

Abstract
Palytoxin is classified as a non-12-O-tetradecanoylphorbol-13-acetate (TPA)-type skin tumor because it does not bind to or activate protein kinase C. Palytoxin is thus a novel tool for investigating alternative signaling pathways that may affect carcinogenesis. We previously showed that palytoxin activates three major members of the mitogen activated protein kinase (MAPK) family, extracellular signal regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38. Here we report that palytoxin also activates another MAPK family member, called ERK5, in HeLa cells and in keratinocytes derived from initiated mouse skin (308 cells). By contrast, TPA does not activate ERK5 in these cell lines. The major cell surface receptor for palytoxin is the Na+,K+-ATPase. Accordingly, ouabain blocked the ability of palytoxin to activate ERK5. Ouabain alone did not activate ERK5. ERK5 thus represents a divergence in the signaling pathways activated by these two agents that bind to the Na+,K+-ATPase. Cycloheximide, okadaic acid, and sodium orthovandate did not mimic the effect of palytoxin on ERK5. These results indicate that the stimulation of ERK5 by palytoxin is not simply due to inhibition of protein synthesis or inhibition of serine/threonine or tyrosine phosphatases. Therefore, the mechanism by which palytoxin activates ERK5 differs from that by which it activates ERK1/2, JNK, and p38. Finally, studies that used pharmacological inhibitors and shRNA to block ERK5 action indicate that ERK5 contributes to palytoxin-stimulated c-Fos gene expression. These results suggest that ERK5 can act as an alternative mediator for transmitting diverse tumor promoter-stimulated signals.
Keywords: Mitogen activated protein kinase; extracellular signal regulated kinase 5; palytoxin; Na+,K+-ATPase
Introduction
Agents identified as tumor promoters in the classic multi-stage mouse skin model of carcinogenesis have helped reveal how the perturbation of signaling pathways contributes to the development of cancer (Yuspa, 1998). Initiation, the first stage of carcinogenesis in this model, typically involves one treatment with a genotoxic agent and is characterized by activation of the oncogene Ras (Balmain and Pragnell, 1983). Tumor promotion, the second stage, involves repeated stimulation over a prolonged period by agents that are typically non-genotoxic, and results in the development of tumors. Interestingly, whereas initiation is irreversible, tumor promotion is reversible if treatment is ceased. This suggests that investigating the action of tumor promoters may aid the development of strategies to block carcinogenesis. The identification of protein kinase C as the receptor for the prototypical skin tumor promoter 12-O- tetradecanoylphorbol-13-acetate (TPA, also called PMA) helped establish that tumor promotion involves the subversion of signal transduction pathways (Nishizuka, 1984). The subsequent identification of non-TPA-type tumor promoters, which do not activate protein kinase C in vitro or require protein kinase C for signal transduction, indicated that protein kinase C-independent signaling pathways also play a role in carcinogenesis (Fujiki et al., 1986; Wattenberg et al., 1987). Accordingly, our laboratory has used the non-TPA type tumor promoter palytoxin to investigate alternative signaling pathways that may play a role in carcinogenesis, but may have been missed by the historical focus on TPA-stimulated signaling.
The major cell surface receptor for palytoxin is the Na+,K+-ATPase (Habermann, 1989). Palytoxin, a large (Mr 2,681) water-soluble polyalcohol, which is isolated from zoanthids (genus Palythoa) (Moore, 1985), binds to the Na+,K+-ATPase and transforms the sodium pump into an ion channel. Palytoxin binding thus typically triggers sodium influx and potassium efflux. Our previous studies indicated that mitogen activated protein kinases (MAPKs) can mediate palytoxin-stimulated signaling (Kuroki et al., 1997; Li and Wattenberg, 1999; Warmka et al., 2002; Warmka et al., 2004).
MAPKs are a family of serine/threonine kinases that transmit a wide variety of signals to the cellular machinery that regulates gene expression, cell fate, and cell function (reviewed in (Turjanski et al., 2007)). Several studies also indicate that aberrant regulation of MAPKs plays a role in carcinogenesis (Dhillon et al., 2007). The three groups of MAPKs that have been studied most extensively are the extracellular signal regulated kinases 1 and 2 (ERK1/2), the c-Jun N-terminal kinases (JNKs) and the p38s. In general ERK1/2 tends to be activated by mitogenic agents, whereas JNK and p38 are typically activated by stress (Turjanski et al., 2007). Our work indicates that MAPKs can mediate the convergence of the different signaling pathways stimulated by palytoxin and TPA, thus providing a mechanism by which these different types of tumor promoters can regulate common biochemical targets that play an important role in carcinogenesis (Warmka et al., 2002; Warmka et al., 2004). For example, we found that palytoxin and TPA both increase ERK1/2 activity, although by different mechanisms, in mouse keratinocytes that are derived from initiated mouse skin and express oncogenic Ras (308 cells); this results in the ability of palytoxin and TPA to regulate common downstream nuclear targets, including the transcription factor AP-1 (Warmka et al., 2002; Warmka et al., 2004; Zeliadt et al., 2004).
ERK5 (also called Big MAP kinase 1 or BMK1) is a MAPK family member that is likely to play a role carcinogenesis, but has not been studied as extensively as ERK1/2, JNK, and p38 (reviewed in (Wang and Tournier, 2006)). Like other MAPKs, activation of ERK5 requires dual phosphorylation on specific threonine and tyrosine residues (Mody et al., 2003). MAPKs are typically activated by a protein kinase cascade, such that a MAPK kinase kinase (MAPKKK or MEKK), phosphorylates and activates a MAPK kinase (MAPKK or MEK), which phosphorylates and activates a MAPK (Wang and Tournier, 2006). For example, Raf is a MAPKKK that phosphorylates and activates MEK1/2, which phosphorylates and activates ERK1/2 (reviewed in (Dhillon et al., 2007)). MEKK2 and MEKK3 have been identified as MAPKKKs, which phosphorylate and activate MEK5, the MAPKK that phosphorylates and activates ERK5 (Chao et al., 1999; Sun et al., 2001). A unique, large C-terminal non-kinase domain makes ERK5 approximately twice the size of ERK1/2 (Lee et al., 1995; Zhou et al., 1995). This C-terminal domain contains a nuclear localization signal (Hayashi and Lee, 2004). ERK5, like other MAPK family members, can translocate from the cytoplasm to the nucleus, where it can regulate transcription factors, including MEF2C (myocyte enhancer factor), Sap1a, and c-Fos (Kato et al., 1997; Kamakura et al., 1999; Terasawa et al., 2003). ERK5 is activated by mitogens, such as epidermal growth factor (EGF), and also by stress, such as osmotic shock and oxidative stress (Abe et al., 1996; Kato et al., 1998). Because palytoxin causes a type of osmotic stress by stimulating ion influx, we wanted to determine whether ERK5 mediates palytoxin-induced signals.
We used two cell culture models to determine whether ERK5 is involved in palytoxin signaling. We used 308 mouse keratinocytes, which were derived from mouse skin initiated in vivo with 7,12-dimethylbenz(a)anthracene and express endogenous oncogenic Ras (Strickland et al., 1988). This is thus an excellent model for studying tumor promoter action. We also used HeLa cells, a human cervical cancer cell line that has been used extensively to study MAPK signaling. HeLa cells, in contrast to keratinocytes, are relatively easy to transfect. This makes HeLa cells very useful for the study of signaling by methods that require the introduction of expression vectors. Our results indicate that ERK5 can mediate the transmission of palytoxin-stimulated signals from the Na+,K+-ATPase to the nucleus. In contrast to palytoxin, we did not detect activation of ERK5 by the prototypical phorbol ester tumor promoter TPA. Altogether, these studies indicate that ERK5 represents an alternative pathway through which diverse tumor promoters can transmit signals.
Materials and methods
Materials
Palytoxin was purchased from the Hawaii Biotechnology Group, Inc. (Aiea, HI). U0126 was purchased from Calbiochem (La Jolla, CA). High glucose Dulbecco’s Modified Eagle Medium, Minimum Essential Medium, and fetal bovine serum were purchased from Invitrogen Corporation (Carlsbad, CA). TPA, cycloheximide, EGF, protein G agarose beads, ouabain, sodium orthovanadate, okadaic acid, phenylmethanesulphonylfluoride (PMSF), NaF, β-glycerophosphate, aprotinin, leupeptin and sorbitol were purchased from Sigma (St. Louis, MO). Calf intestinal alkaline phosphatase was purchased from New England Biolabs (Ipswich, MA).
Cell Culture
HeLa cells were the generous gift of Dr. Audrey Minden (Susan Lehman Cullman Laboratory for Cancer Research, Department of Chemical Biology, Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey), and were grown as described in (Li and Wattenberg, 1998). 308 cells were the generous gift of Dr. Stuart H. Yuspa (Laboratory of Cellular Carcinogenesis and Tumor Promotion, National Cancer Institute), and were grown as described in (Warmka et al., 2004). For experiments, HeLa and 308 cells were plated at a density of approximately 2.4 × 104 cells/cm2 and 5.6 × 104 cells/cm2, respectively. The following day, the cells were switched to serum free media. The cells were incubated in serum free media for approximately 24 hours before treatment, unless otherwise indicated.
Antibodies and immunoblotting
Cell lysates were prepared using the following buffer unless otherwise noted: 50 mM Tris-HCl, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 20 mM β-glycerophosphate, 1 mM Na3VO4, 1mM NaF. Lysates were cleared by centrifugation (16,000 × g, 10 min, 4°C). 20–40 μg of protein was resolved using either 7.5% or 10% SDS-polyacrylamide minigels, and then transferred to Immobilon-P PVDF membrane (Millipore, Bedford, MA). After blocking in either a TBST/5% milk solution or TBST/3% BSA solution, immunoblots were incubated overnight at 4°C using the following primary antibodies and dilutions: phospho-p44/42 MAPK (Thr-202/Tyr-204) (E10) (mouse monoclonal) (1:2000), phospho-ERK5 (Thr-218/Tyr-220) (rabbit polyclonal) (1:1000), ERK5 (rabbit polyclonal) (1:1000), phospho-p38 MAPK (Thr-180/Tyr-182) (rabbit polyclonal) (1:2000), and phospho-SAPK/JNK (Thr-183/Tyr 185) (G9) (mouse monoclonal) (1:2000) from Cell Signaling (Beverly, MA), and ERK2 (C-14) (rabbit polyclonal) (1:2000), JNK1 (FL) (rabbit polyclonal) (1:2000), p38 (C-20) (rabbit polyclonal) (1:2000) from Santa Cruz Biotechnology (Santa Cruz, CA). The following secondary antibodies were used: anti-mouse IgG horseradish peroxidase-linked antibody and anti-rabbit IgG horseradish peroxidase-linked antibody from Cell Signaling. Immunoblots were visualized using the Pierce SuperSignal West Pico substrate.
Densitometry
Protein bands from immunoblots were quantified using a Bio-Rad (Hercules, CA) Fluor-S MultiImager and Bio-Rad Quantity One software.
Quantitative real-time PCR analysis
Total RNA was harvested from cells using the RNeasy Plus Mini Kit from Qiagen (Valencia, CA). RNA quality was verified spectrophotometrically using the A260/A280 ratio. One microgram of RNA was reverse-transcribed using the SuperScript III from Invitrogen (Carlsbad, CA). Negative controls that lacked RNA or reverse transcriptase were included for each primer pair. cDNA was diluted 1:10, and 2 μl of diluted cDNA were used in 25 μl PCR reactions with 200 nM of each primer and SYBR GreenER qPCR SuperMix for iCycler (Invitrogen). PCR was performed on a BioRad iCycler iQ5 using the following primers: human c-Fos (accession no. NM_005252) forward 5′-CGGGCTTCAACGCAGACTA-3′, reverse 5′-CTGGTCGAGATGGCAGTGA-3′; human GAPDH (accession no. NM_0020467) forward 5′-GGGAAGGTGAAGGTCGGAGT-3′, reverse 5′-GAGTTAAAAGCAGCCCTGGTGA-3′; mouse c-Fos (accession no. NM_010234) forward 5′-GGGGCAAAGTAGAGCAGCTA-3′, reverse 5′-GGCTGCCAAAATAAACTCCA-3′; mouse GAPDH (accession no. NM_008084) forward 5′-ATTGTCAGCAATGCATCCTG-3′, reverse 5′-ATGGACTGTGGTCATGAGCC-3′. The amplification program was 50°C for 2 minutes, 95°C for 8 min 30 sec, and 40 cycles of 95°C for 15 s, 60°C for 60 s. A melt curve analysis was performed on all amplification products, revealing a single peak and thus ensuring that products were specific. The software default values for baseline and threshold Ct values were used. The amplification efficiency of each primer pair was tested as described in (Schmittgen and Livak, 2008), and all efficiencies were approximately equal. Amplifications of unknowns were carried out within the dynamic range of each assay. The Ct range of target detection was 17–33. No PCR products were observed in negative controls. Data were analyzed using the comparative Ct method (Schmittgen and Livak, 2008) with GAPDH serving as the internal reference gene. 8
ERK5 knockdown
An ERK5 shRNA clone (V2LHS 202701) and a non-silencing control (RHS4346) were purchased from the Open Biosystems GIPZ Lentiviral shRNAmir library (Huntsville, AL). The lentiviruses were packaged in the 293-FT cell line (Invitrogen, Carlsbad, CA). Supernatants were collected 48 h after transfection, spun at 100 × g for 5 min, filtered through a 0.4 μm membrane, and stored at −80°C. This preparation was used to infect HeLa cells. Selection in 0.2 μg/ml puromycin produced polyclonal stable cell lines. Knockdown of ERK5 was monitored by immunoblot analysis.
Immunoprecipitation
The vector that expresses epitope-tagged ERK5, pcDNA3-FLAG-BMK1, was the generous gift of Dr. Jiing-Dwan Lee (Department of Immunology, The Scripps Research Institute, La Jolla, CA). Cells were transfected with 5 μg of pcDNA3-FLAG-BMK1. The next day, cells were washed twice with cold PBS, solubilized with lysis buffer (20 mM HEPES pH 7.6, 1% Triton X-100, 137 mM NaCl, 0.1 mM Na3VO4, 25 mM β-glycerophosphate, 3 mM EDTA and 1 mM phenylmethylsulfonyl fluoride), incubated for 10 min on ice, and centrifuged at 14,000 × g for 15 min at 4°C. FLAG-BMK1 was immunoprecipitated from precleared lysate (500 μg total protein) by incubation for 2 h at 4°C with anti-FLAG antibody (Sigma, St. Louis, MO) bound to protein G-agarose beads. Phospho-FLAG-BMK1 was detected by immunoblot and an anti-phospho-ERK5 rabbit polyclonal antibody purchased from Cell Signaling (Beverly, MA).
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 4.0 for Macintosh. To compare treatment means, we used a standard 1-way analysis of variance (ANOVA). Where ANOVA main effects were significant (p < 0.05), all pairwise differences between means were assessed using Tukey’s Honestly Significant Differences (HSD) test. Statistical significance of quantitative real-time PCR data was assessed using a 2-way ANOVA and the Bonferroni post-test. Post-hoc tests were conducted with α = 0.05.
Results
Palytoxin, but not TPA, stimulates ERK5 in HeLa and 308 cells
We incubated HeLa cells (Fig. 1) and 308 mouse keratinocytes (Fig. 2) with palytoxin over a 240 minute time course, and monitored ERK5 activation by immunoblot analysis. For these studies, we used the lowest concentrations of palytoxin that stimulated a detectable effect on ERK5 in HeLa and 308 cells, 30 pM and 100 pM, respectively. Several groups have established that ERK5 phosphorylation, which corresponds to ERK5 activity, is indicated by the appearance of a slower migrating form of ERK5 (Kato et al., 1998; Esparis-Ogando et al., 2002; Mody et al., 2003; Scapoli et al., 2004; Woo et al., 2008; Montero et al., 2009). Likewise, we found that palytoxin stimulated the phosphorylation of transiently expressed ERK5 in HeLa cells (Fig. 1, top panel) in a manner that corresponds to the appearance of a slower migrating form of endogenous ERK5 (Fig. 1, second panel). Phosphorylation of transiently expressed ERK5 was detected using antibodies that bind to the dually phosphorylated, active form of ERK5; immunoblot analysis was not sensitive enough to detect endogenous phospho-ERK5 with these antibodies. Altogether, these results indicate that palytoxin triggers the activation of ERK5 in HeLa and 308 cells.
Figure 1. Palytoxin stimulates ERK5 in HeLa cells.

Top panel: HeLa cells transfected with pcDNA-FLAG-BMK1(ERK5) were incubated with 30 pM palytoxin for the indicated times. FLAG-ERK5 was immunoprecipitated from whole cell lysates followed by immunoblotting with an antibody that detects the phosphorylated, active form of ERK5 (pERK5). Other panels: HeLa cells were incubated with 30 pM palytoxin for the indicated times. Whole cell lysates (20 μg protein) were analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2 (pERK2); total ERK2; phosphorylated, active JNK1 and JNK2 (pJNK1 and pJNK2); total JNK1 (JNK1 and JNK2); phosphorylated, active p38 (pp38); and total p38. Note: In HeLa cells, the total JNK1 antibody detects both JNK1 and JNK2. The arrow marks the slower migrating form of ERK5. The data shown are representative of at least three independent experiments.
Figure 2. Palytoxin stimulates ERK5 in 308 cells.

308 cells were incubated with 100 pM palytoxin for the indicated times. Whole cell lysates (20 μg of protein) were analyzed by immunoblot analysis for the following: ERK5; phosphorylated, active ERK1/2 (pERK1 and pERK2); total ERK2; phosphorylated, active JNK1 and JNK2 (pJNK1 and pJNK2); total JNK1; phosphorylated, active p38 (pp38); and total p38. The arrow marks the slower migrating form of ERK5. The data shown are representative of at least three independent experiments.
The time course of palytoxin-stimulated ERK5 phosphorylation differs significantly from the time-courses of palytoxin-stimulated phosphorylation of the three major MAPK family members, ERK1/2, JNK, and p38 (Figs. 3 and 4). In HeLa cells, palytoxin-induced phosphorylation of ERK5 was detected by 60 minutes, remained highly elevated through 120 minutes, and decreased substantially by 240 minutes (Fig. 3, top panel). By contrast, palytoxin-induced ERK1/2 phosphorylation, continued to increase gradually over the 240-minute time course (Fig. 3, second panel). Palytoxin induced a substantial increase in JNK and p38 phosphorylation by 30 minutes, prior to detection of ERK5 activation, and JNK and p38 phosphorylation remained elevated for at least 180 minutes (Fig. 3, third and fourth panels). Palytoxin also stimulated transient ERK5 phosphorylation, the slow accumulation of ERK1/2 phosphorylation, and sustained JNK and p38 phosphorylation in 308 cells (Fig. 4). In 308 cells, palytoxin-induced phosphorylation of ERK5, ERK1/2, JNK, and p38 was first detected at later time points than in HeLa cells (compare Fig. 3 and Fig. 4). The more sensitive cells are to palytoxin, the more rapid the response (Kuroki et al., 1996). Therefore the delay in detection of palytoxin-stimulated MAPK activation in 308 cells suggests that these cells may be less sensitive to palytoxin than HeLa cells. Altogether, the observation that palytoxin stimulates ERK5 activation with a time course that differs from that of ERK1/2, JNK, and p38 activation suggests that the mechanism by which palytoxin modulates ERK5 differs from the mechanisms by which palytoxin modulates the three major MAPK family members.
Figure 3. Palytoxin stimulates ERK5 with a different time course than ERK1/2, JNK, and p38 in HeLa cells.
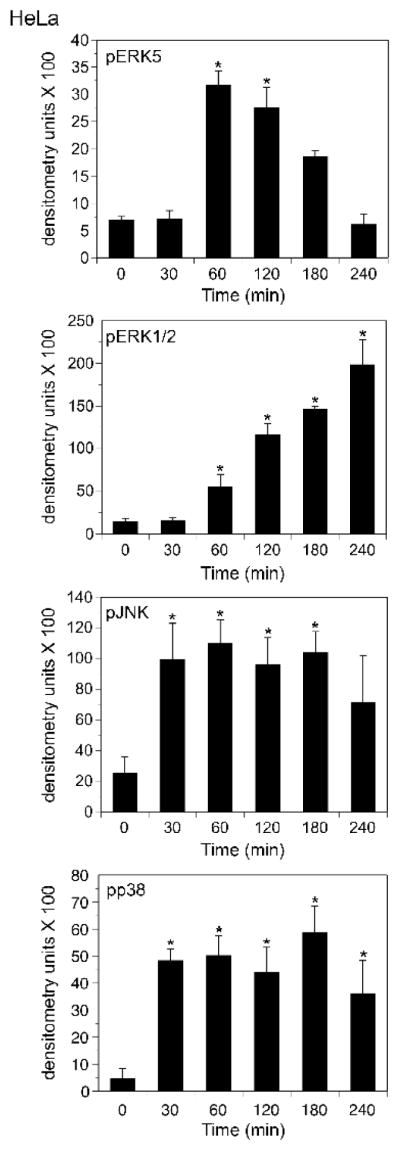
HeLa cells were incubated with 30 pM palytoxin for the indicated times. Whole cell lysates (20 μg of protein) were analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2; phosphorylated, active JNK1 and JNK2; and phosphorylated, active p38. Each time point was run in triplicate. Band intensities were quantitated by densitometry for the graphical representation of the data. The units shown for pERK5 represent the ratio of the shifted ERK5 band to the total of the upper and lower bands as described in (Kato et al., 1998). The bars represent the average of triplicates +/− S.D. Asterisks denote values that were determined to be statistically significantly different from the zero time point (p < 0.01). The data shown are representative of at least three independent experiments.
Figure 4. Palytoxin stimulates ERK5 with a different time course than ERK1/2, JNK, and p38 in 308 cells.
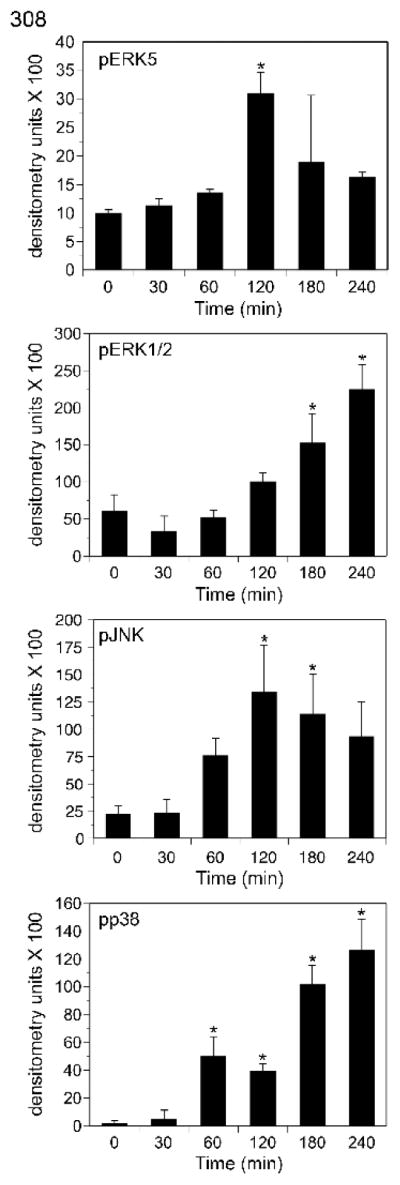
308 cells were incubated with 100 pM palytoxin for the indicated times. Whole cell lysates (20 μg of protein) were analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2; phosphorylated, active JNK1 and JNK2; and phosphorylated, active p38 (pp38). Each time point was run in triplicate. For the graphical representation of the data, band intensities were quantitated by densitometry. The units shown for pERK5 represent the ratio of the shifted ERK5 band to the total of the upper and lower bands as described in (Kato et al., 1998). The bars represent the average of triplicates +/− S.D. Asterisks denote values that were determined to be statistically significantly different from the zero time point (p < 0.01). The data shown are representative of at least three independent experiments.
Next, we determined if the prototypical phorbol ester tumor promoter TPA modulates ERK5 in HeLa and 308 cells. We previously demonstrated that although palytoxin and TPA bind to different cellular receptors, the initially distinct signaling pathways stimulated by palytoxin and TPA can converge to regulate common downstream targets, including ERK1/2 (Warmka et al., 2002). In contrast to palytoxin, however, TPA did not stimulate a detectable increase in ERK5 phosphorylation in either cell line (Fig. 5, top panels) under conditions where it did activate ERK1/2 (Fig. 5, middle panels). This suggests that ERK5 represents a divergence in the signaling pathways activated by palytoxin and TPA in HeLa and 308.
Figure 5. TPA does not stimulate detectable ERK5 activation in HeLa or 308 cells.

HeLa and 308 cells were incubated for the indicated times with 160 nM TPA. Whole cell lysates (20 μg of protein) were analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2 (pERK1 and pERK2); and total ERK2. The HeLa studies represent one experiment. The data shown for the 308 cells are representative of two independent experiments.
Palytoxin stimulates ERK5 through a Na+,K+-ATPase-dependent mechanism that is not mimicked by inhibition protein synthesis or inhibition of serine/threonine or tyrosine phosphatases
The major membrane receptor for palytoxin is the Na+,K+-ATPase (Habermann, 1989). Accordingly, we found that incubation of HeLa cells with ouabain, which binds to the Na+,K+-ATPase, blocked the ability of palytoxin to stimulate the phosphorylation of ERK5 (Figs. 6A and 6C). These studies were not conducted in 308 cells because rodent cells are typically resistant to ouabain (Emanuel et al., 1988). Ouabain did not affect the ability of EGF to stimulate ERK5 phosphorylation (Fig. 6B), which indicates that ouabain does not cause a nonspecific block in ERK5 phosphorylation. Finally ouabain alone did not stimulate a detectable increase in ERK5 phosphorylation under conditions where HeLa cells were incubated with concentrations of ouabain ranging from 100 nM to 1 mM, over time courses that ranged from 2 min to 240 min (Fig. 6B and data not shown). These results indicate that palytoxin activates ERK5 through a mechanism that requires its interaction with the Na+,K+-ATPase.
Figure 6. Palytoxin stimulates ERK5 through a Na+,K+-ATPase-dependent pathway.

A) HeLa cells were incubated for 30 minutes with the indicated concentrations of ouabain. Palytoxin was added to a final concentration of 30 pM, where indicated by the underscore, and the cells were incubated for 120 minutes. Whole cell lysates (20 μg protein) were analyzed for ERK5 by immunoblot. The arrow marks the slower migrating form of ERK5. B) HeLa cells were incubated for 30 minutes in the absence (−) or presence (+) of 100 μM ouabain. EGF was added to a final concentration of 3 nM, where indicated by the underscore, and the cells were incubated for 20 minutes. Whole cell lysates (20 μg protein) were analyzed for ERK5 by immunoblot. The arrow marks the slower migrating form of ERK5. C) HeLa cells were incubated for 30 minutes with the indicated concentrations of ouabain. Palytoxin was not (−) or was (+) added to a final concentration of 30 pM and the cells were incubated for 120 minutes. Whole cell lysates were analyzed for ERK5 by immunoblot. Each point was run in triplicate. The band intensities were quantitated by densitometry. The units shown for pERK5 represent the ratio of the shifted ERK5 band to the total of the upper and lower bands. This method for quantitating the ERK5 band shift was conducted as described in (Kato et al., 1998). The bars represent the average of triplicates +/− S.D. Asterisks denote values that were determined to be statistically significantly different from control (absence of ouabain and palytoxin) (p < 0.05). The palytoxin data shown are representative of at least three independent experiments.
We were interested in determining whether the protein synthesis inhibitor cycloheximide could mimic the effect of palytoxin on ERK5 phosphorylation because we previously showed that palytoxin increases ERK1/2 phosphorylation in 308 cells by stimulating a loss of the phosphatase MKP-3, which is an unstable protein (Warmka et al., 2004). Incubation of HeLa and 308 cells with cycloheximide did not stimulate a detectable increase in ERK5 phosphorylation (Fig. 7, upper panels). Cycloheximide did stimulate the phosphorylation of ERK1/2 in both cell lines, which indicates that the compound is active under the conditions of these studies (Fig. 7, middle panels). Altogether, these results indicate that palytoxin does not stimulate the phosphorylation of ERK5 by blocking the production of an unstable protein.
Figure 7. The protein synthesis inhibitor cycloheximide does not mimic the effect of palytoxin on ERK5.
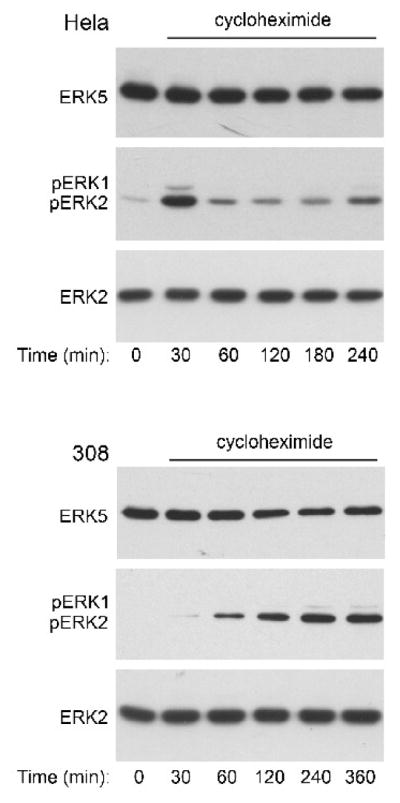
HeLa and 308 cells were incubated for the indicated times with 36 μM cycloheximide. Protein from whole cell lysates (20 μg) was analyzed by immunoblot analysis for the following: ERK5; phosphorylated, active ERK1/2 (pERK1 and pERK2); and total ERK2. The HeLa studies represent one experiment. The data shown for the 308 cells are representative of two independent experiments.
Like other major members of the MAPK family, ERK5 is activated by phosphorylation of specific tyrosine and threonine residues (Mody et al., 2003). Because we previously demonstrated that palytoxin stimulates the phosphorylation of ERK1/2 in 308 cells by stimulating a loss of phosphatase action (Warmka et al., 2004), we wanted to determine if inhibiting major classes of phosphatases could mimic the effect of palytoxin on ERK5. Sodium orthovanadate, which inhibits tyrosine phosphatases, did not stimulate a detectable increase in ERK5 phosphorylation in either HeLa or 308 cells (Fig. 8, upper panels). Sodium orthovanadate stimulated the rapid and sustained phosphorylation of ERK1/2 in both cell lines, however (Fig. 8, middle panels). These results highlight the difference between the regulation of ERK1/2 and ERK5 by protein tyrosine phosphatases, and indicate that palytoxin is unlikely to activate ERK5 by inhibiting this class of phosphatases.
Figure 8. The tyrosine phosphatase inhibitor sodium orthovanadate does not mimic the effect of palytoxin on ERK5.
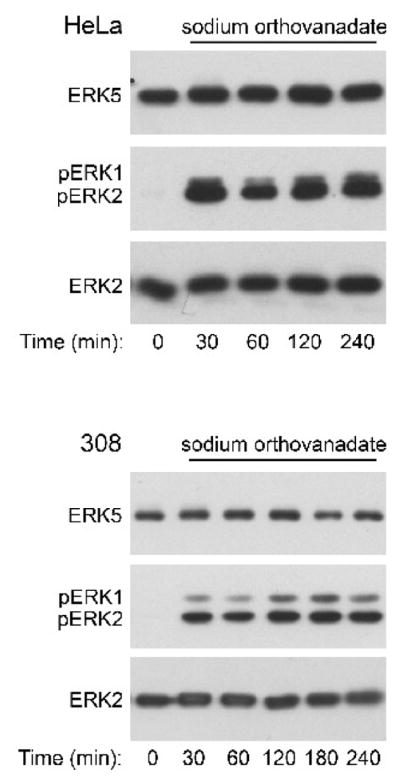
HeLa and 308 cells were incubated for the indicated times with 1 mM sodium orthovanadate. Protein (20 μg) from whole cell lysates was analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2 (pERK1 and pERK2); and total ERK2. The data shown are representative of at least two independent experiments.
Okadaic acid is a selective inhibitor of the serine threonine phosphatase protein phosphatase 2A at the concentration used in these studies (Bialojan and Takai, 1988; Millward et al., 1999). Incubation of HeLa cells with okadaic acid for 240 minutes resulted in a slight increase in ERK5 phosphorylation (Fig. 9, top panel), but a dramatic increase in phospho-ERK1/2 (Fig. 9, middle panel). Incubation of 308 cells with okadaic acid stimulated a detectable increase in both ERK5 and ERK1/2 phosphorylation by 120 minutes and 60 minutes, respectively (Fig. 9). The difference between the sensitivity of HeLa and 308 cells to okadaic acid may be due to cell type-dependent differences in the expression of specific phosphatases. Okadaic acid stimulates the gradual accumulation of both phospho-ERK1/2 and phospho-ERK5 (Fig. 9). Like okadaic acid, palytoxin stimulates the gradual accumulation of phospho-ERK1/2, but in contrast to okadaic acid, palytoxin stimulates transient ERK5 phosphorylation (Figs. 3 and 4). These results indicate that although ERK5 is a common target of these two non-TPA-type tumor promoters, palytoxin and okadaic acid modulate ERK5 through distinct mechanisms.
Figure 9. Okadaic acid stimulates ERK5 phosphorylation with different kinetics than palytoxin in 308 cells.
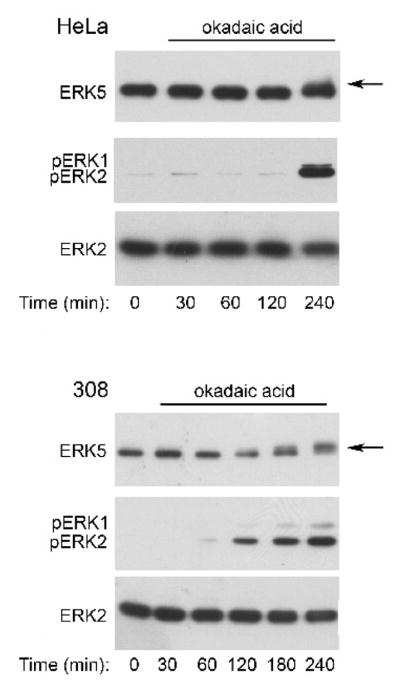
HeLa and 308 cells were incubated for the indicated times with 120 nM okadaic acid. Protein (20 μg) from whole cell lysates was analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2 (pERK1 and pERK2); and total ERK2. The arrow marks the slower migrating form of ERK5. The data shown are representative of at least two independent experiments.
ERK5 contributes to palytoxin-stimulated c-Fos gene expression
We previously reported that palytoxin modulates c-Fos in 308 mouse keratinocytes (Warmka et al., 2002). We concluded that palytoxin modulated c-Fos through an ERK1/2-dependent pathway based on the observation that the ability of palytoxin to increase c-Fos levels was blocked by PD98059, which inhibits MEK1/2, the kinases that phosphorylate and activate ERK1/2. Two lines of evidence led us to investigate the role of ERK5 in the ability of palytoxin to increase c-Fos RNA levels. First, several types of MEK1/2 inhibitors can also inhibit MEK5, the kinase that phosphorylates and activates ERK5 (Kamakura et al., 1999; Mody et al., 2001). Consequently, in some cell types these pharmacological agents can inhibit the activation of ERK5, as well as ERK1/2. Second, studies indicate that both ERK1/2 and ERK5 play a role in serum-stimulated c-Fos gene expression (Sasaki et al., 2006).
To investigate the role of ERK5 in palytoxin-induced c-Fos gene expression, we first identified a concentration of the pharmacological agent U0126 (3 μM) that blocks the ability of palytoxin to activate ERK1/2, but not ERK5 in 308 and HeLa cells (Figs. 10A and 10B). Next, we used real-time PCR to determine how incubation of the cells with 3 μM U0126 affected palytoxin-induced c-Fos gene expression. In 308 cells, 3 μM U0126 partially blocked palytoxin-induced c-Fos gene expression; c-Fos gene expression was inhibited by approximately 80% at 120 minutes and 70% at 180 minutes (Fig. 10C). Incubation of 308 cells with 10 μM U0126, which blocked both palytoxin-stimulated ERK1/2 and ERK5 activation (Fig. 10A), resulted in almost complete inhibition of palytoxin-stimulated c-Fos gene expression (Fig. 10C). These results indicate that although ERK1/2 plays an important role in palytoxin-induced c-Fos gene expression, ERK5 also contributes to the modulation of c-Fos by palytoxin in 308 cells. Likewise, 3 μM U0126 inhibited palytoxin-stimulated c-Fos gene expression by approximately 70% in HeLa cells (Fig. 10D). We were not able to efficiently block palytoxin-stimulated ERK5 activation with U0126 in HeLa cells (Fig. 10B). We were able to achieve efficient knockdown of ERK5 in HeLa cells using shRNA, however (Fig. 11A). Knockdown of ERK5 caused a partial block in palytoxin-induced c-Fos gene expression in HeLa cells at 60 minutes (approximately 50%) (Fig. 11B). There was no detectable effect of ERK5 knockdown on palytoxin-induced in c-Fos gene expression at 180 minutes (Fig. 11B). Knockdown of ERK5 did not affect the ability of TPA to induce c-Fos gene expression (Fig. 11C). This indicates that the effect of ERK5 knockdown on palytoxin-induced c-Fos gene expression is not nonspecific, and furthermore that TPA and palytoxin can modulate c-Fos through activation of different MAPK family members.
Figure 10. U0126 inhibits palytoxin-stimulated increases in c-Fos RNA in HeLa and 308 cells.

(A) 308 and (B) HeLa cells were incubated with the indicated concentrations of U0126 for 30 minutes. Palytoxin was added to a final concentration of 100 pM in 308 cells and 30 pM in HeLa cells. After 120 minutes, the cells were lysed. Protein (20 μg) from whole cell lysates was analyzed by immunoblot for the following: ERK5; phosphorylated, active ERK1/2 (pERK1 and pERK2); and total ERK2. The lane denoted “C” indicates lysates prepared from cells that were not treated with palytoxin or U0126, and therefore represents basal ERK5. The arrow marks the slower migrating form of ERK5. (C) 308 cells were incubated for 30 minutes in the absence (open bars) or presence of either 3 μM U0126 (hatched bars) or 10 μM U0126 (dotted bars) for 30 minutes. Palytoxin was added to a final concentration of 100 pM and the cells were incubated for the indicated times. (D) HeLa cells were incubated in the absence (open bars ) or presence of 3 μM U0126 (hatched bars) for 30 minutes. Palytoxin was added to a final concentration of 30 pM and the cells were incubated for the indicated times. The cells were lysed and RNA levels were determined by quantitative real-time PCR analysis and the comparative Ct method. Data were normalized to GAPDH RNA levels and expressed relative to the zero time point for control. The bars represent the average of triplicates +/− SEM. Asterisks denote values that were determined to be statistically significantly different from the zero time point (p < 0.05). The data shown are representative of at least two independent experiments.
Fig. 11. ERK5 knockdown inhibits palytoxin-induced c-Fos expression.
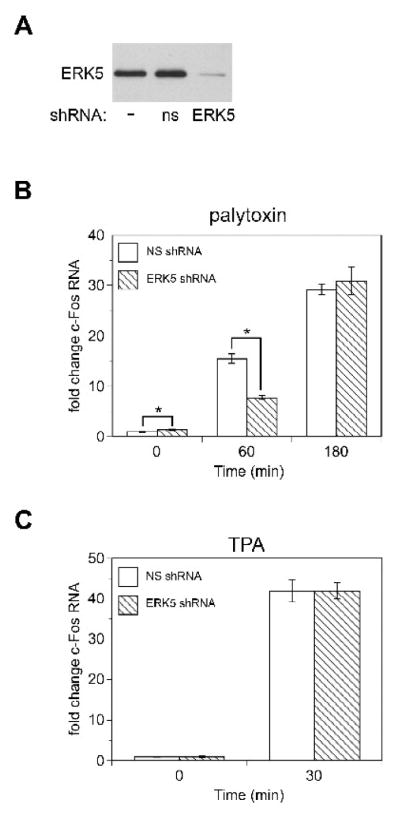
(A) Whole cell lysates were prepared from HeLa cells that were either not transduced (−), that stably express non-silencing shRNA (ns), or that stably express ERK5 shRNA (ERK5). Protein (20 μg) from the whole cell lysates was analyzed by immunoblot for ERK5. HeLa cells that stably express non-silencing shRNA (NS, open bars), or that stably express ERK5 shRNA (ERK5 shRNA, hatched bars) were (B) incubated for the indicated times with 30 pM palytoxin or (C) incubated for 30 minutes with 160 nM TPA. c-Fos RNA levels were determined by quantitative real-time PCR analysis and the comparative Ct method. Data were normalized to GAPDH RNA levels and expressed relative to nontreated HeLa cells that stably express non-silencing shRNA (NS). The bars represent the average of triplicates +/− SEM. Asterisks indicate p < 0.05, ERK5 shRNA compared to NS for each time point. The difference between basal c-Fos gene expression in ERK5 knockdown cells vs. NS cells was never greater than 1.4 fold. The TPA data represent one experiment. The palytoxin data shown are representative of three independent experiments.
Discussion
The studies reported here demonstrate, for the first time, that ERK5 plays a role in the signaling pathways stimulated by the non-TPA-type tumor promoter palytoxin. Moreover, to our knowledge, this is the first report that the interaction of a toxicological or pharmacological agent with the Na+,K+-ATPase results in the modulation of ERK5. We did not detect any effect of ouabain on ERK5, although we previously detected activation of ERK1/2, JNK, and p38 by ouabain in HeLa cells (Li and Wattenberg, 1998). Therefore, activation of ERK5 represents a difference in the signaling pathways stimulated by two agents that bind to the Na+,K+-ATPase. This is also the first report that the non-TPA-type tumor promoter okadaic acid stimulates ERK5 activation. By contrast to palytoxin and okadaic acid, the prototypical phorbol ester tumor promoter TPA did not stimulate detectable ERK5 activation HeLa and 308 cells. Altogether, the results presented here indicate that ERK5 represents an alternative signaling molecule through which different types of tumor promoters can modulate nuclear targets.
ERK5, like the other three major MAPKs, is activated by phosphorylation of specific tyrosine and threonine residues (reviewed in (Dhillon et al., 2007)). MAPK activity is determined by the balance between the phosphorylation and activation by upstream kinases, and the dephosphorylation and inactivation by phosphatases. We have found that palytoxin can modulate the activity of other MAPK family members both through activation of upstream kinases (MAPKKs) and by inhibiting phosphatase action (Kuroki et al., 1997; Li and Wattenberg, 1999; Warmka et al., 2004). For example, we previously showed that palytoxin stimulates the phosphorylation and activation of JNK and p38 through activation of the upstream kinases SEK1, MKK3, and MKK6 (Kuroki et al., 1997; Li and Wattenberg, 1999). We found that U0126, which can inhibit MEK5 (Mody et al., 2001), the kinase that phosphorylates and activates ERK5, inhibits palytoxin-stimulated ERK5 activation in 308 cells and partially inhibits ERK5 activation in HeLa cells. These results indicate that MEK5 plays a role in the activation of ERK5 by palytoxin. The mechanisms by which palytoxin-stimulated ion flux stimulates MEK5 remain to be determined. We previously showed that in 308 cells palytoxin increases ERK1/2 activity not through the activation of MEK1/2, but instead by downregulating MKP-3, an ERK1/2-specific phosphatase that is elevated in this cell line (Warmka et al., 2004). The slow accumulation of phospho-ERK1/2 observed in palytoxin-treated 308 cells is characteristic of phosphatase inhibition. The transient nature of palytoxin-stimulated ERK5 phosphorylation suggests that palytoxin does not increase ERK5 activity by inactivating a phosphatase. Our finding that the phosphatase inhibitors sodium orthovanadate and okadaic acid do not mimic palytoxin action supports this conclusion.
Our studies indicate that ERK5 plays a role in the modulation of c-Fos by palytoxin. We found that a low concentration of U0126, which blocks the stimulation of ERK1/2, but not ERK5, by palytoxin, only partially inhibited c-Fos gene expression. This suggested that other signaling pathways are also be involved in the modulation of this proto-oncogene by palytoxin. Two lines of evidence support the conclusion that ERK5 is involved in the signaling pathways that mediate palytoxin-stimulated c-Fos gene expression. First, a concentration of U0126 that blocks both ERK1/2 and ERK5 further blocked the ability of palytoxin to increase c-Fos RNA in 308 cells. Second, knockdown of ERK5 by shRNA in HeLa cells inhibited palytoxin-stimulated c-Fos gene expression. Knockdown on ERK5 resulted in a significant decrease in c-Fos gene expression at 60 minutes, but did not appear to significantly affect palytoxin-stimulated c-Fos gene expression at 180 minutes. This may be because palytoxin-stimulated ERK1/2 activation can compensate for the loss of ERK5 at this time point. At 60 minutes, palytoxin-stimulated ERK5 activity may predominate over ERK1/2 activity (see Fig. 3). By 180 minutes, ERK5 activity is decreasing, while ERK1/2 activity is continuing to increase; the increase in palytoxin-stimulated ERK1/2 activity by 180 minutes may be sufficiently high that ERK5 is no longer required to maintain elevated c-Fos gene expression. The observation that 3 μM U0126, which blocks the stimulation of ERK1/2 but not ERK5 in HeLa cells, does not totally block palytoxin-induced c-Fos gene expression at 180 minutes (see Fig. 10D), suggests that pathways in addition to ERK1/2 and ERK5 also contribute to palytoxin-induced c-Fos gene expression in HeLa cells. Interestingly, there appears to be considerable redundancy in the action of various MAPK families. For example, the transcription factor Sap1a, which has been implicated in the transcriptional regulation of c-Fos can be modulated by both ERK1/2 and ERK5 (Janknecht et al., 1995; Strahl et al., 1996; Kamakura et al., 1999; Yordy and Muise-Helmericks, 2000). Elk-1, another transcription factor involved in the regulation of c-Fos gene expression, can be modulated by ERK1/2, JNK, and p38 (Cavigelli et al., 1995; Gille et al., 1995; Whitmarsh et al., 1995; Raingeaud et al., 1996). This helps explain why c-Fos is a common target of tumor promoters that trigger diverse signals. Whereas TPA modulates c-Fos through activation of ERK1/2 (Zeliadt et al., 2004), palytoxin can modulate c-Fos through the activation of ERK1/2, ERK5, and perhaps other pathways as well.
Different types of studies suggest that ERK5 is involved in carcinogenesis. For example, it has been reported that ERK5 protein is elevated in tissue from malignant human prostate cancer tissue relative to normal tissue (McCracken et al., 2008). Furthermore, whereas the MEK1 inhibitor PD184352 blocked proliferation of the PC3 prostate cancer cell line at high concentrations that inhibit ERK5, PD184352 did not block proliferation of PC3 cells at low concentrations that inhibit ERK1/2 but not ERK5 (McCracken et al., 2008). Active ERK5 has also been detected in human breast cancer tissue and in tumor tissue from an animal model of breast cancer (Montero et al., 2009). ERK5 also appears to be involved in the signaling pathways that regulate the adhesion and motility of metastatic breast and prostate cancer cell lines (Sawhney et al., 2009). A role for ERK5 in angiogenesis during tumorigenesis has also been suggested by studies conducted using ERK5 knockout mice (Hayashi et al., 2005). ERK5 has also been implicated in the regulation of the proto-oncogenes c-Fos, Fra-1, and c-Jun, which further supports the idea that aberrant regulation of ERK5 may be involved in carcinogenesis (Kato et al., 1997; Terasawa et al., 2003). The observation that ERK5 is involved in asbestos-stimulated proliferation of a murine alveolar type II epithelial cell line (C10) suggests that ERK5 may be a target of environmental carcinogens (Scapoli et al., 2004). Finally, our observation that two non-TPA-type tumor promoters, palytoxin and okadaic acid, activate ERK5 suggests that ERK5 may also be involved in early stages of carcinogenesis.
Establishing the role of ERK5 in palytoxin-stimulated tumor promotion requires further study. The mechanisms by which palytoxin stimulates tumor promotion are not clear. Palytoxin is cytotoxic in cell culture and also caused toxicity when used in the multi-stage mouse skin model (Fujiki et al., 1986). Although palytoxin may stimulate cell proliferation in vivo, palytoxin may also stimulate tumor promotion by other mechanisms. For example, it is possible that initiated cells that express oncogenic Ras are resistant to palytoxin-induced cytoxicity, thus giving a growth advantage to initiated cells. Alternatively, palytoxin may stimulate the release of agents from cells that stimulate the growth of neighboring cells or perhaps induce an inflammatory response that contributes to tumor promotion. For example, palytoxin stimulates arachidonic acid metabolism and the release of prostaglandins in other systems (Levine et al., 1986; Lazzaro et al., 1987; Miura et al., 2006). Our observation that palytoxin stimulates ERK5 activation in two different cell types suggests that the role of ERK5 should be examined in various types of systems in which palytoxinstimulates responses that are likely to be involved in carcinogenesis.
Humans are likely to be exposed to palytoxin through two main routes. Palytoxin is isolated from corals found in Hawaii and the Caribbean Islands (Moore and Scheuer, 1971; Ishida et al., 1983). Humans can be exposed to palytoxin through the food chain (Mebs, 1998); palytoxin has been linked to poisonings due to consumption of crabs, mackerel, and sardines (Alcala et al., 1988; Kodama et al., 1989; Onuma et al., 1999). Another route of exposure to palytoxin is through the maintenance of aquariums (Hoffmann et al., 2008). The corals that produce palytoxin are commonly found in aquariums used in exhibitions as well as aquariums maintained as a hobby. Exposure can occur from inhaling aerosolized water that contains palytoxin and through skin contact. Although palytoxin has been implicated in poisonings from consuming seafood and from cleaning an aquarium, to our knowledge, the amount of palytoxin that humans might be exposed to at nontoxic doses through consumption of seafood and exposure to water the contains palytoxin has not been estimated.
We have used the novel tumor promoter palytoxin to both identify common biochemical targets of proto-typical phorbol ester tumor promoters and non-TPA-type tumor promoters, such as c-Fos, and to reveal alternate signaling pathways by which such potentially critical targets can be modulated. Altogether, these studies may help reveal mechanisms by which a broad range of agents can affect carcinogenesis.
Acknowledgments
We thank Kathleen F. Conklin (Department of Genetics, Cell Biology and Development, Institute of Human Genetics, Masonic Cancer Center, University of Minnesota) for her assistance with the studies that involved the use of lentiviral vectors, and Kathryn L. Schwertfeger (Department of Laboratory Medicine and Pathology) for her assistance with the studies that involved real-time PCR. This work was supported by National Institutes of Health grant RO1-CA104609 (to E.V.W.). The National Institutes of Health was not involved in study design, collection, analysis, or interpretation of data, writing the manuscript, or the decision to submit the manuscript for publication.
Footnotes
Conflict of interest statement
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- Alcala AC, Alcala LC, Garth JS, Yasumura D, Yasumoto T. Human fatality due to ingestion of the crab Demania reynaudii that contained a palytoxin-like toxin. Toxicon. 1988;26:105–107. doi: 10.1016/0041-0101(88)90142-0. [DOI] [PubMed] [Google Scholar]
- Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983;303:72–74. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–290. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavigelli M, Dolfi F, Claret FX, Karin M. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 1995;14:5957–5964. doi: 10.1002/j.1460-2075.1995.tb00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao TH, Hayashi M, Tapping RI, Kato Y, Lee JD. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J Biol Chem. 1999;274:36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Emanuel JR, Schulz J, Zhou XM, Kent RB, Housman D, Cantley L, Levenson R. Expression of an ouabain-resistant Na,K-ATPase in CV-1 cells after transfection with a cDNA encoding the rat Na,K-ATPase alpha 1 subunit. J Biol Chem. 1988;263:7726–7733. [PubMed] [Google Scholar]
- Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002;22:270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki H, Suganuma M, Nakayasu M, Hakii H, Horiuchi T, Takayama S, Sugimura T. Palytoxin is a non-12-O-tetradecanoylphorbol-13-acetate type tumor promoter in two-stage mouse skin carcinogenesis. Carcinogenesis. 1986;7:707–710. doi: 10.1093/carcin/7.5.707. [DOI] [PubMed] [Google Scholar]
- Gille H, Strahl T, Shaw PE. Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr Biol. 1995;5:1191–1200. doi: 10.1016/s0960-9822(95)00235-1. [DOI] [PubMed] [Google Scholar]
- Habermann E. Palytoxin acts through Na+,K+-ATPase. Toxicon. 1989;27:1171–1187. doi: 10.1016/0041-0101(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Fearns C, Eliceiri B, Yang Y, Lee JD. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005;65:7699–7706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Lee JD. Role of the BMK1/ERK5 signaling pathway: lessons from knockout mice. J Mol Med. 2004;82:800–808. doi: 10.1007/s00109-004-0602-8. [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Hermanns-Clausen M, Buhl C, Buchler MW, Schemmer P, Mebs D, Kauferstein S. A case of palytoxin poisoning due to contact with zoanthid corals through a skin injury. Toxicon. 2008;51:1535–1537. doi: 10.1016/j.toxicon.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Takagi K, Takahashi M, Satake N, Shibata S. Palytoxin isolated from marine coelenterates. The inhibitory action on (Na,K)-ATPase. J Biol Chem. 1983;258:7900–7902. [PubMed] [Google Scholar]
- Janknecht R, Ernst WH, Nordheim A. SAP1a is a nuclear target of signaling cascades involving ERKs. Oncogene. 1995;10:1209–1216. [PubMed] [Google Scholar]
- Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274:26563–26571. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- Kodama AM, Hokama Y, Yasumoto T, Fukui M, Manea SJ, Sutherland N. Clinical and laboratory findings implicating palytoxin as cause of ciguatera poisoning due to Decapterus macrosoma (mackerel) Toxicon. 1989;27:1051–1053. doi: 10.1016/0041-0101(89)90156-6. [DOI] [PubMed] [Google Scholar]
- Kuroki DW, Bignami GS, Wattenberg EV. Activation of stress-activator protein kinase/c-Jun N-terminal kinase by the non-TPA-type tumor promoter palytoxin. Cancer Res. 1996;56:637–644. [PubMed] [Google Scholar]
- Kuroki DW, Minden A, Sanchez I, Wattenberg EV. Regulation of a c-Jun amino-terminal kinase/stress-activated protein kinase cascade by a sodium-dependent signal transduction pathway. J Biol Chem. 1997;272:23905–23911. doi: 10.1074/jbc.272.38.23905. [DOI] [PubMed] [Google Scholar]
- Lazzaro M, Tashjian AH, Jr, Fujiki H, Levine L. Palytoxin: an extraordinarily potent stimulator of prostaglandin production and bone resorption in cultured mouse calvariae. Endocrinology. 1987;120:1338–1345. doi: 10.1210/endo-120-4-1338. [DOI] [PubMed] [Google Scholar]
- Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- Levine L, Xiao DM, Fujiki H. Combinations of palytoxin or 12-O-tetradecanoylphorbol-13-acetate and recombinant human insulin growth factor-I or insulin synergistically stimulate prostaglandin production in cultured rat liver cells and squirrel monkey aorta smooth muscle cells. Prostaglandins. 1986;31:669–681. doi: 10.1016/0090-6980(86)90173-5. [DOI] [PubMed] [Google Scholar]
- Li S, Wattenberg EV. Differential activation of mitogen-activated protein kinases by palytoxin and ouabain, two ligands for the Na+,K+-ATPase. Toxicol Appl Pharmacol. 1998;151:377–384. doi: 10.1006/taap.1998.8471. [DOI] [PubMed] [Google Scholar]
- Li S, Wattenberg EV. Cell-type-specific activation of p38 protein kinase cascades by the novel tumor promoter palytoxin. Toxicol Appl Pharmacol. 1999;160:109–119. doi: 10.1006/taap.1999.8754. [DOI] [PubMed] [Google Scholar]
- McCracken SR, Ramsay A, Heer R, Mathers ME, Jenkins BL, Edwards J, Robson CN, Marquez R, Cohen P, Leung HY. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27:2978–2988. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- Mebs D. Occurrence and sequestration of toxins in food chains. Toxicon. 1998;36:1519–1522. doi: 10.1016/s0041-0101(98)00143-3. [DOI] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- Miura D, Kobayashi M, Kakiuchi S, Kasahara Y, Kondo S. Enhancement of transformed foci and induction of prostaglandins in Balb/c 3T3 cells by palytoxin: in vitro model reproduces carcinogenic responses in animal models regarding the inhibitory effect of indomethacin and reversal of indomethacin’s effect by exogenous prostaglandins. Toxicol Sci. 2006;89:154–163. doi: 10.1093/toxsci/kfi342. [DOI] [PubMed] [Google Scholar]
- Mody N, Campbell DG, Morrice N, Peggie M, Cohen P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem J. 2003;372:567–575. doi: 10.1042/BJ20030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001;502:21–24. doi: 10.1016/s0014-5793(01)02651-5. [DOI] [PubMed] [Google Scholar]
- Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS ONE. 2009;4:e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RE. Structure of palytoxin. Fortschr Chem Org Naturst. 1985;48:81–202. doi: 10.1007/978-3-7091-8815-6_2. [DOI] [PubMed] [Google Scholar]
- Moore RE, Scheuer PJ. Palytoxin: a new marine toxin from a coelenterate. Science. 1971;172:495–498. doi: 10.1126/science.172.3982.495. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308:693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- Onuma Y, Satake M, Ukena T, Roux J, Chanteau S, Rasolofonirina N, Ratsimaloto M, Naoki H, Yasumoto T. Identification of putative palytoxin as the cause of clupeotoxism. Toxicon. 1999;37:55–65. doi: 10.1016/s0041-0101(98)00133-0. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Kojima H, Kishimoto R, Ikeda A, Kunimoto H, Nakajima K. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell. 2006;24:63–75. doi: 10.1016/j.molcel.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Sawhney RS, Liu W, Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J Cell Physiol. 2009;219:152–161. doi: 10.1002/jcp.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapoli L, Ramos-Nino ME, Martinelli M, Mossman BT. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene. 2004;23:805–813. doi: 10.1038/sj.onc.1207163. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Strahl T, Gille H, Shaw PE. Selective response of ternary complex factor Sap1a to different mitogen-activated protein kinase subgroups. Proc Natl Acad Sci USA. 1996;93:11563–11568. doi: 10.1073/pnas.93.21.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland JE, Greenhalgh DA, Koceva-Chyla A, Hennings H, Restrepo C, Balaschak M, Yuspa SH. Development of murine epidermal cell lines which contain an activated rasHa oncogene and form papillomas in skin grafts on athymic nude mouse hosts. Cancer Res. 1988;48:165–169. [PubMed] [Google Scholar]
- Sun W, Kesavan K, Schaefer BC, Garrington TP, Ware M, Johnson NL, Gelfand EW, Johnson GL. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J Biol Chem. 2001;276:5093–5100. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- Terasawa K, Okazaki K, Nishida E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells. 2003;8:263–273. doi: 10.1046/j.1365-2443.2003.00631.x. [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Vaque JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18:753–760. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Warmka JK, Mauro LJ, Wattenberg EV. Mitogen-activated protein kinase phosphatase-3 is a tumor promoter target in initiated cells that express oncogenic Ras. J Biol Chem. 2004;279:33085–33092. doi: 10.1074/jbc.M403120200. [DOI] [PubMed] [Google Scholar]
- Warmka JK, Winston SE, Zeliadt NA, Wattenberg EV. Extracellular signal-regulated kinase transmits palytoxin-stimulated signals leading to altered gene expression in mouse keratinocytes. Toxicol Appl Pharmacol. 2002;185:8–17. doi: 10.1006/taap.2002.9519. [DOI] [PubMed] [Google Scholar]
- Wattenberg EV, Fujiki H, Rosner MR. Heterologous regulation of the epidermal growth factor receptor by palytoxin, a non-12-O-tetradecanoylphorbol-13-acetate-type tumor promoter. Cancer Res. 1987;47:4618–4622. [PubMed] [Google Scholar]
- Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- Yordy JS, Muise-Helmericks RC. Signal transduction and the Ets family of transcription factors. Oncogene. 2000;19:6503–6513. doi: 10.1038/sj.onc.1204036. [DOI] [PubMed] [Google Scholar]
- Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17:1–7. doi: 10.1016/s0923-1811(97)00071-6. [DOI] [PubMed] [Google Scholar]
- Zeliadt NA, Warmka JK, Winston SE, Kahler R, Westendorf JJ, Mauro LJ, Wattenberg EV. Tumor promoter-induced MMP-13 gene expression in a model of initiated epidermis. Biochem Biophys Res Commun. 2004;317:570–577. doi: 10.1016/j.bbrc.2004.03.081. [DOI] [PubMed] [Google Scholar]
- Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]