR spectrum was identical to that reported previously (40).
2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(4-methylbenzimidazol-1-yl)-β-d-ribofuranose
The yield was 25 % along with 20 % of other isomer [1′β-d-2′,3′,5′-Tri-O-Acetyl-1′-(4-methylbenzimidazole-3-yl)-ribofuranose].
1′-deoxy-1′-(4-methylbenzimidazol-1-yl)-β-d-ribofuranose
The yield was 90 % and the 1H NMR spectrum was identical to that reported previously (41).
2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(1-deazapurin-9-yl)-β-d-ribofuranose
There were obtained three products from the reaction, 18 % of the desired 2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(1-deazapurin-9-yl)-β-d-ribofuranose (along with 12 % of 2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(1-deazapurin-7-yl)-β-d-ribofuranose and 14 % of alpha-anomer). 1H NMR (400 MHz, CDCl3): 8.40 (dd, 1H, J2,1 = 4.9 Hz, J2,6 = 1.5 Hz, H-2), 8.21 (s, 1H, H-8), 8.07 (dd, 1H, J6,1 = 8.1 Hz, J6,2 = 1.5 Hz, H-6), 7.27 (dd, 1H, J1,6 = 8.1 Hz, J1,2 = 4.8 Hz, H-1), 6.29 (d, 1H, J1′,2′ = 5.2 Hz, H-1′), 6.04 (t, 1H, J2′,1′ = J2′,3′ = 5.3 Hz, H-2′), 5.72 (t, 1H, J3′,2′ = J3′,4′ = 5.1 Hz, H-3′), 4.44 (m, 2H, H-4′ and H-5a′), 4.37 (dd, 1H, Jgem = 13.0 Hz, J5b′,4′ = 5.6 Hz, H-5b′), 2.13 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H, 3xOAc). The structure of this compound was confirmed by COSY, 2D-NOESY, HSQC, and HMBC spectra. 2D-NOESY cross peaks of H-1′ with H-4′ and H-8 with H-2′ confirmed that the compound is β-anomer and HMBC cross peaks of H-1′ with C-5, C-3, C-8 and H-8 with C-5 and C-6 confirmed that the structure is 1-deazapurin-9-yl.
1′-deoxy-1′-(1-deazapurin-9-yl)-β-d-ribofuranose
Was obtained by the deprotection of 2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(1-deazapurin-9-yl)-β-d-ribofuranose in the usual procedure to give 87 % yield. 1H NMR (400 MHz, DMSO-d6): 8.71 (s, 1H, H-8), 8.38 (dd, 1H, J2,1 = 4.8 Hz, J2,6 = 1.3 Hz, H-2), 8.14 (dd, 1H, J6,1 = 8.0 Hz, J6,2 = 1.5 Hz, H-6), 7.34 (dd, 1H, J1,6 = 8.0 Hz, J1,2 = 4.8 Hz, H-1), 6.07 (d, 1H, J1′,2′ = 6.1 Hz, H-1′), 5.50 (d, 1H, J = 6.2 Hz, OH-3′), 5.27 (t, 1H, J = 5.0 Hz, OH-5′), 5.23 (d, 1H, J = 4.8 Hz, OH-2′), 4.68 (m, 1H, H-4′), 4.19 (m, 1H, H-3′), 3.99 (m, 1H, H-2′), 3.69 (ddd, 1H, Jgem= 12.0 Hz, J5a′,4′ = 4.5 Hz, J5a′,OH′ = 4.5 Hz, H-5a′), 3.59 (ddd, 1H, Jgem= 12.0 Hz, J5b′,OH′ = 6.6 Hz, J5b′,4′ = 3.9 Hz, H-5b′).
2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(6-methyl-1-deazapurin-9-yl)-β-d-ribofuranose
Two products were obtained, 33 % of the desired 2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(6-methyl-1-deazapurin-9-yl)-β-d-ribofuranose (along with 12 % of 2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(6-methyl-1-deazapurin-7-yl)-β-d-ribofuranose). 1H NMR (400 MHz, CDCl3): 8.25 (d, 1H, J2,1 = 4.9 Hz, H-2), 8.16 (s, 1H, H-8), 7.07 (dd, 1H, J1,2 = 4.9 Hz, J2 = 0.7 Hz, H-1), 6.27 (d, 1H, J1′,2′ = 5.3 Hz, H-1′), 6.02 (t, 1H, J2′,1′ = J2′,3′ = 5.4 Hz, H-2′), 5.71 (t, 1H, J3′,2′ = J3′,4′ = 5.1 Hz, H-3′), 4.43 (m, 2H, H-4′ and H-5a′), 4.37 (dd, 1H, Jgem = 12.8 Hz, J5b′,4′ = 5.5 Hz, H-5b′), 2.65 (s, 3H, Ar-Me), 2.13 (s, 3H), 2.10 (s, 3H), 2.05 (s, 3H, 3xOAc).
1′-deoxy-1′-(6-methyl-1-deazapurin-9-yl)-β-d-ribofuranose
This compound was obtained by the deprotection of 2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(1-deazapurin-9-yl)-β-d-ribofuranose in the usual procedure to give 84 % yield. 1H NMR (400 MHz, DMSO-d6): 8.64 (s, 1H, H-8), 8.22 (d, 1H, J2,1 = 4.9 Hz, H-2), 7.17 (d, 1H, J1,2 = 4.8 Hz, H-1), 6.03 (d, 1H, J1′,2′ = 6.0 Hz, H-1′), 5.47 (d, 1H, J = 6.1 Hz, OH-2′), 5.37 (dd, 1H, J1 = 6.8 Hz, J2 = 4.8 Hz, OH-5′), 5.22 (d, 1H, J = 4.7 Hz, OH-3′), 4.67 (m, 1H, H-2′), 4.18 (m, 1H, H-3′), 3.99 (m, 1H, H-4′), 3.69 (m, 1H, H-5a′), 3.59 (m, 1H, H-5b′), 2.59 (s, 1H, Me).
2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(9-purinyl)-β-d-ribofuranose
Purine (330 mg, 2.75 mmol) was dissolved in MeCN, (20 mL) and N,O-Bis(trimethylsilyl) acetamide (0.6 mL, 2.95 mmol) was added dropwise. The mixture was heated under reflux for 1h at r.t. After cooling, 1,2,3,5-Tetra-O-Acetyl-β-D-ribofuranose (1g, 3.14 mmol) in MeCN (10 mL) was added. Trimethylsilyl trifluoromethanesulfonate (0.65 mL, 3.59 mmol) was added, and the mixture was heated under reflux for 4h. Upon cooling to r.t., the mixture was extracted to EtOAc twice with saturated NaHCO3 and once with saturated NaCl. The organic layer was dried over Mg2SO4. Solvents were removed under vacuum. The products was purified by chromatography on silica gel (200 mL) eluted with 3:1 EtOAc/hexane and a 0–5% gradient of MeOH. Purification yielded two distinct products; the desired product was a colorless oil (390 mg, 1.18 mmol) in yield of 43%. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H, Ar), 8.97 (s, 1H, Ar), 8.25 (s, 1H, Ar), 6.23 (d, 1H, J1′,2′ = 5.2 Hz, H-1′), 5.95 (t, 1H, J2′,1′ = J2′,3′ = 5.4 Hz, H-2′), 5.66 (t, 1H, J3′,2′ = J3′,4′ = 5.2 Hz, H-3′), 4.30 – 4.70 (m, 3H, H-4′, H-5′a,and H-5′b), 2.12 (s, 3H, Ac), 2.07 (s, 3H, Ac), 2.04 (s, 3H, Ac).
1′-deoxy-1′-(9-purinyl)-β-d-ribofuranose
2′,3′,5′-Tri-O-Acetyl-1′-deoxy-1′-(9-purinyl)-β-d-ribofuranose (390 mg, 1.18 mmol) was deprotected by stirring for 30 min in 0.1M NaOMe in MeOH. The mixture was purified with silica gel chromatography (20 mL) using a gradient of 0–20% MeOH in CH2Cl2. Deprotection and purification yielded 71 mg of product (24% yield). 1H NMR (400 MHz, CD3OD) δ 9.13 (s, 1H, Ar), 8.95 (s, 1H, Ar), 8.81 (s, 1H, Ar), 6.18 (d, 1H, J1′,2′ = 5.6 Hz, H-1′), 4.76 (dd, 1H, J2′,1′ = J2′,3′ 5.3, H-2′), 4.39 (dd, 1H, J3′,2′ = 5.1 Hz, J3′,4′ = 3.7 Hz, H-3′), 4.18 (ddd, 1H, J4′,3′ = J4′,5′a = J4′,5′b = 3.3 Hz, H-4′), 3.90 (dd, 1H, Jgem= 12.3 Hz, J5a’,4′ = 3.0 Hz, H-5′a), 3.79 (dd, 1H, Jgem = 12.3 Hz, J5b’,4′= 3.3 Hz, H-5′b).
1′-deoxy-1′-(2-oxopyridin-1-yl)-β-d-ribofuranose
2-Hydroxypyridine (0.48 g, 5.5 mmol) and 1,2,3,5-Tetra-O-acetyl-β-d-ribofuranose (1.6 g, 5.0 mmol) were dissolved in 25 mL of MeCN and SnCl4 (1.25 mL) was added dropwise. The mixture was stirred for 8 h at r.t. then the solvents were evaporated and the product was purified by the chromatography on silica gel (100 mL) in EtOAc. Oily product was immediately deprotected using 0.1 M MeONa in MeOH, purified by the chromatography on silica gel (30 mL) in gradient from CH2Cl2 to 20 % MeOH in CH2Cl2. The product was crystallized from MeOH/EtOAc to give 280 mg (25 %) of desired compound. 1H NMR (400 MHz, DMSO-d6): 7.99 (dd, 1H, J1 = 7.1 Hz, J2 = 1.6 Hz, H-Pyr), 7.39 (ddd, 1H, J1 = 8.8 Hz, J2 = 6.5 Hz J3 = 2.0 Hz, H-Pyr), 6.34 (ddd, 1H, J1 = 9.2 Hz, J2 = 0.6 Hz J3 = 0.6 Hz, H-Pyr), 6.03 (d, 1H, J1 = 6.8 Hz, J2 = 6.8 Hz J3 = 1.3 Hz, H-Pyr), 6.01 (d, 1H, J1′,2′ = 3.5 Hz, H-1′), 5.41 (d, 1H, J = 4.3 Hz, OH-3′), 5.11 (t, 1H, J = 5.0 Hz, OH-5′), 5.04 (bd, 1H, J = 3.4 Hz, OH-2′), 3.93 (m, 2H, H-4′ and H-3′), 3.88 (m, 1H, H-2′), 3.70 (m, 1H, H-5a’), 3.58 (m, 1H, H-5b’).
1′-deoxy-1′-(4-methylpyridin-2-on-1-yl)-β-d-ribofuranose
This compound was prepared from 4-methyl-2-hydroxypyridine (600 mg, 5.5 mmol) in the same procedure as 2-oxopyridin-1-yl nucleoside with overall yield of 620 mg (44 %). 1H NMR (400 MHz, DMSO-d6): 7.83 (d, 1H, J1 = 7.2 Hz, H-6), 6.15 (bs, 1H, H-3), 6.08 (dd, 1H, J1 = 7.1 Hz, J2 = 1.7 Hz, H-5), 6.01 (d, 1H, J1′,2′ = 3.7 Hz, H-1′), 5.36 (bs, 1H, OH-3′), 5.09 (bs, 1H, OH-5′), 5.00 (bs, 1H, OH-2′), 3.93 (m, 2H, H-4′ and H-3′), 3.87 (m, 1H, H-2′), 3.67 (bd, 1H, Jgem = 11.9 Hz, H-5a’), 3.58 (bd, 1H, Jgem = 12.2 Hz, H-5b’), 2.09 (s, 3H, Me-Ar).
General procedure for the synthesis of DMTr protected nucleosides
Free nucleoside (0.9 mmol) was dissolved in pyridine (10 mL) and dimethoxytritylchloride (880 mg, 2.6 mmol) was added. After the reaction was completed (3 h, analyzed by silica TLC in EtOAc), the mixture was poured into saturated NaHCO3 and the product was extracted into EtOAc. Organic layers were washed with several portions of 1 % NaHCO3, dried over MgSO4 and solvents were evaporated under reduced pressure. The product was purified by silica gel chromatography (0–10 % MeOH in EtOAc). The reaction yielded (60 – 90 %) of 5′-(4,4-dimethoxytrityl)-protected nucleoside.
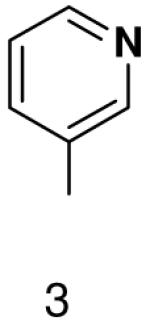
1β-(pyridin-3-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
1H NMR (400 MHz, CDCl3): 8.58 (d, 1H, J2,4 = 2.1 Hz, H-2), 8.51 (dd, 1H, J6,5 = 4.8 Hz, J6,4 = 1.7 Hz, H-6), 7.71 (ddd, J4,5 = 7.9 Hz, J4,2 = 2.1 Hz, J4,6 = 1.6 Hz, H-4), 7.44 (dm, 2H, J = 7.1 Hz, 2 × H-DMTr), 7.18 – 7.36 (m, 8H, 7 × H-DMTr, H-5), 6.81 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 5.20 (dd, 1H, J1′,2′b = 10.3 Hz, J1′,2′a = 5.6 Hz, H-1′), 4.45 (m, 1H, H-3′), 4.10 (m, 1H, H-4′), 3.78 (s, 6H, CH3-DMTr), 3.35 (dd, 1H, Jgem = 9.8 Hz, J5′a,4′ = 4.5 Hz, H-5′a), 3.26 (dd, 1H, Jgem = 9.8 Hz, J5′b,4′ = 5.3 Hz, H-5′b), 2.29 (ddd, 1H, Jgem = 13.0 Hz, J2′a,1′ = 5.6 Hz, J2′a,3′ = 1.9 Hz, H-2′a), 2.02 (ddd, 1H, Jgem = 13.1 Hz, J2′b,1′ = 10.3 Hz, J2′a,3′ = 5.9 Hz, H-2′a).
1β-(6-isobutyrylaminopyridin-3-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
The free nucleoside (100 mg, 0.48 mmol) was dissolved in pyridine (5 mL), trimethylsillyl chloride (300 μL, 3 eq.) was added and the mixture was stirred at r.t. for 5 h. Then Isobutyryl chloride (0.2 mL) was added and the mixture was stirred at r.t. overnight. The product was poured into saturated NaHCO3 and extracted to EtOAc. Organic layers were extracted 3 times with water and then dried over MgSO4. Organic solvents were evaporated to give crude oil of silylated nucleoside, which was deprotected overnight using TBAF in THF (2 mL, 0.1 M). THF was evaporated and the product was chromatographed on silica gel (15 mL) in gradient from CH2Cl2 to 25 % MeOH in CH2Cl2. 1H NMR (400 MHz, CDCl3): 8.81 (bs, 1H, NH), 8.20 (d, 1H, J = 6.1 Hz, H-2), 8.20 (bs, 1H, H-5), 7.70 (dd, 1H, J4,5 = 8.8 Hz, J4,2 = 2.2 Hz, H-4), 5.14 (dd, 1H, J1′,2′b = 9.9 Hz, J1′,2′a = 5.8 Hz, H-1′), 4.45 (dd, 1H, J3′,4′ = 8.8 Hz, J3′2′a = 3.2 Hz, H-3′), 4.06 (m, 1H, H-4′), 3.62 – 3.80 (m, 2H, 2H-5′), 2.50 – 2.64 (m, 1H, CH-iBu), 2.29 (ddd, 1H, Jgem = 13.1 Hz, J2′a,1′ = 5.8 Hz, J2′a,3′ = 2.4 Hz, H-2′a), 2.02 (ddd, 1H, Jgem = 13.1 Hz, J2′b,1′ = 9.9 Hz, J2′a,3′ = 3.4 Hz, H-2′a), 1.23 (d, 1H, J = 7.1 Hz, Me-iBu), 1.23 (d, 1H, J = 7.1 Hz, Me-iBu).
This intermediate was tritylated according to general procedure to give overall yield of 60 mg (22 %) of protected nucleoside. 1H NMR (400 MHz, CDCl3): 8.24 (d, 1H, J2,4 = 2.2 Hz, H-2), 8.99 (d, 1H, J5,4 = 8.6 Hz, H-5), 8.15 (bs, 1H, NH), 7.69 (dd, J4,5 = 8.5 Hz, J4,2 = 2.2 Hz, H-4), 7.44 (dm, 2H, J = 7.2 Hz, 2 × H-DMTr), 7.18 – 7.36 (m, 7H, 7 × H-DMTr), 6.81 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 5.15 (dd, 1H, J1′,2′b = 10.2 Hz, J1′,2′a = 5.4 Hz, H-1′), 4.43 (m, 1H, H-3′), 4.07 (m, 1H, H-4′), 3.79 (s, 6H, CH3-DMTr), 3.34 (dd, 1H, Jgem = 9.8 Hz, J5′a,4′ = 4.7 Hz, H-5′a), 3.25 (dd, 1H, Jgem = 9.8 Hz, J5′b,4′ = 5.3 Hz, H-5′b), 2.54 (septet, 1H, J = 6.8 Hz, CH-iBu), 2.24 (ddd, 1H, Jgem = 13.1 Hz, J2′a,1′ = 5.7 Hz, J2′a,3′ = 1.9 Hz, H-2′a), 2.02 (ddd, 1H, Jgem = 13.1 Hz, J2′b,1′ = 10.3 Hz, J2′a,3′ = 5.9 Hz, H-2′a), 1.24 (d, 3H, J = 7.2 Hz, Me-iBu), 1.24 (d, 3H, J = 7.2 Hz, Me-iBu).
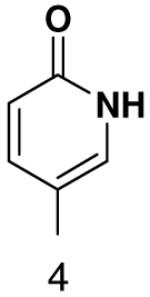
1β-(6-oxopyridin-3-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
1H NMR (400 MHz, CDCl3): 7.69 (dd, J4,5 = 9.4 Hz, J4,2 = 2.5 Hz, H-4), 7.44 (dm, 2H, J = 7.2 Hz, 2 × H-DMTr), 7.18 – 7.36 (m, 7H, 7 × H-DMTr), 7.16 – 7.23 (m, 1H, H-2), 6.82 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 6.52 (d, 1H, J5,4 = 9.4 Hz, H-5), 4.93 (dd, 1H, J1′,2′b = 10.3 Hz, J1′,2′a = 5.4 Hz, H-1′), 4.41 (m, 1H, H-3′), 4.01 (m, 1H, H-4′), 3.79 (s, 6H, CH3-DMTr), 3.31 (dd, 1H, Jgem = 9.8 Hz, J5′a,4′ = 4.5 Hz, H-5′a), 3.20 (dd, 1H, Jgem = 9.8 Hz, J5′b,4′ = 5.4 Hz, H-5′b), 2.24 (ddd, 1H, Jgem = 13.1 Hz, J2′a,1′ = 5.5 Hz, J2′a,3′ = 1.8 Hz, H-2′a), 2.02 (ddd, 1H, Jgem = 13.1 Hz, J2′b,1′ = 10.3 Hz, J2′a,3′ = 6.0 Hz, H-2′a).
1β-(1-deazapurin-9-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
1H NMR (400 MHz, CDCl3): 8.34 (dd, 1H, J1 = 4.7 Hz, J2 = 1.4 Hz, H-2), 8.24 (s, 1H, H-8), 8.07 (dd, 1H, J1 = 8.1 Hz, J2 = 1.5 Hz, H-6), 7.44 (dm, 2H, J = 7.2 Hz, 2 × H-DMTr), 7.18 – 7.36 (m, 8H, 7 × H-DMTr, H-1), 6.82 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 6.64 (t, 1H, J = 6.6 Hz, H-1′), 4.72 (m, 1H, H-3′), 4.23 (m, 1H, H-4′), 3.78 (s, 6H, CH3-DMTr), 3.41 (m, 2H, 2 × H-5′), 2.86 (dt, 1H, Jgem = 13.4 Hz, J2′a,1′ = J2′a,3′ = 5.8 Hz, H-2′a), 2.84 (ddd, 1H, Jgem = 13.4 Hz, J2′b,1′ = 6.3 Hz, J2′b, 3′ = 3.9 Hz, H-2′b).
1β-(1-deaza-6-methylpurin-9-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
1H NMR (400 MHz, CDCl3): 8.17 (d, 1H, J2,3 = 4.9 Hz, H-2), 8.16 (s, 1H, H-8), 7.44 (dm, 2H, J = 7.2 Hz, 2 × H-DMTr), 7.18 – 7.36 (m, 7H, 7 × H-DMTr), 7.03 (d, 1H, J3,2 = 4.9 Hz, H-1), 6.82 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 6.58 (t, 1H, J = 6.7 Hz, H-1′), 4.67 (m, 1H, H-3′), 4.18 (dd, 1H, J4′,5′ = 5.7 Hz, J4′,3′ = 4.9 Hz, H-4′), 3.75 (s, 6H, CH3-DMTr), 3.67 (m, 2H, 2 × H-5′), 2.80 (ddd, 1H, Jgem = 13.5 Hz, J2′a,1′ = J2′a,3′ = 6.4 Hz, H-2′a), 2.65 (s, 3H, Ar-Me), 2.53 (ddd, 1H, Jgem = 13.5 Hz, J2′b,1′ = 6.2 Hz, J2′b, 3′ = 2.3 Hz, H-2′b).
1β-(2-oxopyridin-1-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
1H NMR (400 MHz, CDCl3): 8.94 (dd, 1H, J1 = 6.0 Hz, J2 = 1.8 Hz, H-6), 7.42 (dm, 2H, J = 7.2 Hz, 2 × H-DMTr), 7.14 – 7.37 (m, 8H, 7 × H-DMTr, 1 × H-Pyr), 6.82 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 6.54 (t, 1H, J = 6.1 Hz, H-1′), 6.49 (d, 1H, J = 8.8 Hz, H-Pyr), 6.03 (td, 1H, J1 = 6.9 Hz, J2 = 1.1 Hz, H-5), 4.51 (m, 1H, H-3′), 4.17 (ddd, 1H, J4′,3′ = 7.2 Hz, J4′,5′a = J4′,5′b = 3.7 Hz, H-4′), 3.78 (s, 6H, CH3-DMTr), 3.48 (dd, 1H, Jgem = 10.6 Hz, J5′a,4′ = 3.3 Hz, H-5′a), 3.40 (dd, 1H, Jgem = 10.6 Hz, J5′a,4′ = 3.8 Hz, H-5′b), 2.74 (ddd, 1H, Jgem = 13.7 Hz, J2′a,1′ = 6.0 Hz, J2′a,3′ = 4.3 Hz, H-2′a), 2.19 (dt, 1H, Jgem = 13.0 Hz, J2′b,1′ = J2′b, 3′ = 6.3 Hz, H-2′b).
1β-(4-methyl-2-oxopyridin-1-yl)-1,2-dideoxy-5-(4,4-dimethoxytrityl)-d-ribofuranose
1H NMR (400 MHz, CDCl3): 8.79 (d, 1H, J6,5 = 7.1 Hz, H-6), 7.42 (dm, 2H, J = 7.2 Hz, 2 × H-DMTr), 7.14 – 7.37 (m, 7H, 7 × H-DMTr), 6.82 (dm, 4H, J = 6.9 Hz, 4 × H-DMTr), 6.57 (t, 1H, J1′,2′a = J1′,2′b = 6.3 Hz, H-1′), 6.29 (s, 1H, H-3), 5.89 (dd, 1H, J5,6 = 7.1 Hz, J5,? = 1.7 Hz, H-5), 4.50 (dt, 1H, J3′,4′ = 6.1 Hz, J3′,2′a = J3′,2′b = 3.6 Hz, H-3′), 4.17 (ddd, 1H, J4′,3′ = 7.1 Hz, J4′,5′a = J4′,5′b = 3.5 Hz, H-4′), 3.78 (s, 6H, CH3-DMTr), 3.45 (dd, 1H, Jgem = 10.5 Hz, J5′a,4′ = 3.2 Hz, H-5′a), 3.37 (dd, 1H, Jgem = 10.5 Hz, J5′a,4′ = 4.0 Hz, H-5′b), 2.73 (ddd, 1H, Jgem = 13.7 Hz, J2′a,1′ = 6.0 Hz, J2′a,3′ = 3.7 Hz, H-2′a), 2.19 (m, 1H, H-2′b), 2.13 (s, 3H, Ar-Me).
General procedure for the synthesis of DMTr protected phosphoramidites of nucleosides
Tritylated nucleoside 5 (0.22 mmol) was dissolved in CH2Cl2 (8 mL) and DMAP (catalytical amount) and ethyl-diisopropylamine (0.3 mL) were added. The mixture was cooled to 0 °C and then 2-cyanoethyl diisopropylaminochlorophosphoramidite, (0.3 mL) was added. The mixture was stirred for 30 min at 0 °C and then 30 min while warming to 25 °C. The solvents were evaporated, crude oily product was dissolved in a mixture of ethyl acetate, hexane, triethylamine (49:49:2) and chromatographed over silica gel (10 mL) in the same mixture to give oil of two diastereiosomers of phosphoramidites. The oil was dried by codistillation with MeCN (three times) which yielded 50 – 90 % of phosphoramidite.
[1β- (Pyridin-3-yl)-5′-(dimethoxytrityl)-2′-deoxy- d-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis(isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 82 %. MS (ESI, MeCN, LiCl added, neg.): 733 (M +35 [Cl]) calcd 733. MS (ESI, MeCN, LiCl added, pos.): 705 (M +7 [Li]) calcd 705. 31P NMR (400 MHz, MeCN-d3): 148.37 (s, 1.st diastereoisomer), 148.33 (s, 2.nd diastereoisomer).
[1β- (6-isobutyrylamino-pyridin-3-yl)-5′-(dimethoxytrityl)-2′-deoxy- d-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis(isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 79 %. MS (ESI, MeCN, LiCl added, neg.): 818 (M +35 [Cl]) calcd 818. MS (ESI, MeCN, LiCl added, pos.): 790 (M +7 [Li]) calcd 790. 31P NMR (400 MHz, MeCN-d3): 148.52 (s, 1.st diastereoisomer), 148.49 (s, 2.nd diastereoisomer).
[1β- (6-Oxopyridin-3-yl)-5′-(dimethoxytrityl)-2′-deoxy- d-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis(isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 20 %. MS (ESI, MeCN, LiCl added, neg.): 749 (M +35 [Cl]) calcd 749. MS (ESI, MeCN, LiCl added, pos.): 721 (M +7 [Li]) calcd 721. 31P NMR (400 MHz, MeCN-d3): 148.30 (s, 1.st diastereoisomer), 148.27 (s, 2.nd diastereoisomer).
The yield was small, because there originated 20 % of a byproduct - bis-phosphoramidite (3′-O-phosphoramidite along with and 6-O-phosphoramidite), which was proven by the spectral data: MS (ESI, MeCN, LiCl added, neg.): 948 (M +35 [Cl]) calcd 948. MS (ESI, MeCN, LiCl added, pos.): 920 (M +7 [Li]) calcd 920. 31P NMR (400 MHz, MeCN-d3): 148.31 (s, 1P), 148.28 (s, 1P), 144.59 (s, 1P), 144.56 (s, 1P).
[1β- (1-Deazapurin-9-yl)-5′-(dimethoxytrityl)-2′-deoxy- d-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis (isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 57 %. MS (ESI, MeCN, LiCl added, neg.): 772 (M +35 [Cl]) calcd 772. MS (ESI, MeCN, LiCl added, pos.): 744 (M +7 [Li]) calcd 744. 31P NMR (400 MHz, MeCN-d3): 148.97 (s, 1.st diastereoisomer), 148.87 (s, 2.nd diastereoisomer).
[1β- (6-Methyl-1-deazapurin-9-yl)-5′-(dimethoxytrityl)-2′-deoxy- D-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis (isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 84 %. MS (ESI, MeCN, LiCl added, neg.): 797 (M +35 [Cl]) calcd 797. MS (ESI, MeCN, LiCl added, pos.): 759 (M +7 [Li]) calcd 759. 31P NMR (400 MHz, MeCN-d3): 148.95 (s, 1.st diastereoisomer), 148.84 (s, 2.nd diastereoisomer).
[1β- (2-Oxopyridin-1-yl)-5′-(dimethoxytrityl)-2′-deoxy- d-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis(isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 89 %. MS (ESI, MeCN, LiCl added, neg.): 748 (M +35 [Cl]) calcd 748. MS (ESI, MeCN, LiCl added, pos.): 720 (M +7 [Li]) calcd 720. 31P NMR (400 MHz, MeCN-d3): 149.07 (s, 1.st diastereoisomer), 149.01 (s, 2.nd diastereoisomer).
[1β- (4-Methyl-2-oxopyridin-1-yl)-5′-(dimethoxytrityl)-2′-deoxy- d-ribofuranos-3′-yl]-2-cyanoethyl-N,N-bis(isopropylamino)phosphoramidite
This compound was obtained as a colorless oil, yield 71 %. MS (ESI, MeCN, LiCl added, neg.): 762 (M +35 [Cl]) calcd 762. MS (ESI, MeCN, LiCl added, pos.): 734 (M +7 [Li]) calcd 734. 31P NMR (400 MHz, MeCN-d3): 149.03 (s, 1.st diastereoisomer), 149.02 (s, 2.nd diastereoisomer).
General procedure for the synthesis of triphosphates of nucleosides
Nucleoside (0.27 mmol) was dissolved in trimethyl phosphate (0.6 mL) and the mixture was cooled to 0 °C. Then POCl3 (27 μL, 1.1 equivalent) in 0.2 mL of trimethylphosphate was added dropwise. The mixture was stirred for 3 h and then tetrabutylammonium pyrophosphate (0.8 g, 5 equivalents) in DMF (1 mL) was added. The mixture was stirred for 4 h, while the temperature reached r.t. Crude product was poured into triethylammonium bicarbonate (0.1 M in water), water and TEAB were removed in vacuum and crude product was diluted with water (100 mL) and added directly to a TEAB equilibrated ion-exchange column (Sephadex-DEAE A-25, Aldrich) and eluted using a 0 to 1 M TEAB gradient. Fractions were individually spotted on a MALDI plate with THAP as the matrix and triphosphate fractions were identified by their MALDI- peak (negative ion mode). Fractions were collected, lyophilized and purified by HPLC using gradient 0 to 60 % of MeCN in 20 mM TEAA. Spectrophotometric yields were 2 – 50 %.
1-β-d-(2-Oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose triphosphate
MS (MALDI, neg.): 466 (M -1) calcd 466. 31P NMR (400 MHz, D2O): − 10.00 (d, 1P, J = 40.8 Hz, γ-P), −10.50 (d, 1P, J = 40.0 Hz, α-P), −22.34 (m, 1P, β-P).
1-β-d-(6-Methyl-2-oxopyridine)-3′,5′-di-O-(4-toluoyl)-2′-deoxyribofuranose triphosphate
MS (MALDI, neg.): 481 (M -1) calcd 481. 31P NMR (400 MHz, D2O): − 9.89 (bs, 1P, γ-P), −10.46 (d, 1P, J = 46.4 Hz, α-P), −22.31 (m, 1P, β-P).
1-β-d-(Benzimidazol-1-yl)-β-d-ribofuranose triphosphate
MS (MALDI, neg.): 489 (M -1) calcd 489. 31P NMR (400 MHz, D2O): − 9.67 (d, 1P, J = 46.4 Hz, γ-P), −10.40 (d, 1P, J = 49.2 Hz, α-P), − 22.20 (t, 1P, J = 40.8 Hz, β-P).
1-β-d-(4-Methylbenzimidazol-1-yl)-β-d-ribofuranose triphosphate
MS (MALDI, neg.): 503 (M - 1) calcd 503. 31P NMR (400 MHz, D2O): − 9.44 (m, 1P, γ-P), −10.38 (d, 1P, J = 46.0 Hz, α-P), −22.20 (m, 1P, β-P).
1-β-d-(1-Deazapurin-9-yl)-β-d-ribofuranose triphosphate
MS (MALDI, neg.): 490 (M -1) calcd 490. 31P NMR (400 MHz, D2O): − 9.83 (bs, 1P, γ-P), −10.33 (d, 1P, J = 43.2 Hz, α-P), −22.18 (m, 1P, β-P).
1-β-d-(6-Methyl-1-deazapurin-9-yl)-β -d-ribofuranose triphosphate
MS (MALDI, neg.): 504 (M -1) calcd 504. 31P NMR (400 MHz, D2O): − 9.88 (bs, 1P, γ-P), −10.35 (d, 1P, J = 49.2 Hz, α-P), −22.24 (m, 1P, β-P).
Zebularine triphosphate
MS (MALDI, neg.): 467 (M -1) calcd 467. 31P NMR (400 MHz, D2O): − 9.88 (bs, 1P, γ-P), −10.50 (d, 1P, J = 45.2 Hz, α-P), −22.36 (m, 1P, β-P).
1-β-d-(Purin-9-yl)-β-d-ribofuranose triphosphate
MS (MALDI, neg.): 491 (M -1) calcd 490. 31P NMR (400 MHz, D2O): − 9.87 (bs, 1P, γ-P), −10.46 (d, 1P, J = 43.2 Hz, α-P), −22.45 (m, 1P, β-P).
5′-Dimethoxytrityl-3′-phosphoramidites and oligonucleotides
The 5′ hydroxygroups of the nucleosides were protected with the dimethoxytrityl group as described above and established procedures were used for generation of the phosphoramidites and synthesis of the oligonucleotides on an Applied Biosystems 394 automatic DNA synthesizer (42, 43).
RESULTS
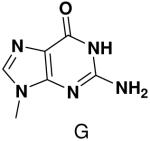
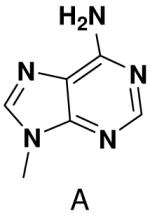
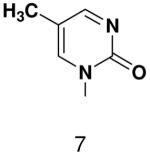
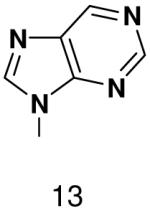
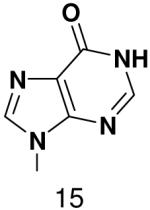
HSV primase misincorporates the natural NTPs at an extremely high frequency (1 every 30 correct NTPs), yet very strongly discriminated against purine NTP analogues that cannot form Watson-Crick hydrogen bonds involving N-3 and N4/O4 of the templating pyrimidine during synthesis of the second and third nucleotide of the primer (33). To better understand how the enzyme discriminates so strongly against NTPs that cannot form a complete set of hydrogen bonds with the template base, we examined a series of templates and NTPs containing modified bases (Figure 1).
Figure 1.

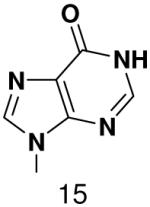
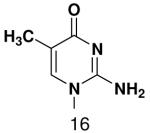
Base analogues used in this study.
We first developed an assay to quantify how efficiently primase incorporates NTPs containing both natural and synthetic bases opposite a natural or synthetic template base. The DNA template 3′-T20GCCCXAT14, where X represents either a natural or synthetic nucleotide, supports high levels of primase activity. Primase exclusively initiates synthesis opposite the two underlined C’s in the canonical GCC initiation site and efficiently incorporates an additional GTP before coming to the variable nucleotide. When assays contain only 800 μM GTP and X is either a natural nucleotide or an analogue, primase synthesizes large amounts of the correct ppp(G)3 trinucleotide, but also readily misincorporates GTP to generate the ppp(G)4 tetranucleotide (Figures 2A and B). Adding a different NTP, either correct or incorrect, decreased the amount of the 5′-ppp(G)4 misincorporation product, and generated a new product of altered electrophoretic mobility due to incorporation of this additional NTP (Figure 2B-2D) 2.
Figure 2.

Experiments contain 400 nM HSV-1 primase, 2 μM template, 0.8 mM [α-32P]GTP and 0–2 mM of investigated NTPs. Panel. The noted concentration of GTP and the template T20GC3[2-pyridone]AT14. Panel B. The noted concentration of CTP and the template T20GC3GAT14.Panel C. The noted concentration of UTP and the template T20GC3GAT14.Panel D. Incorporation of various NTPs opposite purine in the template T20GC3[purine]AT14. Reactions contained 1 mM NTP analogue. Lane 1) no analogue; 2) benzimidazole; 3) 4-methyl-2-pyridone; 4) 7-methylbenzimidazol; 5) 2-pyridone; 6) 6-methyl-1-deazapurine; 7) 1-deazapurine; 8) isoG; 9) purine; 10) 2(1H)-pyrimidinone. Presence of products of altered electrophoretic mobility indicates analog’s incorporation.
We quantified the ability of primase to incorporate a correct versus an incorrect NTP across from X as shown in Scheme 1. Since initiation (i.e., ppp(G)2 dinucleoide synthesis) is the rate-limiting step in primer synthesis, the rate of NTP polymerization onto the trinucleotide cannot be directly measured. Instead, we measured the partitioning of the primase-ppp(G)3 complex to determine the relative efficiency of polymerizing a NTP (38). After generating the trinucleotide ppp(G)3, primase has three choices - i) it can dissociate, thus resulting in the formation of the ppp(G)3 product; ii) it can incorporate a GTP to form ppp(G)4, or; iii) it can incorporate another nucleotide (N) to form the ppp(G)3N product. Thus, by comparing the relative amounts of ppp(G)4 and ppp(G)3N products as a function of NTP concentration at a fixed GTP concentration, we can quantify the relative ability of primase to incorporate GTP versus NTP (38). It should be emphasized that these data do not provide the actual efficiency with which primase incorporates a NTP (analogue). However, by using a constant GTP concentration with the different analogues, the relative efficiency with which primase polymerizes the different NTPs can be determined.
Scheme 1.

Partitioning of the trinucleotide between dissociation, polymerization of GTP opposite X, and polymerization of NTP opposite X. The polymerization of NTPs is effectively irreversible due to the low pyrophosphate concentration in the assays and previous work showed that primase does not elongate short oligonucleotides once they dissociate from the enzyme, thereby making the dissociation step effectively irreversible (1, 56).
Table 1 shows that as expected, producing equal amounts of products due to polymerization of GTP and of the tested NTP required less of the tested NTP when it was correct than when it was incorrect. For example, opposite a template T, primase produced equal amounts of products due to ATP polymerization and GTP polymerization when assays contained only 8 μM of the correct ATP. In contrast, producing equal amounts of product due to CTP polymerization and GTP polymerization required 1900 μM of the incorrect CTP. Consistent with the low fidelity of primase, however, the difference in the amounts of the correct NTP versus the amounts of an incorrect NTP needed to give equal effects was relatively low - varying from 17 in the case polymerization of UTP opposite G as compared to CTP opposite G, to 420 during polymerization of ATP opposite G as compared to CTP opposite G.
Table 1.
Incorporation of natural NTPs and purine NTPs opposite either natural bases or analogue bases lacking one Watson-Crick hydrogen bonding groups. Listed is the concentration (μM) of the NTP that gives equal elongation of the ppp(G)3 trinucleotide with that NTP and with GTP. “ND” means no detectable incorporation
| Triphosphate | |||||
|---|---|---|---|---|---|
 |
 |
 |
 |
||
| Template | 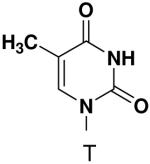 |
240 ± 20 | 1900 ± 50 | 25 ± 10 | 40 ± 5 |
 |
260 ± 20 | 15 ± 1 | 6300 ± 200 | ND | |
 |
8 ± 1 | 920 ± 40 | 2500 ± 100 | 3500 ± 100 | |
 |
1200 ± 50 | ND | 14000 ± 1000 | ND | |
 |
90 ± 10 | ND | 5000 ± 100 | ND | |
 |
30 ± 20 | 23 ± 3 | 6300 ± 300 | ND | |
Effect of removing N4 from CTP
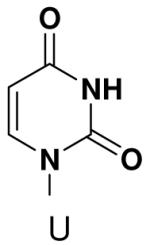
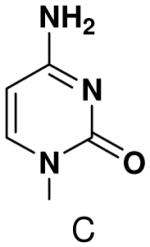
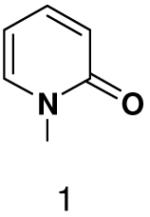
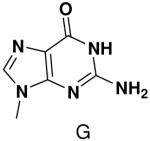
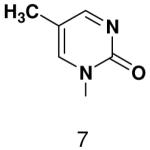
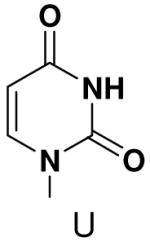
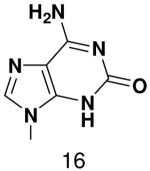
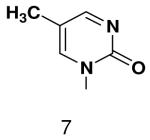
Previously, we showed that removing one of the Watson-Crick hydrogen bonding groups from ATP strongly inhibited polymerization when the template required ATP as either the second or third nucleotide of the primer (33). To test if removing one of the Watson-Crick hydrogen bonding groups also affected pyrimidines, we examined polymerization of 2(1H)-pyrimidinone NTP (zebularine), a C analogue that lacks N4. We found similar results - no detectable polymerization of 2(1H)-pyrimidinone NTP opposite a template G at concentrations up to 2 mM.
Effect of removing a Watson-Crick hydrogen bonding group from the templating base
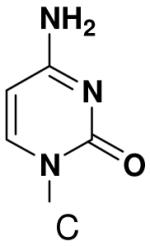
The effect of removing a single Watson-Crick hydrogen bonding group from the template base was explored using templates where X = 2(1H)-pyrimidinone (no N4), X = purine (no N6), and X = hypoxanthine (no N2). Primase very efficiently incorporated CTP opposite hypoxanthine (Table 1), indicating that primase does not require the hydrogen bond normally found between N2 of G and O2 of C. Whereas the loss of N2 did not increase misincorporation of ATP, it significantly increased the misincorporation of UTP. Surprisingly, primase incorporated UTP across from purine only 11-fold less efficiently than across from A, but it discriminated against incorporation of ATP and CTP more effectively than if the template contained A. (Table 1).
Opposite a templating 5-methyl-2(1H)-pyrimidinone, primase did not efficiently polymerize ATP, CTP or UTP (Table 1). To measure the efficiency of GTP polymerization, we measured the fraction of the trinucleotide elongated to the tetranucleotide as a function of GTP concentration. Incorporation of GTP demanded this alternative analysis since the reactions contained only GTP. Table 2 shows that primase incorporated GTP opposite 5-methyl-2(1H)-pyrimidinone much less efficiently than opposite C, and only slightly more efficiently than if the template contained a base completely incapable of Watson-Crick hydrogen bonding (2-pyridone). Thus, primase does not efficiently polymerize GTP across from a templating 5-methyl-2(1H)-pyrimidinone and the loss of Watson-Crick hydrogen bonds clearly interferes with polymerization. As will be described in greater detail below, the low level of GTP misicorporation opposite 2-pyridone and 2(1H)-pyrimidinone likely largely results from template/primer slippage.
Table 2.
Incorporation of GTP opposite the noted template base in the template T20GCCCXAT14. The “Fmax” is the fraction of ppp(G)3 elongated into ppp(G)4 with saturating GTP and the “KM” is the concentration of GTP that gives half maximal elongation. It should be emphasized that Fmax is based on the partitioning of the E-ppp(G)3 complex between addition of another GTP and dissociation of ppp(G)3, and is not Vmax
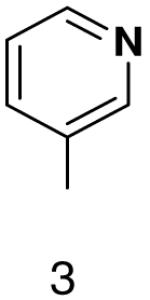
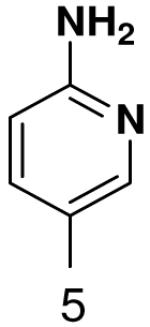
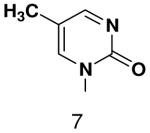
| Template | Vmax | KM (μM) | Vmax/KM | |
|---|---|---|---|---|
| GTP | 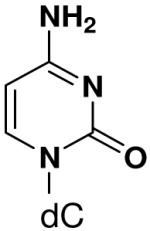 |
0.9 ± 0.025 | 57.1 ± 9 | 0.016 |
 |
0.12 ± 0.002 | 188 ± 14 | 0.00064 | |
 |
0.19 ± 0.016 | 528 ± 121 | 0.00036 | |
 |
0.32 ± 0.009 | 146 ± 17 | 0.0022 | |
 |
0.26 ± 0.006 | 217 ± 29 | 0.0012 |
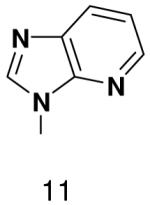
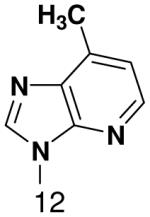
Then, to determine if primase will efficiently incorporate a natural NTP opposite a template base incapable of forming any Watson-Crick hydrogen bonds, we generated a series of templates T20GCCCXGT14, where X was 2-pyridone, 4-methyl-2-pyridone, 1-deazapurine, or 6-methyl-1-deazapurine, and measured incorporation of the natural NTPs (Table 3). As when X was a natural nucleotide, primase misincorporated small amounts of GTP. However, it did not efficiently incorporate any other natural NTPs opposite these hydrophobic template bases. In each case examined, incorporation of a natural NTP was no more efficient than polymerization of a natural NTP opposite an incorrect, natural template base (compare Table 1 and Table 3).
Table 3.
Incorporation of natural NTPs opposite template bases lacking any Watson-Crick hydrogen bonding groups. Listed is the concentration (μM) of the NTP that gives equal elongation of the ppp(G)3 trinucleotide with that NTP and with GTP
| Triphosphate | ||||
|---|---|---|---|---|
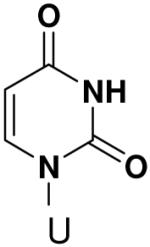 |
 |
 |
||
| Template | 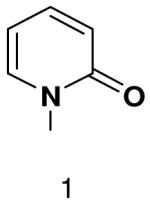 |
490 ± 30 | 2100 ± 20 | 2800 ± 300 |
 |
170 ± 30 | 1900 ± 80 | 4200 ± 250 | |
 |
460 ± 40 | 2200 ± 100 | 5400 ± 400 | |
 |
1800 ± 30 | 1700 ± 30 | 7700 ± 700 | |
Unsatisfied Watson-Crick hydrogen bonding groups
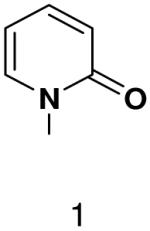
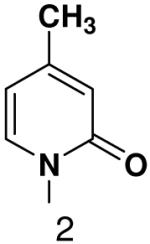
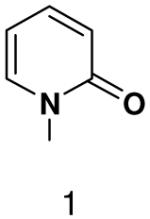
We then tested the hypothesis that primase’s inability to efficiently generate base-pairs between a natural base and a base incapable of forming Watson-Crick hydrogen bonds resulted from the presence of unsatisfied Watson-Crick hydrogen bonding groups on one of the bases. Polymerization of 2-pyridone-, 4-methyl-2-pyridone-, benzimidazole-, 4-methylbenzimidazole-, 1-deazapurine-, and 6-methyl-1-deazapurine-NTPs was measured in assays containing GTP and the template T20GCCCXGT14 where X was 2-pyridone, 4-methyl-2-pyridone, 1-deazapurine, or 6-methyl-1-deazapurine. However, even at elevated concentrations of these hydrophobic NTP analogues (2 mM), primase only detectably incorporated 2-pyridone NTP and 4-methyl-2-pyridone NTP (Figure 3 and 4, and Supplementary Material contains a summary of the hydrophobic dNTP analogues that primase did not incorporate opposite hydrophobic template nucleotides.). Polymerization of 4-methyl-2-pyridone- and 2-pyridone NTP opposite several hydrophobic bases was further examined and quantified as described above (Table 4). Primase incorporated these NTP analogues with similar efficiency to that observed for polymerizing a natural NTP opposite an incorrect, natural template base. Together, these data indicate that the simple lack of unsatisfied H-bonds between the bases of the incoming NTP and the template base does not result in rapid NTP polymerization.
Figure 3.

Incorporation of a variety of NTPs opposite 1-deazapurine using the template T20GCCC[1-deazapurine]AT14. Experiments contained 400 nM HSV-1 primase, 2 μM template, 0.8 mM [α-32P]GTP and 2 mM of the following (analogue) NTPs; 1) no further addition; 2) benzimidazole; 3) 4-methyl-2-pyridone; 4) 7-methylbenzimidazol; 5) 2-pyridone; 6) 6-methyl-1-deazapurine; 7) 1-deazapurine; 8) isoG; 9) 2(1H)-pyrimidinone; 10) ATP. Presence of products of altered electrophoretic mobility in lines 3 and 5 indicates incorporation of 4-methyl-2-pyridone and 2-pyridone, respectively. In case of isoGTP and 2(1H)-pyrimidinone (lines 8 and 9), large amount of these NTPs caused inhibition of the enzyme.
Figure 4.

Detecting primer/template slippage during misincorporation. Reactions were performed as described under Experimental Procedures and contained 400 nM HSV-1 primase and 2 μM template T20GC2TXAT14 and X is 2-pyridone, unless noted otherwise. Primer standards of known composition were generated in assays containing T20GCCCTAAT14 and 0.6 mM [α-32P]GTP (Lane S1). T20GCCTTAAT14 0.6 mM [α-32P]GTP and 0.6 mM ATP (Lane S2) or T20GC2T2A2T14, and 0.6 mM [α-32P]GTP (Lane S3). The identity of the products is listed on the sides of the panels. Panel A. Reactions contained 0.6 mM GTP and 0.2–1mM of [α-32P]ATP. Panel B. Reactions contained 0.6 mM [α-32P]GTP and 0–1mM of ATP. Panel C. Reactions contained 0.6 mM ATP and 0.2–1mM of [α-32P]GTP.
Table 4.
Polymerization of pyridone NTP and 4-methylpyridone NTP opposite various bases. Listed is the concentration (μM) of the NTP that gives equal elongation of the ppp(G)3 trinucleotide with that NTP and with GTP
| Triphosphate | |||
|---|---|---|---|
 |
 |
||
| Template | 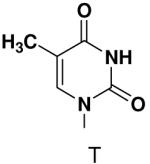 |
520 ± 20 | 2300 ± 30 |
 |
70 ± 15 | 100 ± 30 | |
 |
1300 ± 40 | 2700 ± 100 | |
 |
660 ± 50 | 470 ± 40 | |
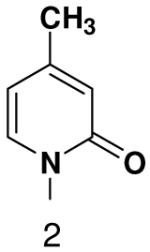 |
580 ± 50 | 810 ± 60 | |
 |
520 ± 30 | 1100 ± 40 | |
 |
660 ± 20 | 800 ± 100 | |
 |
3500 ± 100 | ND | |
 |
ND | ND | |
 |
700 ± 60 | 1100 ± 100 | |
 |
610 ± 40 | 910 ± 80 | |
 |
130 ± 40 | 1800 ± 200 | |
 |
36 ± 30 | 70 ± 30 | |
 |
ND | ND | |
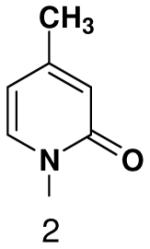
We next tested how efficiently primase incorporated pyridinone- and 4-methyl-2-pyridinone-NTPs opposite the natural bases. Generally, primase incorporated the two pyrimidine analogues much more efficiently than analogous purine analogues, although incorporation opposite A and T was no better than misincorporation (Table 4). Surprisingly, primase incorporated the pyridone NTPs very efficiently across from G, much better than an incorrect natural NTP, but not quite as efficiently as CTP (compare Table 4 and Table 1). The relatively efficient incorporation of the pyridones opposite guanine might have resulted from formation of a hydrogen bond between O2 of the pyridones and N2 of guanine. However, primase also efficiently incorporated the pyridone NTPs opposite hypoxanthine, a guanine analogue that lacks N2, indicating that the efficient incorporation of the pyridones did not result from a hydrogen bond involving N2 (Table 4).
We further examined the role of unsatisfied hydrogen bonding groups by measuring polymerization of purine NTP opposite a templating T. In contrast to previous studies showing that primase strongly discriminated against purine polymerization when T coded for the second or third nucleotide of the primer (33), primase exhibited only modest discrimination (<2-fold) against polymerization of purine NTP when incorporated at the fourth position (Table 1). This result is also consistent with the efficient polymerization of UTP opposite purine described earlier.
Alternative hydrogen bonding patterns
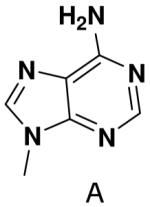
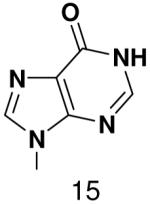
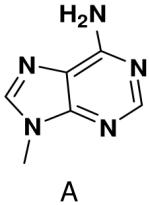
We tested the possibility that primase would incorporate NTPs with a complete set of Watson-Crick hydrogen bonds, albeit in an unusual arrangement. However, primase did not detectably incorporate isoGTP opposite 5-methyl-isoC, even when assays contained 2 mM isoGTP. Likewise, primase polymerized ATP and CTP opposite isoC no better than it misincorporated these natural NTPs opposite a natural template base (Table 5), and polymerized UTP only slightly better.
Table 5.
Polymerization of natural NTPs and iso-GTP opposite iso-C, 3-deazaadenine, and hypoxanthine. Listed is the concentration (μM) of the NTP that gives equal elongation of the ppp(G)3 trinucleotide with that NTP and with GTP. “ND” means no detectable incorporation
| Triphosphate | |||||
|---|---|---|---|---|---|
 |
 |
 |
 |
||
| Template | 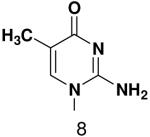 |
100 ± 30 | 3000 ± 100 | 5900 ± 200 | ND |
 |
9 ± 3 | 11 ± 1 | 5000 ± 100 | ND | |
 |
30 ± 20 | 23 ± 3 | 6300 ± 300 | ND | |
Minor groove hydrogen bonding groups
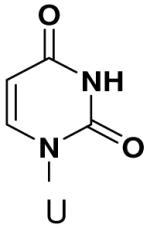
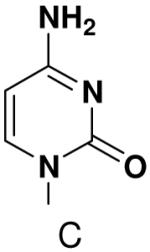
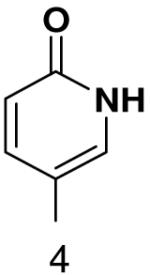
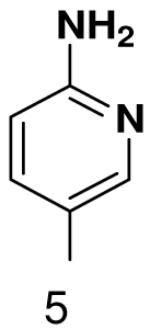
The role of the minor groove hydrogen bond acceptor in the template base was probed using purines that lacked N-3 and pyrimidine analogues that lacked O2. Primase incorporated UTP opposite 3-deazaadenine (Table 5) and adenine (Table 1) with similar efficiency, while the efficiency of misincorporation changed slightly. In contrast to these minimal effects upon removing N-3 from a purine, removing the equivalent minor groove substituent, O2, from pyrimidines had a somewhat greater effect, even when the template base retained the capacity to form two correct Watson-Crick hydrogen bonds (ATP polymerization opposite 6-hydroxypyridin-3-yl and GTP polymerization opposite 6-aminopyridin-3-yl (Table 6 and Table 2)).
Table 6.
Polymerization of natural NTPs opposite pyrimidine analogues. Listed is the concentration (μM) of the NTP that gives equal elongation of the ppp(G)3 trinucleotide with that NTP and with GTP. “N” Dmeans no detectable incorporation
| Triphosphate | ||||
|---|---|---|---|---|
 |
 |
 |
||
| Template | 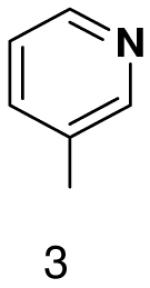 |
1600 ± 100 | 1000 ± 100 | 4200 ± 400 |
 |
120 ± 5 | 870 ± 40 | 120 ± 20 | |
 |
180 ± 20 | ND | ND | |
 |
1200 ± 50 | ND | 14000 ± 1000 | |
Primase can misincorporate NTPs via primer/template slippage
Primase generated the ppp(G)4 tetranucleotide with all of the T20GCCCXAT14 templates. Potentially, this could have occurred via primase actually incorporating GTP opposite X, or via the primer or template slipping after generating the ppp(G)3 trinucleotide. In the slippage mechanism, the formation of ppp(G)4 results from re-replication of the third template C. Two approaches were used to examine this question. First, we examined the efficiency with which primase incorporated GTP opposite X as a function of GTP concentration (Table 2). Using a series of template bases, primase incorporated GTP much less efficiently opposite the analogues than opposite C. Then, we measured misincorporation with the template T20GCCTXAT14. If the polymerization of GTP opposite X on the template T20GCCCXAT14 resulted from a primer/template slippage mechanism, removing the C just before X should reduce or eliminate polymerization of GTP and may give enhanced ATP polymerization opposite X. In contrast, if incorporation of GTP opposite X resulted from primase misreading X and actually polymerizing GTP opposite X, then replacing the GTP prior to X should have little or no effect.
To test for slippage, we examined incorporation of GTP and ATP opposite 2-pyridone in the template T20GCCTXAT14, where X = 2-pyridone (Figure 4). In the presence of ATP and GTP, primase synthesized large amounts of a trinucleotide that contains both A and G, presumably pppGGA, the product coded for by the first 3 nucleotides of the template at the primer initiation site, and two 4-nucleotide long products. Each products contained A and G since including either [α-32P]ATP or [α-32P]GTP in the assays resulted in labeled products. The primary 4-nucleotide product comigrated with pppGGAA, while the minor 4-nucleotide long product migrated slightly slower. Increasing the concentration of ATP significantly increased the amount of the pppGGAA product relative to the slower migrating product and pppGGA. Quantitation of the data showed that 50% conversion of the pppGGA to pppGGAA required only 2.8±0.2 mM ATP. In contrast, when the template nucleotide just before the 2-pyridone was C, 50% conversion of the ppp(G)3 trinucleotide to ppp(G)3A required 41±2 mM ATP. This ability of the template nucleotide just prior to the 2-pyridone to direct polymerization opposite 2-pyridone suggests that this polymerization can occur via a primer-template slippage mechanism. We also measured the effects of increasing GTP concentrations on the synthesis of the two 4-nucleotide products. Figure 4 shows that the amount of the slower migrating 4-nucleotide long product increased relative to pppGGAA. Thus, this slower migrating product likely has the sequence pppGGAG and results from GTP incorporation opposite 2-pyridone. Quantitation of the data showed that 50% elongation of pppGGA to the apparent pppGGAG required 18±1 mM GTP, while only 6.1±0.4 mM was required for 50% elongation of pppGGG to pppGGGG on the template T20GCCCXAT14.
Discussion
We used a series of base analogues to further probe how herpes primase chooses whether or not to polymerize a NTP. Surprisingly, the extent to which the formation of Watson-Crick hydrogen bonds enhances NTP polymerization varied upon both the identity of the base pair and the location of the base pair in the primer (second, third, etc.). In some cases the effects of modifying the base had similar effects when either the template or incoming NTP contained the modified base, whereas in other cases the effects were very asymmetrical.
Primase polymerized purine NTP opposite T only 2-fold worse than it polymerized ATP opposite T as the fourth nucleotide of the primer. Previously, we found that primase strongly discriminated against purine NTP incorporation as the second or third nucleotide of the primer, presumably due to the missing hydrogen bond between N6 of the purine and O4 of T. The decreasing discrimination against purine NTP polymerization with increasing primer length suggests that for polymerization across from T, increased primer:template stability decreases the requirement for Watson-Crick hydrogen bonds. Furthermore, these data also demonstrate that primase does not absolutely require the formation of a complete set of Watson-Crick hydrogen bonds in order to efficiently polymerize a NTP.
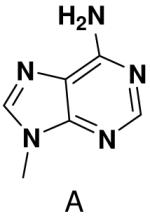
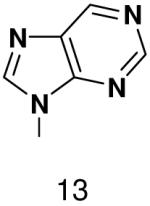
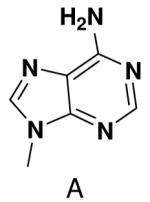
With the exception of 2-pyridone NTP and 4-methyl-2-pyridone NTP, primase very strongly discriminated against polymerizing NTPs whose base cannot form any Watson-Crick hydrogen bonds opposite both natural and analogue bases. This occurred whether or not these analogues contained a minor groove hydrogen bond acceptor (N-3 of a purine). The inability of primase to efficiently generate base pairs between two bases that both lacked Watson-Crick hydrogen bonding groups indicates that the simple absence of unsatisfied Watson-Crick hydrogen bonding groups in the primase active site does not lead to rapid NTP polymerization. Indeed, primase polymerized natural NTPs across from the purine analogues 1-deazapurine and 6-methyl-1-deazapurine more efficiently than it polymerized 1-deazapurine NTP and 6-methyl-1-deazapurine NTP opposite any base, (Tables 1 and 3, and Supplementary Material).
Among the tested NTP analogues incapable of Watson-Crick hydrogen bonding, primase only incorporated the two pyrimidine analogues, 2-pyridone and 4-methyl-2-pyridone NTP. Primase polymerized them surprisingly well opposite both natural bases as well as unnatural bases. In most cases, the incorporation efficiency was similar to that of an incorrect natural NTP across from a natural base. However, polymerization opposite hypoxanthine and G was almost as efficient as generation of a canonical base pair - only 2- to 7-fold less efficient than polymerization of CTP across from hypoxanthine and G, respectively.
The efficient incorporation of 2-pyridone NTP and 4-methyl-2-pyridone NTP clearly indicates that herpes primase does not require the formation of Watson-Crick hydrogen bonds in order to efficiently polymerize a NTP. Yet, primase very potently discriminated against analogous NTPs where the base mimics a purine, 1-deazapurine NTP and 6-methyl-1-deazapurine NTP. Likewise, our previous studies on HSV-1 primase using a larger series of purine NTP analogues indicated a requirement for Watson-Crick hydrogen bonds (33). These contrasting results suggest that the requirements for incoming purine NTPs and incoming pyrimidine NTPs are not identical. Additionally, it raises the intriguing question of how one designs an active site to interact so very differently with these different analogues.
Surprisingly, converting 2-pyridone NTP into 2(1H)-pyrimidinone NTP by adding back N3 greatly impaired incorporation opposite G or any other nucleotide. This occurred even though 2(1H)-pyrimidinone NTP can now form a Watson-Crick hydrogen bond between N3 of 2(1H)-pyrimidinone and H-N3 of G. Likewise, primase also did not efficiently polymerize any natural NTPs opposite a template 5-methyl-2(1H)-pyrimidinone. Indeed, even 2-pyridone NTP and 4-methyl-2-pyridone NTP were not efficiently incorporated across from this base.
Remarkably, converting 2-pyridone into 2(1H)-pyrimidinone had essentially the exact opposite effect of converting 1-deazapurine into purine. The former conversion severely inhibited formation of base pairs whereas the latter conversion greatly stimulated generation of base pairs with U/T. Chemically, however, both changes are remarkably similar in that an electron deficient C-H is converted into an electron rich N that can form a Watson-Crick hydrogen bond in the core of a duplex nucleic acid. Since 2(1H)-pyrimidinone can be considered a C analogue while purine is an A analogue, these differences suggest that herpes primase differentially recognizes A:U/T and G:C base-pairs.
Primase frequently demonstrated a large asymmetry during generation of base-pairs. For example, while it polymerized 2-pyridone NTP opposite G quite efficiently, it did not efficiently incorporate GTP opposite 2-pyridone. Likewise, primase polymerized 2-pyridone NTP across from 1-deazapurine and 6-methyl-1-deazapurine at least 10-fold more efficiently than it polymerized 1-deazapurine NTP and 6-methyl-1-deazapurine NTP opposite 2-pyridone (Primase did not detectably polymerize these NTPs opposite 2-pyridone (Supplemental Information).) Thus, just like with some DNA polymerases (20, 44-48), predicting how altering a template or NTP base will affect activity is problematic at best. Mechanistically, this may result from the interactions of the enzyme with the templating base subtly altering the structure of the active site and, therefore, how the enzyme interacts with the base of the incoming (d)NTP.
This asymmetry in base-pair recognition extends to the minor groove hydrogen bond acceptor found on the canonical bases. Primase does not require a minor groove hydrogen bond acceptor in either an incoming ATP or a templating A, as evidenced by the minimal effects on generation of A:T/U base-pairs upon removal of N-3 from A. In contrast, removing O2 from a templating T or C more significantly impaired polymerization of ATP or GTP, respectively.
Primase likely misincorporates NTPs both by misreading the templating base as well as by a template (or primer) slippage mechanism. Evidence for misreading comes from the ability of primase to polymerize NTPs not coded for by any template base (32). Primase also appears to misincorporate NTPs by rereplicating a template base. Regardless of the identity of X in the template ...GCCCX..., primase always generated significant amounts of ppp(G)4, even if it did not efficiently polymerize ATP, CTP or UTP opposite the base (Ex., 6-methyl-1-deazapurine and isoC). To explicitly test for primer slippage, we examined NTP polymerization opposite X with a template containing the sequence ...GCCTX.... This resulted in slightly decreased polymreization of GTP along with 20-fold increased polymerization of ATP. Thus, the template nucleotide immediately before the analogue can direct what NTP primase incorporates opposite X. Potentially, this could occur by primase allowing the primer to slide backwards or the template strand to slip forwards, thereby resulting in re-replication of a template base.
Misincorporation of dNTPs by DNA polymerases, including high fidelity polymerases, can also occur by a primer-template slippage mechanism (49-52). With DNA polymerases, this problem appears to be most acute in highly repetitive sequences. Indeed, triplet expansion diseases may result from primer-template slippage during DNA synthesis (49, 52). Thus, this mechanism of misincorporation is clearly not limited to herpes primase and presents a general potential problem for nucleotide polymerizing enzymes. In the case of primase, however, slippage can occur even in the absence of highly repetitive sequences.
Incorporation of NTPs by herpes primase cannot be described by any one simple model. Primase clearly does not require the formation of Watson-Crick hydrogen bonds, as evidenced by the efficient incorporation of 2-pyridone and 4-methyl-2-pyridone NTPs opposite both natural bases and bases incapable of Watson-Crick hydrogen bonding. The simple absence of unsatisfied Watson-Crick hydrogen bonding groups is also not sufficient to allow rapid NTP polymerization, both in the presence and absence of minor groove hydrogen bond acceptors. For example, primase did not efficiently polymerize benzimidazole NTP or 1-deazapurine NTP opposite a templating 2-pyridone. Likewise, a correctly shaped base-pair is not critical as shown by the efficient misincorporation of wrong natural NTPs as well as the efficient incorporation of 2-pyridone and 4-methyl-2-pyridone NTPs opposite a variety of differently shaped bases. Hydrophobicity by itself cannot drive efficient incorporation of NTPs since converting ATP or GTP into benzimidazole NTP or 4-methylbenzimidazole NTP greatly interferes with incorporation, whereas converting UTP or CTP into 2-pyridone NTP or 4-methyl-2-pyridone NTP greatly stimulates some polymerization events. Finally, the presence or absence of specific functional groups on either the templating base or the NTP base did not predictably enhance or inhibit NTP polymerization, indicating that herpes primase does not use a simple positive/negative selectivity model as proposed for B-family DNA polymerases (14, 17).
Rather, primase may have an extremely plastic active site that can relatively efficiently generate many unnatural base-pairs. Multiple chemical solutions exist for generating a base-pair that primase efficiently synthesizes. For example, UTP, CTP and 2-pyridone NTP all satisfy the requirements for efficient polymerization opposite a templating hypoxanthine, even though the bases on the NTPs are very different chemically. Similarly, primase polymerizes UTP opposite iso-C, purine, and 6-hydroxypyridin-3-yl with virtually identical efficiencies and only slightly worse than a correct, natural base-pair, even though the structures and chemistry of any resulting base-pairs must vary substantially. Elucidating the chemical features of the incipient base-pair that enhance or inhibit polymerization will likely require elucidating the structure of primase with multiple different base-pairs in the active site.
In a general sense, these results with primase also demonstrate the difficulties inherent in drawing conclusion about the mechanism of any nucleotide polymerase based on a limited set of substrate analogues. It will be interesting to determine for other polymerases that have been posited to require Watson-Crick hydrogen bonds to polymerize a dNTP whether this is generally true or a consequence of the specific base(s) tested.
Herpes primase is a particularly tempting target for the development of novel anti-herpes therapeutics given its essential role in herpes replication. Indeed, various groups have recently described several potent non-nucleotide inhibitors of this enzyme (53-55). Unfortunately, the unique and unpredictable features of herpes primase with respect to how it interacts with nucleotide analogues suggest that it will be difficult to rationally design novel nucleotide-based primase inhibitors containing modified bases. Of the bases tested, the most promising are clearly the 2-pyridones in light of their relatively efficient polymerization.
Supplementary Material
Footnotes
This work was supported by a grant to R. D. K. from the NIH (AI059764) and grants to M. H. from the Academy of Sciences of the Czech Republic (Z4 055 905), the Ministry of Education (LC 512) and the Grant Agency of the ASCR (IAA400550902).
- HSV
- Herpes simplex virus 1
- pyr
- pyrimidine
Many of the short products appear as doublets due to the helicase removing the γ-phosphate from ppp(G)n to generate pp(G)n (1) .
References
- 1.Ramirez-Aguilar KA, Low-Nam NA, Kuchta RD. Key role of template sequence for primer synthesis by the herpes simplex virus 1 helicase-primase. Biochemistry. 2002;41:14569–14579. doi: 10.1021/bi026680v. [DOI] [PubMed] [Google Scholar]
- 2.Crute JJ, Lehman IR. Herpes simplex virus-1 helicase-primase. Physical and catalytic properties. J. Biol. Chem. 1991;266:4484–4488. [PubMed] [Google Scholar]
- 3.Nimonkar AV, Boehmer PE. Reconstitution of recombination-dependent DNA synthesis in herpes simplex virus 1. Proc Natl Acad Sci U S A. 2003;100:10201–10206. doi: 10.1073/pnas.1534569100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehmer PE, Lehman IR. Herpes simplex virus DNA replication. Ann. Rev. Biochem. 1997;66:347–384. doi: 10.1146/annurev.biochem.66.1.347. [DOI] [PubMed] [Google Scholar]
- 5.Crute JJ, Tsurumi T, Zhu L, Weller SK, Olivo PD, Challberg MD, Mocarski ES, Lehman IR. Herpes-Simplex Virus-1 Helicase Primase - a Complex of 3 Herpes-Encoded Gene-Products. Proc, Natl, Acad, Sci, U S A. 1989;86:2186–2189. doi: 10.1073/pnas.86.7.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein DJ, Weller SK. An ICP6::lacZ insertional mutagen is used to demonstrate that the UL52 gene of herpes simplex virus type 1 is required for virus growth and DNA synthesis. J. Virol. 1988;62:2970–2977. doi: 10.1128/jvi.62.8.2970-2977.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dodson MS, Lehman IR. Association of DNA helicase and primase activities with a subassembly of the herpes simplex virus 1 helicase-primase composed of the UL5 and UL52 gene products. Proc Natl Acad Sci U S A. 1991;88:1105–1109. doi: 10.1073/pnas.88.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klinedinst DK, Challberg MD. Helicase-primase complex of herpes simplex virus type 1: a mutation in the UL52 subunit abolishes primase activity. Journal of virology. 1994;68:3693–3701. doi: 10.1128/jvi.68.6.3693-3701.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dracheva S, Koonin EV, Crute JJ. Identification of the Primase Active-Site of the Herpes-Simplex Virus Type-1 Helicase-Primase. J. Biol. Chem. 1995;270:14148–14153. doi: 10.1074/jbc.270.23.14148. [DOI] [PubMed] [Google Scholar]
- 10.Sherman G, Gottlieb J, Challberg MD. The UL8 subunit of the herpes simplex virus helicase-primase complex is required for efficient primer utilization. J. Virol. 1992;66:4884–4892. doi: 10.1128/jvi.66.8.4884-4892.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuchta RD, Beckman J, Kincaid K, Moore C, Loi M, Timblin G. Mechanisms by which DNA and RNA polymerases discriminate between right and wrong (d)NTPs. Faseb J. 2006;20:A512–A513. [Google Scholar]
- 13.Patro JN, Urban M, Kuchta RD. Role of the 2-Amino Group of Purines during dNTP Polymerization by Human DNA Polymerase alpha. Biochemistry. 2009;48:180–189. doi: 10.1021/bi801823z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavanaugh NA, Urban M, Beckman J, Spratt TE, Kuchta RD. Identifying the Features of Purine dNTPs that Allow Accurate and Efficient DNA Replication by Herpes Simplex Virus I DNA Polymerase. Biochemistry. 2009;48:3554–3564. doi: 10.1021/bi8022202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kincaid K, Beckman J, Zivkovic A, Halcomb RL, Engels JW, Kuchta RD. Exploration of factors driving incorporation of unnatural dNTPS into DNA by Klenow fragment (DNA polymerase I) and DNA polymerase alpha. Nucleic Acids Res. 2005;33:2620–2628. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiaramonte M, Moore CL, Kincaid K, Kuchta RD. Facile polymerization of dNTPs bearing unnatural base analogues by DNA polymerase alpha and Klenow fragment (DNA polymerase I) Biochemistry. 2003;42:10472–10481. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]
- 17.Beckman J, Kincaid K, Hocek M, Spratt T, Engels J, Cosstick R, Kuchta RD. Human DNA polymerase alpha uses a combination of positive and negative selectivity to polymerize purine dNTPs with high fidelity. Biochemistry. 2007;46:448–460. doi: 10.1021/bi061243s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henry AA, Olsen AG, Matsuda S, Yu CZ, Geierstanger BH, Romesberg FE. Efforts to expand the genetic alphabet: Identification of a replicable unnatural DNA self-pair. J. Am. Chem. Soc. 2004;126:6923–6931. doi: 10.1021/ja049961u. [DOI] [PubMed] [Google Scholar]
- 19.Leconte AM, Hwang GT, Matsuda S, Capek P, Hari Y, Romesberg FE. Discovery, characterization, and optimization of an unnatural base pair for expansion of the genetic alphabet. J. Am. Chem. Soc. 2008;130:2336–2343. doi: 10.1021/ja078223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trostler M, Delier A, Beckman J, Urban M, Patro JN, Spratt TE, Beese LS, Kuchta RD. Discrimination between Right and Wrong Purine dNTPs by DNA Polymerase I from Bacillus stearothermophilus. Biochemistry. 2009;48:4633–4641. doi: 10.1021/bi900104n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee HR, Helquist SA, Kool ET, Johnson KA. Importance of hydrogen bonding for efficiency and specificity of the human mitochondrial DNA polymerase. J. Biol. Chem. 2008;283:14402–14410. doi: 10.1074/jbc.M705007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim TW, Delaney JC, Essigmann JM, Kool ET. Probing the active site tightness of DNA polymerase in subangstrom increments. Proc Natl Acad Sci U S A. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunkel TA, Bebenek K. DNA replication fidelity. Ann. Rev. Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 24.Kool ET. Active site tightness and substrate fit in DNA replication. Ann. Rev. Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 25.Guckian KM, Kool ET. Highly precise shape mimicry by a difluorotoluene deoxynucleoside, a replication-competent substitute for thymidine. Angew Chem Int Edit. 1997;36:2825–2828. doi: 10.1002/anie.199728251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moran S, Ren RXF, Kool ET. A thymidine triphosphate shape analog lacking Watson-Crick pairing ability is replicated with high sequence selectivity. Proc, Natl, Acad, Sci, U S A. 1997;94:10506–10511. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moran S, Ren RXF, Rumney S, Kool ET. Difluorotoluene, a nonpolar isostere for thymine, codes specifically and efficiently for adenine in DNA replication. J. Am. Chem. Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore CL, Zivkovic A, Engels JW, Kuchta RD. Human DNA primase uses Watson-Crick hydrogen bonds to distinguish between correct and incorrect nucleoside triphosphates. Biochemistry. 2004;43:12367–12374. doi: 10.1021/bi0490791. [DOI] [PubMed] [Google Scholar]
- 29.Arezi B, Kuchta RD. Eukaryotic DNA primase. Trends Biochem Sci. 2000;25:572–576. doi: 10.1016/s0968-0004(00)01680-7. [DOI] [PubMed] [Google Scholar]
- 30.Choi JY, Lim S, Eoff RL, Guengerich FP. Kinetic Analysis of Base-Pairing Preference for Nucleotide Incorporation Opposite Template Pyrimidines by Human DNA Polymerase iota. J Mol Biol. 2009;389:264–274. doi: 10.1016/j.jmb.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfle WT, Washington MT, Kool ET, Spratt TE, Helquist SA, Prakash L, Prakash S. Evidence for a Watson-Crick hydrogen bonding requirement in DNA synthesis by human DNA polymerase kappa. Mol Cell Biol. 2005;25:7137–7143. doi: 10.1128/MCB.25.16.7137-7143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramirez-Aguilar KA, Kuchta RD. Herpes simplex virus-1 primase: a polymerase with extraordinarily low fidelity. Biochemistry. 2004;43:9084–9091. doi: 10.1021/bi049335+. [DOI] [PubMed] [Google Scholar]
- 33.Ramirez-Aguilar KA, Moore CL, Kuchta RD. Herpes simplex virus 1 primase employs watson-crick hydrogen bonding to identify cognate nucleoside triphosphates. Biochemistry. 2005;44:15585–15593. doi: 10.1021/bi0513711. [DOI] [PubMed] [Google Scholar]
- 34.Cullis PM, Wolfenden R. Affinities of Nucleic-Acid Bases for Solvent Water. Biochemistry. 1981;20:3024–3028. doi: 10.1021/bi00514a006. [DOI] [PubMed] [Google Scholar]
- 35.Broom AD, Amarnath V, Vince R, Brownell J. Poly(2-Fluoroadenylic Acid) - Role of Basicity in the Stabilization of Complementary Helices. Biochim Biophys Acta. 1979;563:508–517. doi: 10.1016/0005-2787(79)90069-8. [DOI] [PubMed] [Google Scholar]
- 36.Major DT, Laxer A, Fischer B. Protonation studies of modified adenine and adenine nucleotides by theoretical calculations and N-15 NMR. Journal of Organic Chemistry. 2002;67:790–802. doi: 10.1021/jo0107554. [DOI] [PubMed] [Google Scholar]
- 37.Joubert N, Pohl R, Klepetarova B, Hocek M. Modular and practical synthesis of 6-substituted pyridin-3-yl C-nucleosides. Journal of Organic Chemistry. 2007;72:6797–6805. doi: 10.1021/jo0709504. [DOI] [PubMed] [Google Scholar]
- 38.Kuchta RD, Ilsley D, Kravig KD, Schubert S, Harris B. Inhibition of DNA Primase and Polymerase-Alpha by Arabinofuranosylnucleoside Triphosphates and Related-Compounds. Biochemistry. 1992;31:4720–4728. doi: 10.1021/bi00134a027. [DOI] [PubMed] [Google Scholar]
- 39.Leconte AM, Matsuda S, Hwang GT, Romesberg F. Efforts towards Expansion of the Genetic Alphabet: Pyridone and Methyl Pyridone Nucleobases. Angew. Chem. Int. Ed. 2006;45:4326–4329. doi: 10.1002/anie.200601272. [DOI] [PubMed] [Google Scholar]
- 40.Parsch J, Engels JW. C-F...H-C hydrogen bonds in ribonucleic acids. J Am Chem Soc. 2002;124:5664–5672. doi: 10.1021/ja012116g. [DOI] [PubMed] [Google Scholar]
- 41.Ji Q, Li JL, Huang F, Han J, Meng JB. Regioselective synthesis of 2-substituted-4-methylbenzimidazole nucleosides. Synlett. 2005:1301–1305. [Google Scholar]
- 42.Beaucage SL, Caruthers MH. Deoxunucleoside Phosphoramidites-A New Class of Key Intermediates for Deoxypolynucleotide Synthesis. Tetrahedron Letters. 1981;22:1859–1862. [Google Scholar]
- 43.McBride LJ, Caruthers MH. An Investigation of Several Deoxynucleoside Phosphoramidites Useful for Synthesizing Deoxynucleotides. Tetrahedron Letters. 1983;24:245–248. [Google Scholar]
- 44.Hirao I, Harada Y, Kimoto M, Mitsui T, Fujiwara T, Yokoyama S. A two-unnatural-base-pair system toward the expansion of the genetic code. J. Am. Chem. Soc. 2004;126:13298–13305. doi: 10.1021/ja047201d. [DOI] [PubMed] [Google Scholar]
- 45.Mitsui T, Kimoto M, Harada Y, Yokoyama S, Hirao L. An efficient unnatural base pair for a base-pair-expanded transcription system. J. Am. Chem. Soc. 2005;127:8652–8658. doi: 10.1021/ja0425280. [DOI] [PubMed] [Google Scholar]
- 46.Horlacher J, Hottiger M, Podust VN, Hubscher U, Benner SA. Recognition by viral and cellular DNA polymerases of nucleosides bearing bases with nonstandard hydrogen bonding patterns. Proc Natl Acad Sci U S A. 1995;92:6329–6333. doi: 10.1073/pnas.92.14.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henry AA, Yu C, Romesberg FE. Determinants of unnatural nucleobase stability and polymerase recognition. J Am Chem Soc. 2003;125:9638–9646. doi: 10.1021/ja035398o. [DOI] [PubMed] [Google Scholar]
- 48.Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 49.Petruska J, Hartenstine MJ, Goodman MF. Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease. The J. Biol. Chem. 1998;273:5204–5210. doi: 10.1074/jbc.273.9.5204. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell. 2006;124:331–342. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 51.Kunkel TA, Patel SS, Johnson KA. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc Natl Acad Sci U S A. 1994;91:6830–6834. doi: 10.1073/pnas.91.15.6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruggiero BL, Topal MD. Triplet repeat expansion generated by DNA slippage is suppressed by human flap endonuclease 1. J. Biol. Chem. 2004;279:23088–23097. doi: 10.1074/jbc.M313170200. [DOI] [PubMed] [Google Scholar]
- 53.Crute JJ, Grygon CA, Hargrave KD, Simoneau B, Faucher AM, Bolger G, Kibler P, Liuzzi M, Cordingley MG. Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nature medicine. 2002;8:386–391. doi: 10.1038/nm0402-386. [DOI] [PubMed] [Google Scholar]
- 54.Kleymann G, Fischer R, Betz UA, Hendrix M, Bender W, Schneider U, Handke G, Eckenberg P, Hewlett G, Pevzner V, Baumeister J, Weber O, Henninger K, Keldenich J, Jensen A, Kolb J, Bach U, Popp A, Maben J, Frappa I, Haebich D, Lockhoff O, Rubsamen-Waigmann H. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nature medicine. 2002;8:392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 55.Spector FC, Liang L, Giordano H, Sivaraja M, Peterson MG. Inhibition of herpes simplex virus replication by a 2-amino thiazole via interactions with the helicase component of the UL5-UL8-UL52 complex. J. Virol. 1998;72:6979–6987. doi: 10.1128/jvi.72.9.6979-6987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramirez-Aguilar KA, Kuchta RD. Mechanism of primer synthesis by the herpes simplex virus 1 helicase-primase. Biochemistry. 2004;43:1754–1762. doi: 10.1021/bi035519x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.