

Abstract
The catalytic mechanism of the hydrogen-mediated coupling of acetylene to carbonyl compounds and imines has been examined using three techniques: (a) ESI-MS and ESI-CAD-MS analyses, (b) computational modeling, and (c) experiments wherein putative reactive intermediates are diverted to alternative reaction products. ESI-MS analysis of reaction mixtures from the hydrogen-mediated reductive coupling of acetylene to α-ketoesters or N-benzenesulfonyl aldimines corroborate a catalytic mechanism involving C=X (X = O, NSO2Ph) insertion into a cationic rhodacyclopentadiene obtained by way of acetylene oxidative dimerization with subsequent Brønsted acid cocatalyzed hydrogenolysis of the resulting oxa- or azarhodacycloheptadiene. Hydrogenation of 1,6-diynes in the presence of α-ketoesters provides analogous coupling products. ESI mass spectrometric analysis again corroborates a catalytic mechanism involving carbonyl insertion into a cationic rhodacyclopentadiene. For all ESI-MS experiments, the structural assignments of ions are supported by multi-stage collisional activated dissociation (CAD) ESI-MS analyses. Further support for the proposed catalytic mechanism derives from experiments aimed at the interception of putative reactive intermediates and their diversion to alternate reaction products. For example, rhodium catalyzed coupling of acetylene to aldehyde in the absence of hydrogen or Brønsted acid cocatalyst provides the corresponding (Z)-butadienyl ketone, which arises from β-hydride elimination of the proposed oxarhodacycloheptadiene intermediate, as corroborated by isotopic labeling. Additionally, the putative rhodacyclopentadiene intermediate obtained from the oxidative coupling of acetylene is diverted to the product of reductive [2+2+2] cycloaddition when N-p-toluenesulfonyl-dehydroalanine ethyl ester is used as the coupling partner. The mechanism of this transformation also is corroborated by isotopic labeling. Computer model studies based on density functional theory (DFT) support the proposed mechanism and identify Brønsted acid cocatalyst assisted hydrogenolysis to be the most difficult step. The collective studies provide new insight into the reactivity of cationic rhodacyclopentadienes, which should facilitate the design of related rhodium catalyzed C-C couplings.
Introduction
The Fischer-Tropsch1 reaction and alkene hydroformylation2 rank among the largest volume catalytic processes practiced in the chemical industry3 and may be viewed as prototypical C-C bond forming hydrogenations. Despite the impact of these processes, systematic efforts toward hydrogen-mediated C-C bond formations that extend beyond carbon monoxide coupling have begun to emerge only recently.4,5 We have found that diverse unsaturates couple to carbonyl compounds and imines under the conditions of catalytic hydrogenation and transfer hydrogenation, offering an alternative to stoichiometrically preformed organometallics in a range of classical C=X (X = O, NR) addition processes, including aldol and Mannich addition,6 carbonyl allylation,7 and carbonyl and imine vinylation.8 Remarkably, under transfer hydrogenation conditions, an alcohol serves dually as hydrogen donor and precursor to the carbonyl electrophile, enabling carbonyl addition from the alcohol oxidation level, a formal hydrohydroxyalkylation.7b-d,f,g,8n,9
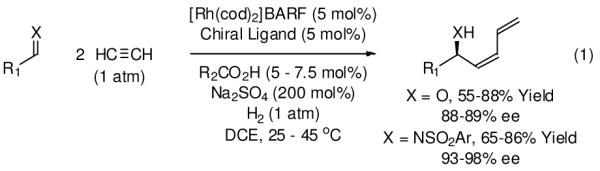
A broad goal of these investigations resides in the development of hydrogen-mediated couplings applicable to basic chemical feedstocks. Accordingly, it was found that rhodium catalyzed hydrogenation of acetylene (2 cents/mol, annual US production > 500 metric kilotons)10 in the presence of carbonyl compounds or imines promotes formation of (Z)-butadienyl allylic alcohols and (Z)-butadienyl allylic amines, respectively (eqn. 1).11 In these multicomponent couplings, two molecules of acetylene combine with elemental hydrogen and a molecule of carbonyl compound or imine.
 |
In the present account, we disclose ESI-mass spectrometric12 and computational modeling studies of these transformations that corroborate a catalytic mechanism involving oxidative coupling of acetylene to generate a cationic rhodacyclopentadiene, which engages in carbonyl or imine insertion, followed by Brønsted acid assisted hydrogenolysis of the resulting oxa- or azarhodacycloheptadienes to furnish the products of (Z)-butadienylation. Structural assignments of ions observed in the ESI-mass spectra are supported by multi-stage collisional activated dissociation (CAD). Intervention of the purported rhodacyclopentadiene and the purported oxa- and azarhodacycloheptadiene as reactive intermediates is supported further through the design of related processes in which these transient species are diverted to products of [2+2+2] cycloaddition and carbonyl insertion, respectively. The collective studies demonstrate that cationic rhodacyclopentadienes engage in carbonyl and imine insertion, providing a foundation for the development of related rhodium catalyzed C-C couplings.13
Results and Discussion
In our initial studies on the hydrogen-mediated reductive coupling of acetylene to carbonyl compounds,11a a catalytic mechanism was proposed involving carbonyl insertion into a cationic rhodacyclopentadiene obtained upon oxidative dimerization of acetylene, followed by Brønsted acid assisted hydrogenolysis of the resulting oxarhodacycloheptadiene,14 as shown in Cycle A (Scheme 1). This interpretation of the catalytic mechanism is consistent with the results from isotopic labeling studies. Rhodium catalyzed reductive coupling of α-ketoester 1a and acetylene under an atmosphere of elemental deuterium provides deuterio-1b, which incorporates a single deuterium atom at the diene terminus as the (Z)-stereoisomer. Rhodacyclopentadienes that are catalytically competent species in acetylene cyclotrimerization to form benzene, have been isolated and characterized by single crystal X-ray diffraction analysis.15 Further, carbonyl insertion into a Rh-C bond followed by protonolytic cleavage or β-hydride elimination of the resulting rhodium alkoxide also has been documented.16 Nevertheless, a related catalytic mechanism Cycle B (Scheme 1) involving acetylene-carbonyl oxidative coupling followed by insertion of a second acetylene molecule to form an identical oxarhodacycloheptadiene cannot be excluded.
Scheme 1.

Hydrogen-mediated coupling of acetylene to pyruvate 1a (TPAA = Ph3CCO2H) and plausible catalytic cycles A and B consistent with the results of deuterium labeling.
We began our mechanistic investigation using ESI-MS to analyze the products observed for various combinations of reactants, in addition to analyzing the individual reactants. Supplemental experiments in which individual reactants were replaced by other analogs or excluded entirely also were conducted. Multi-stage CAD experiments were used to provide corroborating fragmentation fingerprints of the resulting products. For example, in an effort to discriminate between cycles A and B, an aliquot of the crude reaction mixture from the hydrogenative coupling of gaseous acetylene to α-ketoester 2a (Figure 1) was diluted in methanol prior to ESI-MS analysis. The observation of ions consistent with the masses of putative reactive intermediates corroborates their intervention, and while the composition of transient species in solution and in the gas phase may differ, good correlation has been documented using soft ionization mass spectrometric techniques such as ESI.17 Whereas these MS techniques are capable of providing invaluable mechanistic clues, it is always difficult to assign a mechanistic role to a detectable species with confidence. Mechanistic computational molecular modeling is a powerful complement to these experimental techniques, as will be highlighted below.
Figure 1.

Hydrogen-mediated coupling of gaseous acetylene to α-ketoester 2a (Ar = p-NO2Ph) using triphenylacetic acid as co-catalyst. (A): ESI mass spectrum with proposed structural assignment of observed ions. (B): CAD mass spectrum of the ion of m/z 1188 with proposed structural assignment of observed ions.
A representative ESI-mass spectrum of the reaction mixture sampled at one hour (Figure 1A) shows that the most abundant ion matches the molecular weight of Rh(BIPHEP) (m/z 625). Of particular interest is the ion that matches the molecular weight of the rhodacyclopentadiene of Cycle A (m/z 677). This key intermediate of cycle A also is identified to be a stable intermediate by computational modeling (vide infra). Ions matching the molecular weights of the oxarhodacycloheptadiene (m/z 900) and the corresponding chloride adduct (ion of m/z 935, assigned based on theoretical isotope ratio) were observed, as well as an ion matching the molecular weight of the intermediate postulated to arise upon protonolytic cleavage of the oxarhodacycloheptadiene by triphenylacetic acid (m/z 1188). Finally, an ion corresponding to the molecular weight of Rh(BIPHEP)2(C4H4) was observed (m/z 1199). In contrast, ions corresponding to neither the oxarhodacyclopentene (calculated M.W. = 874 Da) of Cycle B nor the monohydride, vinyl- or dienylrhodium species are observed, thus offering no direct support for Cycle B. The penultimate intermediate postulated in both Cycles A and B, the vinyl hydride, is likewise not observed. Our computer model indicates that the hydride species should have a short lifetime due to rapid C-H reductive elimination, as discussed in greater detail below. The key ion of m/z 1188 was subjected to CAD (Figure 1B). This ion dissociates by loss of 288 Da, consistent with the elimination of triphenylacetic acid to regenerate the oxarhodacycloheptadiene intermediate (m/z 900). When the latter species is subjected to a second stage of CAD (data not shown), it dissociates to an ion of m/z 677 which again matches the molecular weight of the rhodacyclopentadiene species shown in Cycle A, suggesting a retro-carbonyl insertion process.
These MS/MS/MS data offer support for catalytic cycle A. Nevertheless, the structural assignments of the ions observed using ESI-MS must be considered tentative, as constitutionally isomeric species cannot be excluded on the basis of this data alone. Therefore, we turned to quantum chemical modeling in pursuit of a more complete assessment of the energetics and structures of species in the proposed catalytic mechanism. Whereas highly efficient quantum chemical methods, such as density functional theory,21 have allowed computational models to become relatively large in size, the current systems are too large to be treated without structural simplifications. Potential simplifications were explored and it was found that the BIPHEP ligand cannot be truncated without introducing significant artificial effects, although the general mechanistic pattern can be qualitatively reproduced with a smaller model. These model calculations are presented in the supporting information and we limit our discussion here to the model that utilizes the untruncated BIPHEP ligand. The α-ketoester 1a was modeled as methyl-2-oxoacetate and the acid cocatalyst triphenylacetic acid, TPAA, was represented as acetic acid. While these simplifications are significant and, hence, the present model is not anticipated to be quantitatively reliable, there is strong evidence that this model captures the main features of the mechanism and constitutes a reasonable compromise between computational cost and accuracy.
Figures 2 and 3 summarize the salient features of the consensus reaction mechanism determined after extensive sampling of Cycles A and B. Our model mechanism begins with the initial reactant complex 3, where the RhI-d8 center forms a [(BIPHEP)Rh(acetylene)2]+ π-complex. The computed lowest energy structure of 3 is shown in Figure 4. Not surprisingly, the rhodium center adopts a square planar geometry with the acetylene ligands aligned approximately orthogonal to the coordination plane. The first step of the catalytic cycle is an oxidative coupling of the two acetylene moieties to furnish the rhodacyclopentadiene 4, which traverses the transition state 3-TS at 15.9 kcal mol−1. This key intermediate, containing a RhIII-d6 center, is 21.6 kcal mol−1 lower in free energy than the reactant complex and is a stable intermediate that should be accessible to experimental detection. This result is in good agreement with the ESI-MS data mentioned above and assigns a key role to this rhodacyclopentadiene complex in the catalytic cycle. Species 4 is a 14-electron complex that can bind the carbonyl substrate to deliver complex 5. The formation of this complex is energetically uphill by only 4.8 kcal mol−1. The ketoester undergoes migratory insertion to give the ring-expanded intermediate 6 that is lower in energy compared to 5 by 17.7 kcal mol−1 (34.5 kcal mol−1 relative to 3). The transition state that connects these two intermediates, 5-TS, has an activation barrier of 10.1 kcal mol−1 measured from the intermediate 4 (Figure 2). Extensive exploratory calculations reveal that intermediate 6, formally a 14-electron complex with a RhIII-d6 center, is too electron-deficient to engage in direct hydrogenolysis of the Rh-O bond. The Rh-O bond cleavage must be facilitated by the carboxylic acid cocatalyst.
Figure 2.
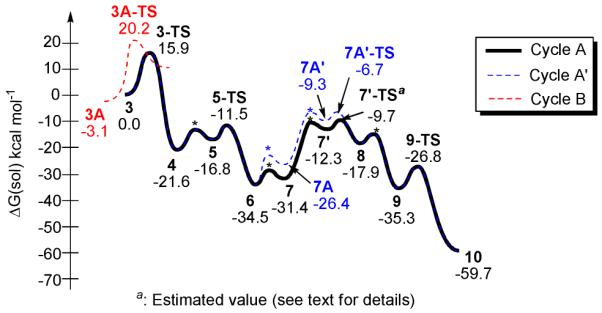
Computed reaction energy profile.
Figure 3.

Lowest energy reaction pathway depicted in Figure 2.
Figure 4.
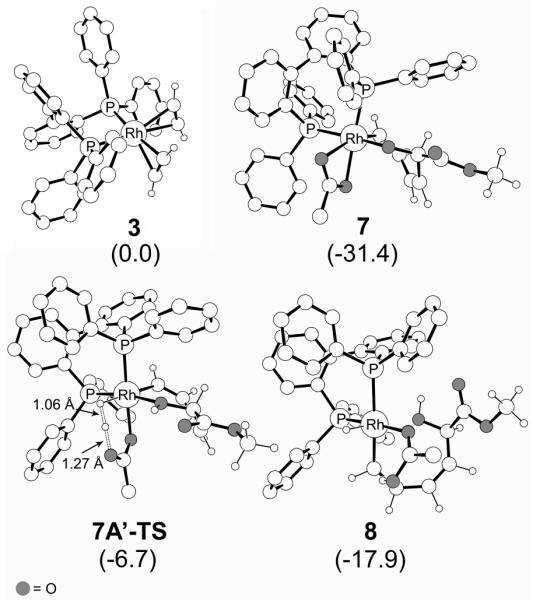
Computed structures of 3, 7, 7Aߣ-TS and 8. Non-essential hydrogens are deleted for clarity. Solution phase free energies in kcal mol−1 are given in parenthesis.
The carboxylate moiety of the cocatalyst binds to rhodium in a bidentate fashion and the proton adds to the oxo moiety of the rhodacycle to furnish the 18-electron intermediate 7A (Cycle Aߣ in Figure 2, blue in Figure 3). Addition of H2 to this complex is mediated by removal of one of the arms of the carboxylate ion, leading to a high energy intermediate 7Aߣ, with a free energy that is 25.2 kcal mol−1 higher than 6. From this intermediate, the barrier for heterolytic cleavage of the H-H bond, resulting in a hydride on the Rh center and protonation of the non-coordinating oxygen atom of the carboxylate ligand, is associated with a barrier of only 2.6 kcal mol−1 (7Aߣ-TS). Thus, the calculations suggest that the overall barrier for dihydrogen activation is 27.8 kcal mol−1. This could possibly be the rate determining step of the reaction (Figure 2, blue profile), under the reasonable assumption that the barrier for the H2 adduct formation is lower than or comparable to the H-H bond cleavage step. A slightly lower pathway involves the protonation of the oxarhodacycloheptadiene 6 after dihydrogen activation, i.e. the binding of carboxylate to rhodium is not accompanied by protonation, leading to the neutral complex 7 (Cycle A in Figure 2). H2 addition to this intermediate 7′ is 22.3 kcal mol−1 higher in free energy compared to 6. Despite several attempts, we were unable to locate a transition state for H2 activation from this intermediate. We expect however, that the barrier will be quite small, and very similar to that obtained for H2 activation from 7Aߣ. Adding the same barrier to the energy of 7′ gives us an estimated overall activation free energy of 24.8 kcal mol−1 (7′-TS). Note, however, that the H-H bond cleavage could in reality be almost barrierless if tunneling is involved. Thus, the activation energy of 24.8 kcal mol−1 should be considered to be the upper limit for dihydrogen activation. Subsequent addition of a proton to the oxygen of the rhodacycle leads to the cationic hydride intermediate 8, in which the Rh-O bond is nearly cleaved at a distance of 2.37 Å. Whereas this interpretation of our computed results is self-consistent and plausible, we must be careful not to overinterpret these model results. Our explorative calculations show that the calculated energies can shift notably with model size (see Supporting Information), in addition to intrinsic concerns about the accuracy of DFT and the continuum solvation models used in this work. These concerns demonstrate the importance of combining clues from various techniques of scientific inquiry, both experimental and computational, as highlighted in this work. We propose that the cocatalyst plays a dual role: (a) it acts as a Brønsted acid and assists Rh-O bond cleavage, as previously proposed, and (b) the conjugate base acts as a ligand, increasing the electron-count at the Rh-center to directly assist H-H bond cleavage.14 Having completed its catalytic function, the carboxylic acid dissociates, resulting in the formation of intermediate 9. It is instructive to recognize that loss of the carboxylic acid decreases the electron count at the metal center, which predisposes complex 9 (formally a 14-electron complex) toward reductive elimination to deliver the final product complex 10 traversing the transition state 9-TS with a barrier of 8.5 kcal mol−1.
Cycle B, which involves initial acetylene-carbonyl oxidative coupling also was explored. Calculations show that the initial reactant complex 3A is slightly lower in energy than the bisalkyne adduct 3 by 3.1 kcal mol−1. The oxidative addition barrier however, at 20.2 kcal mol−1 relative to 3, is almost 7 kcal mol−1 higher than the barrier for the coupling of two alkynes. Such a high barrier for the initial step, combined with the inability to detect the oxarhodacyclopentene intermediate by mass spectrometry (vide supra) or infer its presence through its diversion to alternate reaction products makes this pathway, which is otherwise plausible based on the deuterium labeling experiments, highly unlikely.
Analogous Reactions
If catalytic cycle A is operative, one would expect related cationic rhodacyclopentadienes to display similar reactivity and participate in carbonyl insertion processes. It is known that 1,6-diynes react with rhodium(I) salts to furnish isolable rhodacyclopentadienes, which have been characterized in detail.18 Accordingly, 1,6-diyne 11a (100 mol%) was hydrogenated in the presence of α-ketoester 2a (100 mol%) using a cationic rhodium catalyst. The product of tandem reductive cyclization-carbonyl coupling 11b is obtained in 58% yield as a single alkene geometrical isomer. Thus, the cationic rhodacyclopentadiene presumed to arise upon oxidative coupling of 1,6-diyne 11a does display reactivity analogous to that observed in the reductive couplings of gaseous acetylene. Interestingly, for nonsymmetric 1,6-diyne 12a, carbonyl insertion occurs such that α-ketoester 2a couples at the substituted terminus of the rhodacyclopentadiene intermediate to furnish 12b as a single geometrical and constitutional isomer. The modest yield of tandem reductive cyclization-carbonyl insertion product 12b is due to competitive alkyne [2+2+2] cycloaddition (Scheme 2).
Scheme 2.

Proposed catalytic mechanism for the hydrogen-mediated of 1,6-diynes 11a and 12a to α-ketoester 2a using triphenylacetic acid as co-catalyst.
An aliquot of the crude reaction mixture from the hydrogenative coupling of 1,6-diyne 12a to α-ketoester 2a was diluted 5000-fold in methanol and subjected to ESI-MS analysis (Figure 5A). The two most abundant ions include one that matches the mass of the purported cationic rhodacylcopentadiene (m/z 909) and another that matches the mass of the purported cationic oxarhodacycloheptadiene (m/z 1132). Analogous intermediates are postulated in Cycle A. Upon CAD, the ion of m/z 1132 dissociates by loss of 223 Da, consistent with elimination of 2a to regenerate the rhodacyclopentadiene (m/z 909) (Figure 5B). The additional loss of 284 Da, which is consistent with elimination of 12a, regenerates cationic Rh(BIPHEP) (m/z 625). An ion of m/z 1195 corresponding to the mass of [Rh(BIPHEP)(12a)(2a)(OMe)(HOMe)] is also observed, which may correspond to the methanol-methoxide adduct of the cationic oxarhodacycloheptadiene
Figure 5.

Hydrogen-mediated of 1,6-diyne 12a to α-ketoester 2a (Ar = p-NO2Ph) using triphenylacetic acid as co-catalyst. (A): ESI mass spectrum with proposed structural assignment of observed ions. (B): CAD mass spectrum of the ion of m/z 1132 with proposed structural assignment of observed ions.
The hydrogen-mediated coupling of acetylene to N-arylsulfonyl imines using chirally modified rhodium catalysts provides optically enriched (Z)-butadienyl allylic amines.11b An analogous mechanism involving imine insertion into a cationic rhodacyclopentadiene derived via acetylene oxidative dimerization, followed by Brønsted acid cocatalyzed hydrogenolysis of the resulting azarhodacycloheptadiene was postulated. To probe the mechanism, the crude reaction mixture from the hydrogenative coupling of N-benzenesulfonyl aldimine 13a and gaseous acetylene was subjected to ESI-MS analysis. As noted above for the corresponding carbonyl couplings, ions matching the molecular weight of Rh(BIPHEP) (m/z 625), the BIPHEP ligated rhodacyclopentadiene (m/z 677), and Rh(BIPHEP)2(C4H4) (m/z 1199) are seen (Figure 6A). Two ions are of particular interest: that corresponding to the molecular weight of the azarhodacycloheptadiene (m/z 967), which would be derived upon insertion of imine 13a into the rhodacyclopentadiene, and the ion matching the molecular weight of the intermediate postulated to arise upon protonolytic cleavage of the azarhodacycloheptadiene induced by triphenylacetic acid (m/z 1255). Ions consistent with the molecular weight of Rh(BIPHEP)2 (m/z 1147) and, interestingly, Rh(BIPHEP)(Ph3CCO2H)(C2H4) (m/z 939) also are observed.
Figure 6.

Hydrogen-mediated coupling of gaseous acetylene to N-benzenesulfonyl aldimine 13a (Ar = p-NO2Ph, Bs = Benzenesulfonyl) using triphenylacetic acid as co-catalyst. (A): ESI mass spectrum with proposed structural assignment of observed ions. (B): CAD mass spectrum of the ion of m/z 1255 with proposed structural assignment of observed ions.
The ion of m/z 1255 was subjected to multi-stage CAD (Figure 6B). This ion dissociates by loss of 288 Da, consistent with elimination of triphenylacetic acid to regenerate the azarhodacycloheptadiene (m/z 967). Ions corresponding to ligand degradation also are observed (m/z 783, 707 and 625). Notably, triphenylacetic acid is eliminated upon CAD of the analogous oxarhodacycloheptadiene in the aforementioned coupling of acetylene to α-ketoester 2a (Figure 1).
Interception of Putative Reactive Intermediates
As described above, compounds 11a and 12a were converted to adducts 11b and 12b, thereby corroborating the ability of rhodacyclopentadienes to engage in carbonyl insertion processes (Scheme 2). To further test the proposed mechanism, additional experiments were designed aimed at the interception of putative reactive intermediates and their diversion to alternate reaction products.
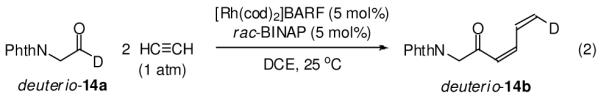
In one experiment, the rhodium catalyzed coupling of acetylene to aldehyde 14a was performed in the absence of both hydrogen and the Brønsted acid cocatalyst. Here, the putative oxarhodacycloheptadiene is potentially intercepted via β-hydride elimination to deliver the (Z)-butadienyl ketone 14b.16 Remarkably, upon exposure of 14a to gaseous acetylene in the absence of hydrogen and Brønsted acid the anticipated product of β-hydride elimination 14b is obtained in 52% isolated yield. If the reaction is performed under identical conditions, but using triphenylacetic acid as cocatalyst (5 mol%), the product of β-hydride elimination 14b is not formed, presumably due to protonolysis of the intermediate oxarhodacycloheptadiene (Scheme 3). Additional evidence supporting the intervention of the oxarhodacycloheptadiene intermediate is found in the conversion of deuterio-14a to deuterio-14b. Here, the deuterium located at the aldehydic position is stereoselectively transferred to the vinyl terminus of deuterio-14b (eqn. 2).
Scheme 3.
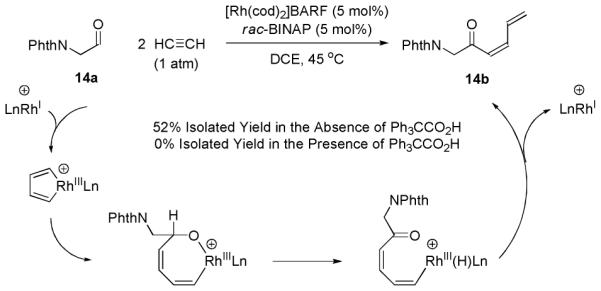
Rhodium catalyzed coupling of acetylene to aldehyde 14a in the absence of hydrogen and Brønsted acid cocatalyst delivers ketone 14b, corroborating intervention of the proposed oxarhodacycloheptadiene intermediate.
 |
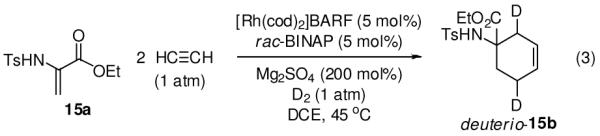
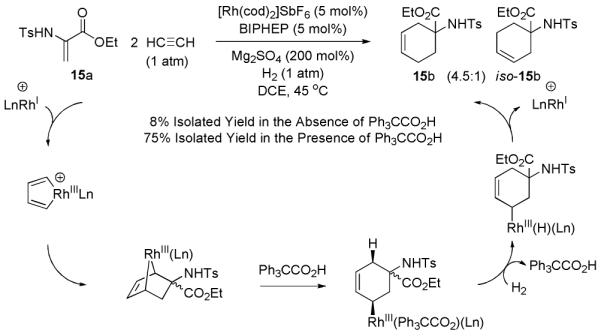
Further support for the intervention of rhodacyclopentadiene intermediates in the hydrogen-mediated couplings of acetylene is found in reductive cycloadditions of N-p-toluenesulfonyl-dehydroalanine ethyl ester 15a.19 Specifically, hydrogenation of acetylene in the presence of 15a results in the formation of the reductive [2+2+2] cycloaddition products 15b and iso-15b in a combined 75% isolated yield (Scheme 4).20 The structural assignment of 15b was confirmed by single crystal X-ray diffraction analysis. If the reaction is performed under identical conditions, but in the absence of triphenylacetic acid, 15b and iso-15b are formed in only 8% isolated yield, again signifying the key role of Brønsted acids as cocatalysts for the hydrogenolysis of organorhodium intermediates.14 Consistent with intervention of an allyl rhodium intermediate, isomeric alkenes 15b and iso-15b are generated. Hydrogenation of 15b and iso-15b results in convergence to a common cyclohexane derivative, as described in the experimental section. Under an atmosphere of deuterium, 15a provides deuterio-15b as the major reaction product, which incorporates two deuterium atoms, as is consistent with the proposed mechanism (eqn. 3).
 |
Scheme 4.
Rhodium catalyzed hydrogenation of acetylene in the presence of dehydroalanine 15a delivers the product of reductive [2+2+2] cycloaddition 15b, corroborating intervention of the proposed rhodacyclopentadiene intermediate.
Conclusion
In summary, the catalytic mechanism of the hydrogen-mediated coupling of acetylene to carbonyl compounds and imines has been illuminated using three powerful techniques: (a) ESI-MS and ESI-CAD-MS analyses, (b) computational modeling, and (c) experiments wherein putative reactive intermediates are diverted to alternate reaction products. The collective data provide strong evidence for Cycle A (Scheme 1), which involves oxidative coupling of acetylene to furnish a rhodacyclopentadiene that engages in carbonyl and imine insertion.13 Additionally, all three methods of analysis point to the key role of Brønsted acid additives as co-catalysts in the hydrogenolysis of organorhodium intermediates.4,14 These studies provide further insight into the structural and interactional features of catalytic systems for hydrogen-mediated C-C bond formation and should facilitate the design of related processes.
Supplementary Material
Acknowledgment
Acknowledgment is made to the Research Corporation Cottrell Scholar Program, the Sloan Foundation, the Dreyfus Foundation, Johnson & Johnson, Merck, the NIH-NIGMS (RO1-GM69445), the Robert A. Welch Foundation (F1466 to MJK and F1155 to JSB), and the NSF (JSB, CHE-0718320; MHB, CHE-0645381) for partial support of this research. Oliver Briel of Umicore is thanked for a generous donation of Rh(cod)2OTf and Rh(cod)2BF4. Fernando Pedraza is acknowledged for initial experiments on the conversion of 14a to 14b.
References
- (1).For a review, see:Cornils Boy, Herrmann A, Rasch Manfred. Angew. Chem., Int. Ed. Engl. 1994;33:2144.
- (2).For recent reviews on alkene hydroformylation, see:Cornils B, Herrmann WA, Kohlpaintner CW. Angew. Chem., Int. Ed. Engl. 1994;33:2144.Beller M, Cornils B, Frohning CD, Kohlpaintner CW. J. Mol. Catal. 1995;104:17.Eilbracht P, Barfacker L, Buss C, Hollmann C, Kitsos-Rzychon BE, Kranemann CL, Rische T, Roggenbuck R, Schmidt A. Chem. Rev. 1999;99:3329. doi: 10.1021/cr970413r.Nozaki K. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 1. Springer-Verlag; Berlin: 1999. p. 381.Breit B. Acc. Chem. Res. 2003;36:264. doi: 10.1021/ar0200596.
- (3).Baade William F., Parekh Uday N., Raman Venkat S. Kirk-Othmer’s Encyclopedia of Chemical Technology. 5th ed. Vol. 13. Wiley; Hoboken: 2004. Hydrogen; p. 759. [Google Scholar]
- (4).For recent reviews on hydrogen-mediated C-C coupling, see:Ngai M-Y, Kong J-R, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m.Iida H, Krische MJ. Top. Curr. Chem. 2007;279:77.Shibahara F, Krische MJ. Chem. Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938.
- (5).Prior to systematic studies from our laboratory, two isolated examples of hydrogen-mediated C-C bond formation not involving the couplings of carbon monoxide were reported:Molander GA, Hoberg JO. J. Am. Chem. Soc. 1992;114:3123.Kokubo K, Miura M, Nomura M. Organometallics. 1995;14:4521.
- (6).For hydrogen-mediated reductive aldol and Mannich couplings, see:Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2002;124:15156. doi: 10.1021/ja021163l.Huddleston RR, Krische MJ. Org. Lett. 2003;5:1143. doi: 10.1021/ol0300219.Koech PK, Krische MJ. Org. Lett. 2004;6:691. doi: 10.1021/ol030136c.Marriner GA, Garner SA, Jang H-Y, Krische MJ. J. Org. Chem. 2004;69:1380. doi: 10.1021/jo030310a.Jung C-K, Garner SA, Krische MJ. Org. Lett. 2006;8:519. doi: 10.1021/ol052859x.Han SB, Krische MJ. Org. Lett. 2006;8:5657. doi: 10.1021/ol0624023.Jung C-K, Krische MJ. J. Am. Chem. Soc. 2006;128:17051. doi: 10.1021/ja066198q.Garner SA, Krische MJ. J. Org. Chem. 2007;72:5843. doi: 10.1021/jo070779w.Bee C, Han SB, Hassan A, Iida H, Krische MJ. J. Am. Chem. Soc. 2008;130:2747. doi: 10.1021/ja710862u.
- (7).For hydrogen-mediated carbonyl allylation, see:Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u.Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b.Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b.Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x.Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705. doi: 10.1021/ol800836v.Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han S-B, Krische MJ. J. Am. Chem. Soc. 2009;131:2514. doi: 10.1021/ja808857w.
- (8).For hydrogen-mediated vinylation of carbonyl compounds and imines, see:Huddleston RR, Jang H-Y, Krische MJ. J. Am. Chem. Soc. 2003;125:11488. doi: 10.1021/ja030415v.Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2004;126:4664. doi: 10.1021/ja0316566.Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269. doi: 10.1021/ja051104i.Cho C-W, Krische MJ. Org. Lett. 2006;8:891. doi: 10.1021/ol052976s.Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718. doi: 10.1021/ja056474l.Cho C-W, Krische MJ. Org. Lett. 2006;8:3873. doi: 10.1021/ol061485k.Rhee J-U, Krische MJ. J. Am. Chem. Soc. 2006;128:10674. doi: 10.1021/ja0637954.Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448. doi: 10.1021/ja0673027.Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:280. doi: 10.1021/ja0670815.Barchuk A, Ngai M-Y, Krische MJ. 2007;129:8432. doi: 10.1021/ja073018j.Cho C-W, Skucas E, Krische MJ. Organometallics. 2007;26:3860.Hong Y-T, Cho C-W, Skucas E, Krische MJ. Org. Lett. 2007;9:3745. doi: 10.1021/ol7015548.Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:12644. doi: 10.1021/ja075438e.Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066. doi: 10.1021/ja809456u.
- (9).For related hydroaminoalkylations (amine-unsaturate C-C coupling), see:Maspero F, Clerici MG. Synthesis. 1980:305.Nugent WA, Ovenall DW, Homes SJ. Organometallics. 1983;2:161.Herzon SB, Hartwig JF. J. Am. Chem. Soc. 2007;129:6690. doi: 10.1021/ja0718366.Herzon SB, Hartwig JF. J. Am. Chem. Soc. 2008;130:14940. doi: 10.1021/ja806367e.Kubiak R, Prochnow I, Doye S. Angew. Chem. Int. Ed. 2009;48:1153. doi: 10.1002/anie.200805169.Bexrud JA, Eisenberger P, Leitch DC, Payne PR, Schafer LL. J. Am. Chem. Soc. 2009;131:2116. doi: 10.1021/ja808862w.
- (10).Gannon RE, Manyik RM, Dietz CM, Sargent HB, Schaffer RP, Thribolet RO. Kirk-Othmer’s Encyclopedia of Chemical Technology. 5th ed. Vol. 1. Wiley; Hoboken: 2004. p. 216. [Google Scholar]
- (11).For hydrogen-mediated couplings of acetylene to carbonyl compounds and imines, see:Kong J-R, Krische MJ. J. Am. Chem. Soc. 2006;128:16040. doi: 10.1021/ja0664786.Skucas E, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2007;129:7242. doi: 10.1021/ja0715896.Han SB, Kong J-R, Krische MJ. Org. Lett. 2008;10:4133. doi: 10.1021/ol8018874.
- (12).For reviews covering use of ESI mass spectrometric analysis as applied to the characterization of catalytic reaction mechanisms, see:Plattner D. Int. J. Mass. Specrom. 2001;207:125.Chen P. Angew. Chem. Int. Ed. 2003;42:2832. doi: 10.1002/anie.200200560.
- (13).Following disclosure of our work (reference 11), other rhodium catalyzed C-C bond formations believed to proceed by way of carbonyl insertion into transient rhodacyclopentadienes were reported:Tanaka K, Otake Y, Wada A, Noguchi K, Hirano M. Org. Lett. 2007;9:2203. doi: 10.1021/ol0707721.Tsuchikama K, Yoshinami Y, Shibata T. Synlett. 2007:1395.
- (14).As previously observed (see references 8e,g,h), carboxylic acid cocatalysts enhance rate and conversion, presumably by circumventing highly energetic 4-centered transition structures for σ-bond metathesis, as required for direct hydrogenolysis of oxametallacyclic intermediates, with 6-centered transition structures for hydrogenolysis of iridium carboxylates derived upon protonolytic cleavage of the nitrogen-iridium bond. This interpretation finds support in recent theoretical studies on the hydrogenolysis of rhodium formates:Musashi Y, Sakaki S. J. Am. Chem. Soc. 2002;124:7588. doi: 10.1021/ja020063c.
- (15).Bianchini C, Caulton KG, Chardon C, Eisenstein O, Folting K, Johnson TJ, Meli A, Peruzzini M, Rauscher DJ, Streib WE, Vizza F. J. Am. Chem. Soc. 1991;113:5127.and references cited therein.
- (16).For insertion of carbonyl moieties into Rh-C bonds followed by protonolytic cleavage or β-hydride elimination of the incipient rhodium alkoxide, see:Krug C, Hartwig JF. J. Am. Chem. Soc. 2002;124:1674. doi: 10.1021/ja017401e.Fujii T, Koike T, Mori A, Osakada K. Synlett. 2002:298.
- (17).(a) Daniel JM, Friess SD, Rajagopalan S, Wendt S, Zenobi R. Int. J. Mass Spec. 2002;216:1. [Google Scholar]; (b) Schrader W, Handayani PP, Zhou J, List B. Angew. Chem. Int. Ed. 2009;48:1463. doi: 10.1002/anie.200804353. [DOI] [PubMed] [Google Scholar]; (c) Paz-Schmidt RA, Bonrath W, Plattner DA. Anal. Chem. 2009;81:3665. doi: 10.1021/ac802754q. [DOI] [PubMed] [Google Scholar]
- (18).(a) Müller E, Thomas R, Zountsas G. Liebigs Ann. Chem. 1972:16. [Google Scholar]; (b) Müller E, Winter W. Chem. Ber. 1972;105:2523. [Google Scholar]; (c) Müller E, Winter W. Liebigs Ann. Chem. 1975:41. [Google Scholar]; (d) Scheller A, Winter W, Müller E. Liebigs Ann. Chem. 1976:1448. [Google Scholar]
- (19).Rhodacyclopentadienes also are believed to participate in the insertion of substituted alkynes, as exemplified by catalytic [2+2+2] cycloaddition processes. For reviews, see:Lautens M, Klute W, Tam W. Chem. Rev. 1996;96:49. doi: 10.1021/cr950016l.Kotha S, Brahmachary E, Lahiri K. Eur. J. Org. Chem. 2005;22:4741.Chopade PR, Louie J. Adv. Synth. Catal. 2006;348:2307.
- (20).Metal catalyzed reductive cycloadditions are highly uncommon. For examples, see:Herath A, Montgomery J. J. Am. Chem. Soc. 2006;128:14030. doi: 10.1021/ja0660249.Chang H-T, Jayanth TT, Cheng C-H. J. Am. Chem. Soc. 2007;129:4166. doi: 10.1021/ja0710196.
- (21).Parr RG, Yang W. Density Functional Theory of Atoms and Molecules. Oxford University Press; New York: 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.