

Abstract
Drug carrier particles composed of poly(ethylene glycol)-co-poly(sebacic acid) (PEG-PSA) have been shown capable of efficient aerosolization into model lungs and the ability to rapidly penetrate human mucus. Here, we develop PEG-PSA particles (Etop/PEG-PSA) that encapsulate up to 40% etoposide by weight in a one step process, release it continuously for 6 days in vitro, and maintain its cytotoxic activity against a human lung tumor cell line in vitro. We further show that Etop/PEG-PSA injected intratumorally effectively suppress human lung tumor growth in a xenograft mouse model, with 100% survival after 31 days. In contrast, 0% survival was observed by day 24 in animals that received free etoposide (either intratumoral or intraperitoneal administration) or placebo particles intratumorally. These findings support PEG-PSA as a drug delivery platform for improved local therapy of cancer.
Keywords: Drug delivery, Mucus penetrating particles, Cancer, Controlled release, Biodegradable polymers
Introduction
Lung cancer is the leading cause of cancer-related deaths in the United States and throughout the world, with a higher annual death rate than prostate, colon, and breast cancers combined and a dismal 5-year survival rate of 15% [1]. Despite more than 30 years of advancements in cancer detection and treatment, the primary modality of treatment for lung cancer patients remains systemic chemotherapy. The limited effectiveness of systemic chemotherapy for lung cancer has often been attributed to dose-limiting side effects [2, 3]. Local sustained therapy, as could potentially be achieved by inhalation of drug-loaded controlled-release particles, offers the potential to reduce side effects and improve efficacy by maintaining drug concentrations at effective levels for extended time periods within the lungs.
Biodegradable polymer drug carriers, especially polyesters and polyanhydrides, are widely used to encapsulate therapeutics and deliver them in a sustained fashion [4-7]. Polyanhydrides are a class of biodegradable polymers that undergo surface erosion to provide continuous drug release rates that are roughly proportional to polymer erosion rate [7]. Our lab has developed poly(ether-anhydrides) composed of poly(ethylene glycol) and sebacic acid (PEG-PSA) for the delivery of therapeutics following injection [8] or inhalation [9, 10], and demonstrated that PEG-PSA particles can provide sustained release of a variety of molecules [11]. We also recently demonstrated that PEG-PSA nanoparticles diffuse rapidly through undiluted human mucus (Tang et al., unpublished results), a robust barrier that typically traps and quickly removes foreign particles from the lung airways [12, 13]. Here, we sought to test whether PEG-PSA particles could provide efficient encapsulation and sustained release of a relevant chemotherapeutic for the potential treatment of lung cancer. Etoposide, a front line chemotherapeutic drug for lung, ovarian and testicular cancers, was used as a model drug.
Materials and Methods
PEG-PSA synthesis and characterization
PEG-PSA polymer (10% w/w PEG) was synthesized by melt polymerization of poly(ethylene glycol) and sebacic acid prepolymers, as previously described [10]. Weight-average molecular weights were determined relative to polystyrene standards (Fluka, Milwaukee, WI) by gel permeation chromatography [10]. Polymer structure was verified by 1H NMR in CDCl3 (Bruker, Madison, WI) and FT-IR in potassium bromide pellets (Perkin-Elmer, Wellesley, MA). The mass ratio between PEG and SA was estimated by the area ratios of PEG protons to SA protons from NMR. 1H NMR (CDCl3): δ 3.65 (s, OCH2CH2), 2.45 (t, CH2), 1.67 (m, 4H, CH2), 1.33 (m, 8H, CH2). IR (KBr, cm-1): 1807, 1741 (anhydride).
Etoposide particle preparation and characterization
Etoposide (NetQem, Durham, NC) and PEG-PSA were dissolved in chloroform and emulsified into a 1% w/w poly(vinyl alcohol) (Mw = 25 kDa, PolySciences, Warrington, PA) aqueous solution using a Silverson homogenizer at 8,000 RPM for 2 minutes (L4RT, Silverson Machines, East Longmeadow, MA). Particles were hardened by allowing chloroform to evaporate at room temperature while stirring for 2 hr. Particles were collected and washed three times with double distilled water via centrifugation at 2,600 × g for 15 min (International Equipment Co., Needham Heights, MA). Particle size distribution was determined using a Coulter Multisizer IIe (Beckman). Particles were resuspended in double distilled water and added dropwise to 100 ml of ISOTON II diluents (Beckman) until the coincidence of the particles was between 8% and 10%. Greater than 100,000 particles were sized for each batch of microparticles to determine the mean particle size and size distribution. Particle morphology was evaluated by scanning electron microscopy (SEM) using a cold cathode field emission SEM (JEOL JSM-6700F, Peabody, MA). Particles were lyophilized, resuspended in 100% ethanol and dried onto SEM mounts at room temperature. Particles were then sputter coated with platinum (300 s at 1500 amps, Quorum Technologies, Newhaven, England).
Loading and release of etoposide
Etoposide loading into PEG-PSA particles was determined by dissolving particles in chloroform and measuring absorbance at 284 nm in triplicate (Cary 50 Bio Spectrophotometer, Varian, Palo Alto, CA), the maximum absorption of pure etoposide in chloroform [14]. Absorbance of blank particles dissolved in chloroform at the same polymer concentration was subtracted to account for polymer interference. Release kinetics were obtained by suspending 5.0 mg of particles in 1.0 mL phosphate buffered saline (PBS, pH 7.4) and incubating at 37°C on a platform shaker (140 rpm). Supernatant was collected at predetermined intervals by centrifugation at 13,500 × g for 5 minutes (Micro A Marathon centrifuge, Fisher Scientific, Pittsburgh, PA). Drug-containing supernatant was collected and particles were resuspended in 1 mL of fresh PBS. Etoposide concentration in the collected supernatant was assayed via absorbance at 284 nm (Cary 50 Bio Spectrophotometer) in triplicate for each sample (n=3).
Cytotoxicity assay
NCI-H82 human small cell lung cancer cells (ATCC, Manassas, VA) were cultured in RPMI 1640 (Gibco, Carlsbad, CA) supplemented with 10% FBS (Invitrogen, Carlsbad, CA) and penicillin/streptomycin (Sigma) in a humidified environment (37°C, 5% CO2). Cells were seeded into 96 well plates at 1×105 cells per well and treated for 5 days before assaying viability with a colorimetric 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma). Briefly, 50 μl of 2 mg/ml MTT in PBS was added to each well and incubated at 37°C for 2 hr. Cells were collected (92 × g for 5 min), dissolved in 200 μl DMSO and absorbance at 570 nm was measured, n=6 wells per condition (SpectraMax Plus Spectrophotometer, Molecular Devices, Sunnyvale, CA). Controls included untreated cells (no cell death) and wells containing only media (complete cell death), n=6 wells per condition. Cell viability was determined from a linear fit between the absorbance of the two controls. Released etoposide was collected from particles as described above and lyophilized before dissolving in whole media and adding to cells. Stock etoposide was prepared by dissolving etoposide directly in whole media.
Animals
Animal protocols were approved by the Institutional Animal Care and Use Committee at The Johns Hopkins Medical Institutions. Four to six week old female Hsd:Athymic Nude-Foxn1nu mice (Harlan, Indianapolis, IN) were allowed to acclimate for 1 week prior to handling. Animals were injected subcutaneously in the right flank with 1×106 NCI-H82 cells suspended in 50% Matrigel (BD Biosciences, San Jose, CA) and tumors were allowed to establish to 6-7 mm in diameter before treatment [15]. Etoposide was administered locally (intratumoral, IT) and systemically (intraperitoneal, IP) at 12 mg/kg/day in PBS for 3 consecutive days. Etoposide-loaded polymer particles and placebo (blank) particles were injected once intratumorally after resuspension in PBS at 36 mg/kg or equivalent polymer load for placebo particles. Tumor sizes in two dimensions were measured with digital calipers, and volumes were calculated with the formula (0.5 × L × W × W), where L is length and W is width [16]. Tumor volumes were normalized by initial tumor volume. The Kaplan-Meier log-rank test was used to compare survival between mice that received Etop/PEG-PSA treatment versus those that received placebo particles or free drug. A p value less than 0.05 was considered statistically significant.
Results and Discussion
PEG-PSA synthesis and characterization
Poly(ethylene glycol)-co-poly(sebacic acid) (PEG-PSA) copolymers were prepared via melt polycondensation of prepolymers of sebacic acid and polyethylene glycol. The resulting polymer was characterized by 1H NMR and FT-IR spectroscopy. The NMR spectra exhibited a single peak at 3.65 ppm, indicative of the presence of PEG in the polymer, and peaks at 2.45, 1.67, and 1.33 ppm corresponding to sebacic anhydride. The weight fraction of PEG in the polymer (10.2%) matched the feed fraction of 10.0%, as determined by the area ratios of PEG to SA protons. The typical anhydride IR double peaks appeared at ~1807 and 1741 cm-1. The weight-average molecular weight was 18 kDa (determined by GPC), with no peak corresponding to the weight of free PEG, thereby confirming PEG incorporation into the polymer.
Loading and release kinetics of etoposide
Etoposide was loaded into spherical PEG-PSA particles (Etop/PEG-PSA, Figure 1A) by a single oil/water emulsion method [17]. Etop/PEG-PSA particles possessed rough surfaces, which has been correlated with improved aerosolization efficiency of PEG-PSA particles compared to particles with smooth surfaces (due to decreased particle-particle contact area, leading to reduced interparticle adhesive forces) [11]. Particles possessed fairly narrow size distributions (Figure 1B), with a mass mean diameter of 1.8 μm for both polymer-only (placebo) and etoposide-loaded particles. The geometric standard deviation (GSD) of particle size was calculated for Etop/PEG-PSA and placebo particles as described previously [11]. GSD=1.7 for both Etop/PEG-PSA and placebo particles, which is within the typical range for aerosol particles (1.3-3.0) [18, 19]. High etoposide encapsulation efficiency (71% of initial drug was encapsulated into particles) and high drug loading (41% of particle mass was drug) were achieved.
Figure 1.

(A) Scanning electron micrographs of etoposide-loaded particles (Etop/PEG-PSA); bars represent 1 μm. (B) size distribution by volume of blank particles (○) and etoposide loaded particles (●).
Poly(lactic-co-glycolic acid) (PLGA) and polycaprolactone (PCL) microspheres (~45 μm) can entrap etoposide with high efficiency, however, usually only at low drug:polymer ratios [20, 21]. Low drug:polymer ratios (e.g., 5% w/w drug) result in encapsulation efficiencies >80% in PLGA and PCL, whereas encapsulation efficiency decreases as the drug:polymer ratio is increased (<30% encapsulation efficiency for formulations of 33% w/w drug:polymer in feed) [21]. Lipid nanocarriers composed of hydroxystearates and glycerides have also been used to entrap etoposide, with particle sizes ranging from 27 to 400 nm [20, 22]. Reported encapsulation efficiencies for these lipid nanocarriers are high (>85%), but the drug loading is low, ranging only from 0.1% to 5%.
In addition to efficiently encapsulating drug with high loading, drug delivery systems must be able to release active drug in a controlled manner in order to achieve improved efficacy. Controlled release formulations have the potential to maintain drug concentrations within the therapeutic window, which can increase efficacy and minimize harmful side effects while protecting the drug against hydrolytic and enzymatic degradation [23]. PEG-PSA particles released encapsulated etoposide in vitro at physiological pH and 37°C in a continuous fashion over 6 days with no burst release (Figure 2). Several other chemotherapeutics, including doxorubicin, gemcitabine and paclitaxel were also efficiently encapsulated and released in a sustained and controlled manner from PEG-PSA particles (data not shown). Etoposide release occurred as a result of polymer erosion; no particles remained after etoposide was completely released. Released etoposide exhibited equivalent cytotoxicity compared to freshly prepared drug in vitro against NCI-H82, a human small cell lung cancer cell line (Figure 3). Differences in cytotoxicity between freshly prepared etoposide and released etoposide were determined to be statistically insignificant. Placebo PEG-PSA particles and degraded polymer were not toxic to cells (data not shown).
Figure 2.

Etoposide release kinetics from PEG-PSA particles. Etoposide release was obtained by suspending particles in PBS (pH 7.4), incubating at 37°C, and assaying supernatant at various time points using UV absorbance. Error bars representing standard error (n=3) are smaller than the symbol.
Figure 3.

Cytotoxicity of released etoposide (●) compared to freshly prepared stock etoposide (○) on H82 cells. H82 cells were exposed to treatment for 5 days before assaying with MTT for viability. Difference between treatments is not statistically significant at any concentration (p > 0.05 by Students t-test). Error bars represent standard error (n=6).
Controlled release of etoposide (half life of ~4 hr [24]) has been demonstrated with PLGA and PCL microspheres designed for intramuscular depot administration (~45 μm) [21]. With low etoposide loading (<5%), the drug could be released over a period spanning several weeks. However, these formulations were not designed for potential inhalation (they are too large) and they provided an initial burst of up to 14% of the drug load within the first day. Polyesters, such as PLGA and PCL, typically degrade on the order of weeks to years, but release drugs in shorter time frames, which can lead to undesired accumulation of polymer upon repeat administration [25-27]. Second, an acidic core can develop in PLGA particles as they undergo bulk degradation [28], which accelerates the degradation of pH sensitive drugs. PEG-PSA particles undergo surface erosion as a result of their highly water labile anhydride bonds and high diffusion rates of soluble fragments from the particle, which lessens the effect of acidic build-up [29, 30]. Particle erosion times for polyanhydrides can be tuned from hours to weeks, with steady release of drug as the particle erodes [9].
Efficacy in lung tumor mouse model
We next tested the efficacy of Etop/PEG-PSA particles in a subcutaneous xenograft model of an aggressive human small cell lung cancer (NCI-H82). NCI-H82 small cell lung cancer cells were inoculated ~14 days prior to treatment and animals were treated when tumor volume reached ~50 mm3 (Day 0). Treatment groups included: (i) single intratumoral injection of Etop/PEG-PSA particles, (ii) single intratumoral injection of blank particles (negative control), (iii) multiple intraperitoneal injections of free drug (systemic delivery, positive control) and (iv) multiple intratumoral injections of free drug (local delivery, positive control). A single intratumoral dose of Etop/PEG-PSA particles reversed tumor growth and significantly reduced the volume of established tumors by Day 3, and then suppressed tumor growth for over 20 days (Figure 4). During this period, the tumor volume remained at or below its size at the time of treatment. In contrast, fractionated dosing of etoposide (daily for 3 days) locally or systemically (gold standard positive control) did not suppress tumor growth appreciably. Likewise, tumor size was not affected by treatment with blank PEG-PSA particles administered intratumorally. All animals receiving Etop/PEG-PSA treatment survived at least 31 days, whereas all animals in control groups required humane sacrifice by day 24 (Figure 5). The median survival times increased from 11-16 days for local or systemic delivery of free etoposide to 47 days for mice receiving a single Etop/PEG-PSA bolus treatment (Table 1).
Figure 4.
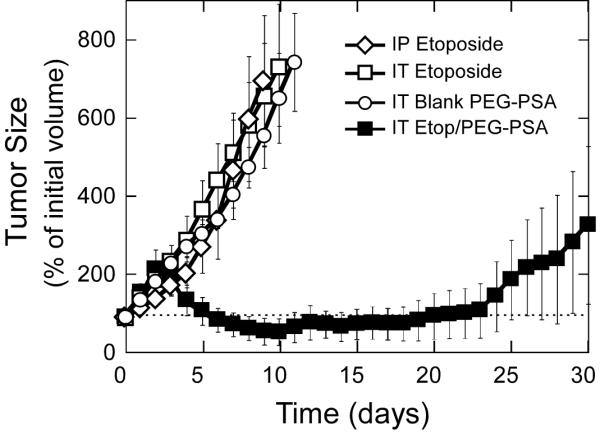
Response of subcutaneous H82 xenograft in nude mice to treatment. 5-6 animals per treatment group. IT - intratumoral; IP - intraperitoneal. Particle mass-mean diameter is 1.5 μm. Tumor inoculation was on day 0; non-encapsulated etoposide was administered on days 0, 1, and 2. Polymer particles administered once, on day 0. IT etoposide particles provide significantly improved tumor control (p<0.05) compared to other treatments starting day 4.
Figure 5.
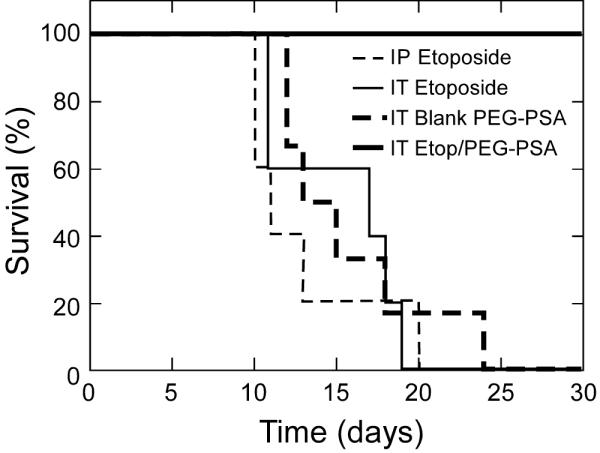
Kaplan-Meier curves for survival of mice bearing subcutaneous H82 xenografts. 5-6 animals per treatment set. IT - intratumoral; IP - intraperitoneal. Particle mass-mean diameter is 1.5 μm. Animals were sacrificed when tumor volume reached 1 cm3 per guidelines for humane treatment.
Table 1.
Median survival times from date of human small cell lung tumor inoculation. Free etoposide was administered on days 0, 1, and 2, whereas particles were administered on day 0 only. Mice were sacrificed when tumors reached 1 cm3
| Treatment | Administration Route# | Median survival time (days) | n |
|---|---|---|---|
| Blank PEG-PSA particles | IT | 13 | 5 |
| Free etoposide | IT | 16 | 5 |
| Free etoposide | IP | 11 | 5 |
| Etoposide in PEG-PSA particles | IT | 47* | 6 |
IT = intratumoral; IP = intraperitoneal
statistically significant compared to other treatments (p < 0.05).
The substantial improvement in suppressing tumor growth is likely due to sustained concentrations of etoposide at the tumor site achieved with Etop/PEG-PSA particles. Despite multiple dosing, free etoposide is rapidly absorbed systemically and thus cleared from the tumor site, leading to inadequate drug exposure and negligible tumor shrinkage. In contrast, the continuous drug release provided by Etop/PEG-PSA maintains effective drug levels within the tumor over periods long enough to attack tumor cells as they asynchronously undergo DNA synthesis, the point in the cell cycle at which etoposide exerts cytotoxic activity [31]. Despite the prolonged suppression of tumor growth, a single dose of Etop/PEG-PSA did not mediate complete tumor eradication, as evident by the re-proliferation of tumor cells after Day 25. Tumor relapse may be attributed to incomplete killing of the existing tumor due to insufficient dosing, incomplete distribution within the tumor, or to a subpopulation of etoposide-resistant tumor cells. Repeated administration of drug loaded particles may lead to prolonged suppression or possibly tumor eradication, but was not tested here.
In addition to high drug loading and sustained release kinetics, PEG-PSA may offer a number of advantages for pulmonary delivery. First, PEG-PSA particles have been shown to efficiently aerosolize into an Anderson Cascade Impactor model of the lungs [11]. The hydrophilic nature of PEG enables it to partition to the particle surface during particle formulation [8, 11, 32], which decreases interparticle adhesion leading to improved deaggregation upon aerosolization compared to particles that do not contain PEG [11]. Second, mucus barriers efficiently trap and remove most foreign particles from the eyes, respiratory tract, gastrointestinal tract, and female reproductive tract [33]. Particles are typically immobilized upon contact with mucus by forming adhesive interactions with the hydrophobic “naked protein” domains on mucins, leading to their clearance from the mucosal organ on the order of seconds in the eyes to a few hours in the lungs or gastrointestinal tract [34]. Recently, we demonstrated that a dense coating of low MW PEG on latex nanoparticle surfaces effectively minimizes particle-mucus adhesive interactions and, thereby, allows particles as large as 500 nm to diffuse rapidly through human mucus [35, 36]. We then showed that PEG-PSA particles also possess a dense enough surface PEG coating to allow them to readily diffuse in fresh human mucus at rates comparable to densely coated latex beads; PEG-PSA nanoparticles diffused >250-fold faster than nanoparticles composed of PLGA or PSA (Tang et al., unpublished). Efficient aerosolization and the ability to diffuse through mucus barriers may allow PEG-PSA particles to provide prolonged local release of drugs in the lung airways.
An orthotopic lung cancer model provides the most ideal setting to evaluate local sustained therapies for lung cancer, with physiologically relevant barriers against efficient and effective pulmonary delivery as well as appropriate tumor-stroma interactions for cancer growth [37, 38]. Non-small cell lung cancer represents the majority of current orthotopic lung cancer models [39, 40]; however, the tumors establish in the lower airways, where the mucus barrier is typically not present. In contrast, small cell lung cancer (SCLC) predominantly establishes in the major airways where access to the tumor is limited by the mucus barrier [41]. Thus, an orthotopic small cell lung cancer model would be ideal for the evaluation of potential inhaled sustained therapies; unfortunately, such SCLC models do not exist in the literature. Future studies are aimed at encapsulating etoposide within mucus-penetrating PEG-PSA nanoparticles for testing against an orthotopic SCLC tumor model recently developed by our labs (unpublished).
The potential benefits of localized therapies have led to increased testing of inhalation-mediated delivery of chemotherapeutics to the lungs. For example, the inhalation of free doxorubicin, paclitaxel and 5-fluorouracil in dogs and lung cancer patients all demonstrated reduced systemic toxicity and minimal local toxicity compared to systemic administration [42, 43]. In a preliminary study, 6 of 10 advanced stage lung cancer patients responded positively to inhaled 5-fluorouracil, with two complete responses [43]. In contrast, the highest response rates of advanced lung cancer patients with systemic therapy using cytostatic agents such as 5-fluorouracil and doxorubicin lie around 20% [44]. However, the efficacy of inhalation of free drug is limited by rapid absorption and mucus clearance [45], so inhalation therapy of lung cancer may be improved in the future by sustained drug release from mucus penetrating particle systems.
Conclusion
PEG-PSA particles were shown capable of efficient encapsulation and sustained delivery of etoposide in a fashion that suppressed growth of a lung cancer xenograft in mice. Drug-loaded PEG-PSA nanoparticles offer a potential new method for localized sustained drug delivery to treat lung cancer.
Acknowledgements
The authors thank Dr. Samuel Lai for expert advice and assistance in preparation of the manuscript. This work was supported in part by the NIH R21HL089816 (J.H), Flight Attendant Medical Research Institute (N.W), and NSF GK-12 BIGSTEP Fellowship (B.C.T). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute, or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Thun MJ, Ries LA, Howe HL, Weir HK, Center MM, et al. Annual report to the nation on the status of cancer, 1975-2005, featuring trends in lung cancer, tobacco use, and tobacco control. J Natl Cancer Inst. 2008;100(23):1672–1694. doi: 10.1093/jnci/djn389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol. 1991;9(3):491–498. doi: 10.1200/JCO.1991.9.3.491. [DOI] [PubMed] [Google Scholar]
- 3.Otterson GA, Villalona-Calero MA, Sharma S, Kris MG, Imondi A, Gerber M, et al. Phase I study of inhaled Doxorubicin for patients with metastatic tumors to the lungs. Clin Cancer Res. 2007;13(4):1246–1252. doi: 10.1158/1078-0432.CCR-06-1096. [DOI] [PubMed] [Google Scholar]
- 4.Winet H, Hollinger JO, Stevanovic M. Incorporation of polylactide-polyglycolide in a cortical defect: neoangiogenesis and blood supply in a bone chamber. J Orthop Res. 1995;13(5):679–689. doi: 10.1002/jor.1100130507. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa Y, Okada H, Yamamoto M, Shimamoto T. In vivo release profiles of leuprolide acetate from microcapsules prepared with polylactic acids or copoly(lactic/glycolic) acids and in vivo degradation of these polymers. Chem Pharm Bull (Tokyo) 1988;36(7):2576–2581. doi: 10.1248/cpb.36.2576. [DOI] [PubMed] [Google Scholar]
- 6.Menei P, Daniel V, Montero-Menei C, Brouillard M, Pouplard-Barthelaix A, Benoit JP. Biodegradation and brain tissue reaction to poly(D,L-lactide-co-glycolide) microspheres. Biomaterials. 1993;14(6):470–478. doi: 10.1016/0142-9612(93)90151-q. [DOI] [PubMed] [Google Scholar]
- 7.Kumar N, Langer RS, Domb AJ. Polyanhydrides: an overview. Adv Drug Deliv Rev. 2002;54(7):889–910. doi: 10.1016/s0169-409x(02)00050-9. [DOI] [PubMed] [Google Scholar]
- 8.Deosarkar SP, Malgor R, Fu J, Kohn LD, Hanes J, Goetz DJ. Polymeric particles conjugated with a ligand to VCAM-1 exhibit selective, avid, and focal adhesion to sites of atherosclerosis. Biotechnol Bioeng. 2008;101(2):400–407. doi: 10.1002/bit.21885. [DOI] [PubMed] [Google Scholar]
- 9.Fu J, Fiegel J, Hanes J. Synthesis and characterization of PEG-based ether-anhydride terpolymers: Novel polymers for controlled drug delivery. Macromolecules. 2004;37(19):7174–7180. [Google Scholar]
- 10.Fu J, Fiegel J, Krauland E, Hanes J. New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials. 2002;23(22):4425–4433. doi: 10.1016/s0142-9612(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 11.Fiegel J, Fu J, Hanes J. Poly(ether-anhydride) dry powder aerosols for sustained drug delivery in the lungs. J Control Release. 2004;96(3):411–423. doi: 10.1016/j.jconrel.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Contreras L, Hickey AJ. Aerosol treatment of cystic fibrosis. Crit Rev Ther Drug Carrier Syst. 2003;20(5):317–356. doi: 10.1615/critrevtherdrugcarriersyst.v20.i5.10. [DOI] [PubMed] [Google Scholar]
- 13.Laube BL. The expanding role of aerosols in systemic drug delivery, gene therapy, and vaccination. Respir Care. 2005;50(9):1161–1176. [PubMed] [Google Scholar]
- 14.Oh KT, Bronich TK, Bromberg L, Hatton TA, Kabanov AV. Block ionomer complexes as prospective nanocontainers for drug delivery. J Control Release. 2006;115(1):9–17. doi: 10.1016/j.jconrel.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 15.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422(6929):313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 16.Park BH, Vogelstein B, Kinzler KW. Genetic disruption of PPARdelta decreases the tumorigenicity of human colon cancer cells. Proc Natl Acad Sci U S A. 2001;98(5):2598–2603. doi: 10.1073/pnas.051630998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 18.Ferin J, Oberdorster G, Penney DP. Pulmonary retention of ultrafine and fine particles in rats. Am J Respir Cell Mol Biol. 1992;6(5):535–542. doi: 10.1165/ajrcmb/6.5.535. [DOI] [PubMed] [Google Scholar]
- 19.O’Hara P, Hickey AJ. Respirable PLGA microspheres containing rifampicin for the treatment of tuberculosis: manufacture and characterization. Pharm Res. 2000;17(8):955–961. doi: 10.1023/a:1007527204887. [DOI] [PubMed] [Google Scholar]
- 20.Lamprecht A, Benoit JP. Etoposide nanocarriers suppress glioma cell growth by intracellular drug delivery and simultaneous P-glycoprotein inhibition. J Control Release. 2006;112(2):208–213. doi: 10.1016/j.jconrel.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Vivek K, Reddy LH, Murthy RS. Comparative study of some biodegradable polymers on the entrapment efficiency and release behavior of etoposide from microspheres. Pharm Dev Technol. 2007;12(1):79–88. doi: 10.1080/10837450601168581. [DOI] [PubMed] [Google Scholar]
- 22.Reddy LH, Murthy RS. Etoposide-loaded nanoparticles made from glyceride lipids: formulation, characterization, in vitro drug release, and stability evaluation. AAPS PharmSciTech. 2005;6(2):E158–166. doi: 10.1208/pt060224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428(6982):487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 24.Beijnen JH, Holthuis JJM, Kerkdijk HG, Vanderhouwen OAGJ, Paalman ACA, Bult A, et al. Degradation Kinetics of Etoposide in Aqueous-Solution. Int J Pharm. 1988;41(12):169–178. [Google Scholar]
- 25.Batycky RP, Hanes J, Langer R, Edwards DA. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J Pharm Sci. 1997;86(12):1464–1477. doi: 10.1021/js9604117. [DOI] [PubMed] [Google Scholar]
- 26.Shenoy D, Little S, Langer R, Amiji M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs. 1 In vitro evaluations. Mol Pharm. 2005;2(5):357–366. doi: 10.1021/mp0500420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pitt CG, Chasalow FI, Hibionada YM, Klimas DM, Schindler A. Aliphatic Polyesters .1. The Degradation of Poly(Epsilon-Caprolactone) In vivo. J Appl Polym Sci. 1981;26(11):3779–3787. [Google Scholar]
- 28.Zhu G, Mallery SR, Schwendeman SP. Stabilization of proteins encapsulated in injectable poly (lactide- co-glycolide) Nat Biotechnol. 2000;18(1):52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 29.Shieh L, Tamada J, Chen I, Pang J, Domb A, Langer R. Erosion of a new family of biodegradable polyanhydrides. J Biomed Mater Res. 1994;28(12):1465–1475. doi: 10.1002/jbm.820281212. [DOI] [PubMed] [Google Scholar]
- 30.Gopferich A, Tessmar J. Polyanhydride degradation and erosion. Adv Drug Deliv Rev. 2002;54(7):911–931. doi: 10.1016/s0169-409x(02)00051-0. [DOI] [PubMed] [Google Scholar]
- 31.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 32.Sakhalkar HS, Dalal MK, Salem AK, Ansari R, Fu J, Kiani MF, et al. Leukocyte-inspired biodegradable particles that selectively and avidly adhere to inflamed endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2003;100(26):15895–15900. doi: 10.1073/pnas.2631433100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2008;61(2):75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang YY, Cone R, et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A. 2007;104(5):1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang YY, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew Chem Int Ed. 2008;47(50):9726–9729. doi: 10.1002/anie.200803526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39(1):29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- 38.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6(6):425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 39.Onn A, Isobe T, Itasaka S, Wu W, O’Reilly MS, Ki Hong W, et al. Development of an orthotopic model to study the biology and therapy of primary human lung cancer in nude mice. Clin Cancer Res. 2003;9(15):5532–5539. [PubMed] [Google Scholar]
- 40.McLemore TL, Liu MC, Blacker PC, Gregg M, Alley MC, Abbott BJ, et al. Novel intrapulmonary model for orthotopic propagation of human lung cancers in athymic nude mice. Cancer Res. 1987;47(19):5132–5140. [PubMed] [Google Scholar]
- 41.Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113(1):5–21. doi: 10.1002/cncr.23542. [DOI] [PubMed] [Google Scholar]
- 42.Hershey AE, Kurzman ID, Forrest LJ, Bohling CA, Stonerook M, Placke ME, et al. Inhalation chemotherapy for macroscopic primary or metastatic lung tumors: proof of principle using dogs with spontaneously occurring tumors as a model. Clin Cancer Res. 1999;5(9):2653–2659. [PubMed] [Google Scholar]
- 43.Tatsumura T, Koyama S, Tsujimoto M, Kitagawa M, Kagamimori S. Further study of nebulisation chemotherapy, a new chemotherapeutic method in the treatment of lung carcinomas: fundamental and clinical. Br J Cancer. 1993;68(6):1146–1149. doi: 10.1038/bjc.1993.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorensen JB, Clerici M, Hansen HH. Single-agent chemotherapy for advanced adenocarcinoma of the lung. A review. Cancer Chemother Pharmacol. 1988;21(2):89–102. doi: 10.1007/BF00257354. [DOI] [PubMed] [Google Scholar]
- 45.Patton JS, Fishburn CS, Weers JG. The lungs as a portal of entry for systemic drug delivery. Proc Am Thorac Soc. 2004;1(4):338–344. doi: 10.1513/pats.200409-049TA. [DOI] [PubMed] [Google Scholar]