

Abstract
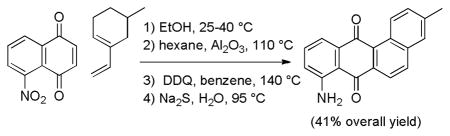
An efficient four-step route to the tetracyclic aminoquinone moiety of marmycin A that proceeds in 41% overall yield from 5-nitronaphthoquinone and 5-methyl-1-vinylcyclohexene will facilitate preparation of marmycin A analogues for biological evaluation. The Diels-Alder reaction gave exclusively the desired adduct that is favored by steric considerations rather than the regioisomeric adduct that is favored by electronic considerations.
Fenical and coworkers recently isolated two cytotoxic quinones of the angucycline family, marmycins A (1a) and B (2), from a marine sediment-derived actinomycete related to the genus Streptomycetes.1 The structures were determined by spectroscopic analysis and X-ray crystallography. C-glycosidic linkages are quite common in angucyclines,2 but the C- and N-glycosidic linkages resulting in a hexacyclic skeleton is unique to the marmycins. Marmycin A (1a) showed potent activity against 12 human tumor cell lines with a mean IC50 value of 22 nM. The combination of the novel skeleton and potent biological activity of 1a prompted us to undertake its synthesis.
We envisioned that condensation of glycal 2a with tetracyclic aminoquinone 3 would provide the acetate of 1a. Yadav has extensively studied related condensations of simple anilines with glycals such as 2b that contain hydrogen on C-3 to form the tricyclic ABC ring system of the marmycins with a hydrogen rather than a methyl group at C-3′.3 While our work was in progress, Yao and Zhang reported a ten-step synthesis of tetracyclic aminoquinone 3 and the InBr3-catalyzed condensation of 3 and 2b to afford C-3′-desmethyl marmycin A (1c).4
Our approach to tetracyclic aminoquinone 3 by the Diels-Alder reaction of 5-nitronaphthoquinone (4)5 with 5-methyl-1-vinylcyclohexene (5)6 will form the complete carbon skeleton in a single step. However, the regiochemistry of the proposed Diels-Alder reaction was of some concern. Naphthoquinones with electron withdrawing substituents on C-5 react preferentially with nucleophiles, including the nucleophilic end of a diene, at C-2, not C-3, which should lead to the undesired regioisomer in the Diels-Alder reaction (see Scheme 2).7 Nitroquinone 4 reacts with 1,1-dimethoxyethylene preferentially (2.7:1) at C-2.8 Oda studied the Diels-Alder reactions of 4 with isoprene, which gave a 9.2:1 mixture favoring the expected major isomer 6.9 However, with piperylene the expected major isomer 8 was favored by only 1.4:1. Molecular mechanics calculations of the Diels-Alder transition states provide a possible explanation for this loss of selectivity.10 The formation of 6 is preferred sterically by 0.34 kcal/mol in addition to the electronic preference. However, the formation of 9 is preferred sterically by 0.37 kcal/mol due to repulsion between the nitro and methyl groups in the transition state that leads to the electronically preferred adduct 8. Competing electronic and steric preferences should result in reduced selectivity. Calculations for the Diels-Alder reaction of 4 with 1-vinylcyclohexene suggest that the transition state for the desired adduct 11 is favored over that for the electronically preferred adduct 10 by 1.02 kcal/mol due to steric repulsion between the nitro group and cyclohexene ring (see Figure 1). We therefore decided to investigate this route to aminoquinone 3.
Scheme 2.

Diels-Alder Reactions of 4 with Unsymmetrical Dienes
Figure 1.
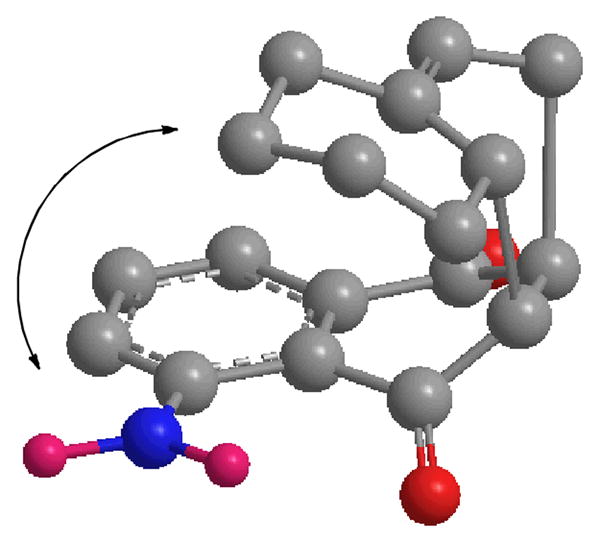
MMX calculated structure for the Diels-Alder reaction leading to 11 with arrow showing steric repulsion between the nitro group and cyclohexene ring.
Addition of vinylmagnesium bromide to 3-methyl-cyclohexanone (12) followed by dehydration of the resulting tertiary allylic alcohol in 90:1 THF/H2SO4 for 72 hours at 50 °C afforded a 1.5:1 mixture of the desired diene 5 and 13 in 70% yield, which was used directly because previous studies of Diels-Alder reactions with other naphthoquinones indicated that the more hindered minor isomer 13 was much less reactive than 5 (see Scheme 3).6 Nitration of naphthoquinone with sodium nitrate in sulfuric acid afforded 4 in 75% yield.5 Treating 4 with 2 equiv of the 1.5:1 mixture of 5 and 13 in EtOH for 12 hours at 25 °C and 2 hours at 40 °C afforded a complex mixture of stereo and regioisomeric Diels-Alder adducts 14 and 15 that was oxidized to the quinone and aromatic E ring prior to purification.
Scheme 3.

Diels-Alder Reaction to Form 17
Oda oxidized 6–9 to anthraquinones by aeration in ethanolic potassium hydroxide or by heating in hexane containing alumina.9 Stirring the mixture of 14 and 15 in 0.1 M KOH in EtOH at 25 °C for 24 hours in a tube sealed under air gave a 1:1 mixture of the desired nitroquinone 17 and the unexpected aminoquinone 16 in low yield. The structure of amine 16 was confirmed by reduction of 17 with aqueous Na2S at 95 °C for 3 hours to give 16 in 98% yield.11 This suggested that both oxygen and the nitro group could function as oxidants for the aromatization of the E ring. Bubbling air into the reaction through a dispersion tube decreased the formation of 16, giving a 10:1 mixture of 17 and amine 16, but in only 24% yield from naphthoquinone 4. A possible mechanism for the formation of 16 is presented below in Scheme 6.
Scheme 6.

Oxidation to the Anthraquinone by the Nitro Group
Much better results were obtained by heating the mixture of 14 and 15 in 90:10 hexane/benzene containing alumina in a sealed tube under oxygen (~0.7 equiv) for 2 hours, followed by replacing the oxygen and heating for an additional two hours. This gave a 9:1 mixture of the desired adduct 17 and adduct 18 (formed from the undesired diene 13) that contained only 2–4% of amine 16. Recrystallization from CHCl3 afforded pure nitroquinone 17 in 63% overall yield from naphthoquinone 4. The oxidation was not complete and more amine was formed if the reaction was heated for 4 h without replacement of the oxygen after 2 h. The structure of 17 was established by X-ray crystal structure determination of 3 as described below. We were delighted to find that the Diels-Alder reaction was very selective for the desired product 17 that was expected on steric grounds rather than the undesired regioisomer corresponding to 10 that was expected on the basis of electronic considerations.
Oxidation of 17 with 10 equivalents of DDQ in benzene at 140 °C in a microwave oven for 20 minutes provided fully aromatic nitroquinone 19 in 71% yield (see Scheme 4).12 Although only 2 equivalents of DDQ are required stoichiometrically, the reaction did not go to completion with 6 equivalents, even after heating for 30 min at 140 °C, suggesting that DDQ decomposes at a rate competitive with the oxidation of 17. Oxidation with DDQ at lower temperatures was less effective. The DDQ byproducts are very polar and easily removed by flash chromatography. Reduction of the nitro group of 19 with aqueous sodium sulfide for 2 hours at 95 °C for 2 hours provided the desired fully aromatic tetracyclic aminoquinone 3 in 91% yield.11 The structure of 3 was established by X-ray crystallography. The spectral data are identical to those reported by Yao and Zhang.4 This sequence provides tetracyclic aminoquinone 3 in only 4 steps from naphthoquinone 4 in 41% overall yield.
Scheme 4.

Synthesis of Aminoquinone 3
Yao and Zhang reported that the reaction of 3 with 2b was very sluggish with many catalysts but gave a 2:1 mixture of 20 (56%) and 21 (28%) by reaction for 12hours at 25 °C with 10% InBr3 in CH2Cl2. In our hands, this reaction with InBr3 appeared to be exceedingly moisture sensitive. Reaction with In (OTf)3 was less moisture sensitive; use of 10% In (OTf)3 in CH2Cl2 for 12 hours at 45 °C provided 20 (27%) and 21 (14%).
Yadav proposed that these reactions proceed by a Friedel-Crafts reaction (C-glycosylation) to give 22 followed by an intramolecular hydroamination to form 20 and 21 (see Scheme 5).3a,4 The formation of C-glycoside 22 is well precedented.13 Although hydroaminations of unactivated alkenes catalyzed by acid or lanthanum triflates have recently been reported, they require temperatures between 135–160 °C.14 Therefore, the intramolecular hydroamination of 22 to give 20 and 21 is unlikely to take place at 25 °C. It seems more likely that the first step involves loss of acetate from C-3 of 2b to give an allylic cation that reacts with the amine to give 23. There is ample precedent for the formation of compounds analogous to 23 from glycals and other nucleophiles. The BF3•OEt2-catalyzed reaction of 2b with NaN3 occurs initially at C-1, but the major product formed after equilibration is the C-3 azide corresponding to 23.15a Similarly, the SnCl4-catalyzed reaction of glycals with aliphatic thiols occurs initially at C-1, but gives mainly 3-alkylthio glycals corresponding to 23 under equilibrium conditions.15b Protonation of glycal 23 and an intramolecular Friedel-Crafts reaction will then form 20 and 21. The intramolecular Friedel-Crafts reaction of 23 appears to be more likely for the cyclization step than the intramolecular hydroamination of unactivated alkene 22.
Scheme 5.
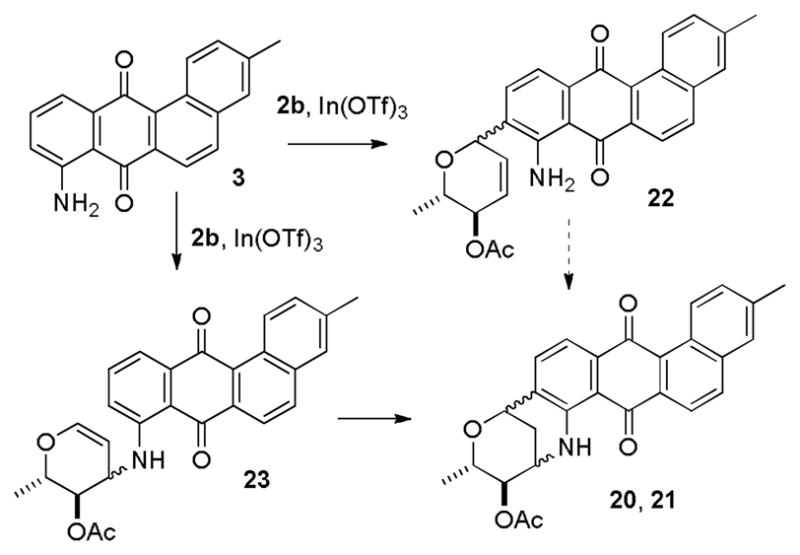
Mechanistic Considerations
There is limited precedent for a nitro group functioning as an oxidant in the conversion of 14 to 16.16 A possible mechanism involves enolization of 14 to give 24 followed by addition of the enolate to the nitro group, which will give 25 after protonation and loss of water (see Scheme 6). Deprotonation and elimination will convert 25 to nitrosoquinone 26. Enolization of 26 followed by attack of the enolate on the nitroso group will give 27 after protonation. Deprotonation and elimination will give anthraquinone 28 at the hydroxylamine oxidation state which must then be reduced to give 16.
In conclusion, we have developed an efficient four-step route to aminoquinone 3 from naphthoquinone 4 that proceeds in 41% overall yield. This will facilitate the preparation of marmycin analogues for biological evaluation. The Diels-Alder reaction of 4 and 5 gave exclusively the desired Diels-Alder adduct 14 that is favored by steric considerations rather than the adduct corresponding to 10 that is favored by electronic considerations.
Supplementary Material
Scheme 1.
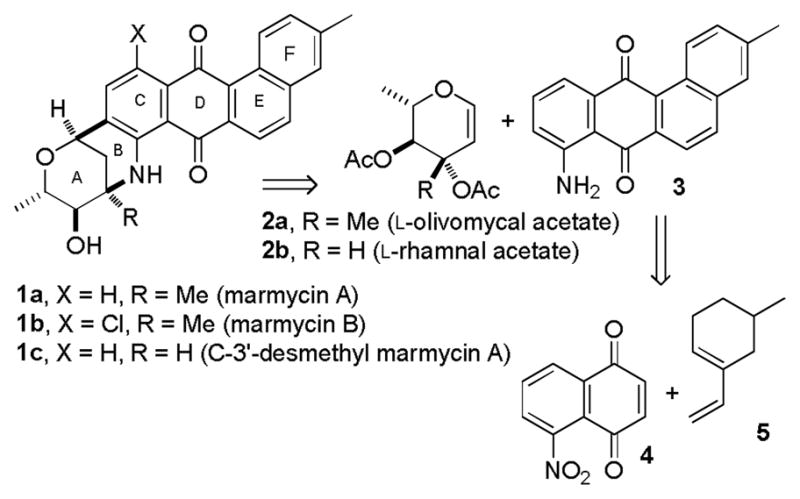
Retrosynthesis of Marmycin A
Acknowledgments
We are grateful to the National Institutes of Health (GM-50151) for support of this work. We thank the National Science Foundation for partial support of this work through grant CHE-0521047 for the purchase of a new X-ray diffractometer.
Footnotes
Supporting Information Available: Complete experimental procedures, copies of 1H and 13C NMR spectral data, details of the structure determination of 3, and CIF file of 3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Martin GDA, Tan LT, Jensen PR, Dimayuga RE, Fairchild CR, Raventos-Suarez C, Fenical W. J Nat Prod. 2007;70:1406–1409. doi: 10.1021/np060621r. [DOI] [PubMed] [Google Scholar]
- 2.(a) Rohr J, Thierike R. Nat Prod Rep. 1992:103–137. doi: 10.1039/np9920900103. [DOI] [PubMed] [Google Scholar]; (b) Krohn K, Rohr J. Topics in Current Chemistry. 1997;188:127–195. [Google Scholar]; (c) Carreño MC, Urbano A. Synlett. 2005:1–25. [Google Scholar]
- 3.(a) Yadav JS, Reddy BVS, Rao KV, Raj KS, Prasad AR, Kumar SK, Kunwar AC, Jayaprakash P, Jagannath B. Angew Chem Int Ed. 2003;42:5198–5201. doi: 10.1002/anie.200351267. [DOI] [PubMed] [Google Scholar]; (b) Yadav JS, Reddy BVS, Parimala G, Raju AK. Tetrahedron Lett. 2004;45:1543–1546. [Google Scholar]; (c) Yadav JS, Reddy BVS, Srinivas M, Padmavani B. Tetrahedron. 2004;60:3261–3266. [Google Scholar]; (d) Yadav JS, Reddy BVS, Padmavani B. Synthesis. 2004:405–408. [Google Scholar]; (e) Yadav JS, Reddy BVS, Meraj S, Vishnumurthy P, Narsimulu K, Kunwar AC. Synthesis. 2006:2923–2926. [Google Scholar]; (f) Rafiee E, Azad A. Bioorg Med Chem Lett. 2007;17:2756–2759. doi: 10.1016/j.bmcl.2007.02.070. [DOI] [PubMed] [Google Scholar]; (g) Yadav JS, Subba Reddy BV, Srinivas M, Divyavani Ch, Kunwarb AC, Madavi Ch. Tetrahedron Lett. 2007;48:8301–8305. [Google Scholar]; (h) Narasimhulu M, Reddy SM, Rajesh K, Suryakiran N, Ramesh D, Venkateswarlu Y. Heteroatom Chem. 2008;19:429–433. [Google Scholar]
- 4.Ding C, Tu S, Li F, Wang Y, Yao Q, Hu W, Xie H, Meng L, Zhang A. J Org Chem. 2009;74:6111–6119. doi: 10.1021/jo9011078. [DOI] [PubMed] [Google Scholar]
- 5.Ivashikina NV, Romanov VS, Moroz AA, Shvartsberg MS. Bull Acad Sci USSR, Ser Chem. 1984;33:2345–2348. [Google Scholar]; Izv Akad SSSR, Ser Khim. 1984;33:2261–2265. [Google Scholar]
- 6.(a) Motoyoshiya J, Masue Y, Iwayama G, Yoshioka S, Nishii Y, Aoyama H. Synthesis. 2004:2099–2102. [Google Scholar]; (b) Krohn K, Md Sohrab H, Flörke U. Tetrahedron: Asymmetry. 2004;15:713–718. [Google Scholar]
- 7.(a) Rozeboom MD, Tegmo-Larsson IM, Houk KN. J Org Chem. 1981;46:2338–2345. [Google Scholar]; (b) Pyrek J, St Achmatowicz O, Jr, Zamojski A. Tetrahedron. 1977;33:673–680. [Google Scholar]
- 8.Cameron DW, Feutrill GI, Patti AF. Aust J Chem. 1980;33:1805–1816. [Google Scholar]
- 9.Oda N, Kobayashi K, Ueda T, Ito I. Chem Pharm Bull. 1978;26:2578–2581. [Google Scholar]
- 10.PCMODEL version 8.0 from Serena Software was used. MMX calculations were performed with transition state bond formation of 30%.
- 11.Ghaieni H, Sharifi M, Fattollahy M. Dyes Pigm. 2007;72:97–100. [Google Scholar]
- 12.Mal D, Dey S. Tetrahedron. 2006;62:9589–9602. [Google Scholar]
- 13.Ferrier RJ, Zubkov OA. Org React. 2003;62:569–736. [Google Scholar]
- 14.(a) Anderson LL, Arnold J, Bergman RG. J Am Chem Soc. 2005;127:14542–14543. doi: 10.1021/ja053700i. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marcseková K, Doye S. Synthesis. 2007:145–154. [Google Scholar]; (c) Yin P, Loh TP. Org Lett. 2009;11:3791–3793. doi: 10.1021/ol9012074. [DOI] [PubMed] [Google Scholar]
- 15.(a) Jaunzems J, Kashin D, Schonberger A, Kirschning A. Eur J Org Chem. 2004:3435–3446. [Google Scholar]; (b) Priebe W, Zamojski A. Tetrahedron. 1980;36:287–297. [Google Scholar]
- 16.(a) Rosenblum M. J Am Chem Soc. 1960;82:3796–3798. [Google Scholar]; (b) Srinivasan PS, Mahesh R, Venkateswara Rao G, Kalyanam N. Synth Commun. 1996;26:2161–2164. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.