

Abstract
Objective
The objective of this study was to determine the effects and molecular mechanisms of eotaxin, a newly discovered chemokine (CCL11), on endothelial permeability in the human coronary artery endothelial cells (HCAECs).
Methods and Results
Cells were treated with eotaxin, and the monolayer permeability was studied by using a costar transwell system with a Texas-Red-labeled dextran tracer. Eotaxin significantly increased monolayer permeability in a concentration-dependent manner. In addition, eotaxin treatment significantly decreased the mRNA and protein levels of endothelial junction molecules including zonula occludens-1 (ZO-1), occludin and claudin-1 in a concentration-dependent manner as determined by real time RT-PCR and Western blot analysis, respectively. Increased oxidative stress was observed in eotaxin-treated HCAECs by analysis of cellular glutathione levels. Furthermore, eotaxin treatment substantially activated the phosphorylation of MAPK p38. HCAECs expressed CCR3. Consequently, antioxidants (ginkgolide B and MnTBAP), specific p38 inhibitor SB203580 and anti-CCR3 antibody effectively blocked the eotaxin-induced permeability increase in HCAECs. Eotaxin also increased phosphorylation of Stat3 and nuclear translocation of NF-κB in HCAECs.
Conclusions
Eotaxin increases vascular permeability through CCR3, the down regulation of tight junction proteins, increase of oxidative stress and activation of MAPK p38, Stat3 and NF-kB pathways in HCAECs.
Keywords: Endothelial permeability, HCAEC, eotaxin, tight junction molecules, oxidative stress, ginkgolide B, MAPK p38
Introduction
The vascular endothelium forms a barrier between the circulation and the interstitium. Aberration of endothelial barrier function leads to an abnormal extravasation of blood components and accumulation of fluid in the extravascular space, resulting in organ dysfunction. This injurious process has been implicated in inflammation,1 trauma, sepsis, ischemia-reperfusion, diabetes, atherosclerosis,2 and tumor development and metastasis.3,4 The endothelial barrier function is predominantly maintained by the interendothelial junction structures including tight junctions, adherens junctions and gap junctions.5,6 Tight junctions include transmembrane proteins such as occludin, claudin and junctional adhesion (JAM) molecules. These transmembrane molecules are linked intracellularly to the cytoskeleton via zonula occludens (ZO-1, ZO-2 and ZO-3). Adherens junctions are mainly composed of cadherins and β-catenin and provide strong mechanical attachments between adjacent cells. Gap junctions are communication structures, which allow the passage of small molecular weight solutes between neighboring cells. Several inflammation cytokines such as tumor necrosis factor-alpha (TNF-α) can significantly induce endothelial permeability by changing these junction structures.7,8
Eotaxin is a recently discovered chemokine (CCL11) characterized by its high chemotactic selectivity for eosinophils.9,10 The position of four cysteine residues in eotaxin places it in the C-C family of chemokines, along with other chemokines such as RANTES, MCP-3, and MIP-1 alpha. Human eotaxin is an 8.3 kDa protein containing 74 amino acid residues. The CC chemokine eotaxin signals through the CCR3 receptor. It is produced by IFN-gamma stimulated endothelial cells and TNF-α-activated monocytes. A growing body of evidence indicates that eotaxin may participate in the atherosclerotic process, For example, eotaxin levels were increased in human atherosclerotic plaques, indicating a potential role of eotaxin in vascular inflammation.11 Circulating eotaxin levels were increased in patients with coronary artery disease12-14 and the presence of the non-conservative polymorphism in eotaxin gene is associated with increased risk for myocardial infarction.15 Also, eotaxin is involved in endothelial inflammation16 and vascular smooth muscle cell migration.17 Vascular smooth muscle cells in human atheroma prominently express eotaxin suggesting that eotaxin may contribute to the progression of atherosclerosis.11 These emerging studies indicate that eotaxin plays a crucial role in the cardiovascular system. However, the exact roles and mechanisms of eotaxin in the vascular system are largely unknown.
In the current study, we determined whether eotaxin could affect endothelial monolayer permeability. Human coronary artery endothelial cells (HCAECs) were treated with eotaxin, and monolayer permeability was investigated. In addition, potential molecular mechanisms such as the role of CCR3 receptors, endothelial junction molecules, oxidative stress, MAPK signal transduction and Stats and NF-κB transcription factors were also studied. These experiments, for the first time, explore the molecular mechanisms of eotaxin-induced endothelial dysfunction, thereby having clinical relevance and therapeutic potential.
Methods
Endothelial Permeability
HCAECs were obtained from Gelantis (San Diego, CA) and cultured in HCAEC Growth medium (Gelantis). Human recombinant eotaxin was obtained from Peprotech (Rocky Hill, NJ). Paracellular permeability was studied in a Coaster Transwell system as previously described.18
Real-time RT-PCR Analysis
HCAECs were treated with different concentrations of eotaxin (50, 100 and 200 ng/mL) for 24 h. Total RNA extraction and cDNA reverse transcription were done as previously described.19 Primers for VE-cadherin, ZO-1, claudin-1, occludin, and JAM-1 were described in our previous publication.19-21
Western Blot Analysis
Equal amount of proteins (40 μg) was loaded onto 10% SDS-PAGE, fractionated by electrophoresis, and transferred to nitrocellulose membranes (BioRad). Primary antibodies against ZO-1, claudin-1, occludin, JAM-1, VE cadherin, CCR3, p38, NF-κB p65, Stat3, and β-actin were used.
Cellular Glutathione (GSH) Assay
HCAECs were treated with either eotaxin (100 ng/mL) or pretreated with MnTBAP (2 μM) for 30 min followed by eotaxin treatment for 45 min. Antimycin A (10 μg/mL) and TNF-α (2 ng/mL) were used for the positive controls. Cellular GSH was measured as per manufacture's instructions by following a GSH-Glo Glutathione assay kit (Promega, Madison, WI).
Statistical Analysis
Data were expressed as the mean ± SD. Comparisons were made using the Student's t-test. A p value <0.05 was considered statistically significant.
Online Supplemental Materials
Online supplemental materials include the background information about gap junction, detail materials and methods, and 4 supplemental figures.
Results
Eotaxin Increases Endothelial Monolayer Permeability in HACECs
To determine whether eotaxin could affect the paracellular permeability of the endothelial cell monolayer, HCAECs were treated with eotaxin in a concentration-dependent manner, and endothelial permeability was analyzed by a Costar transwell system with a Texas Red-labeled dextran tracer. Treatment of HCAEC monolayer with increasing concentrations (100 and 200 ng/mL) of eotaxin for 24 h significantly increased endothelial permeability by 43% and 59%, respectively, compared with untreated cells (n=3, p<0.05, Figure 1A). TNF-α (2 ng/mL) was used as a positive control since it can significantly increase monolayer permeability.18 Heat inactivated eotaxin did not increase the permeability through HCAECs indicating that the eotaxin effect is specific (Figure 1B). Interestingly, eotaxin-induced permeability increase in HCAECs was significantly blocked by the treatment with antioxidant ginkgolide B (Figure 1A). As a positive control, antimycin A (ROS generator) also increased endothelial permeability and this effect was effectively blocked by MnTBAP (cell permeable SOD mimetic) (n=3, p<0.05, Figure 1C). Similarly to HCAECs, eotaxin treatment also increased the paracellular permeability in human umbilical vein endothelial cells (HUVECs) in a concentration dependent manner (Figure S1). Eotaxin treatment (100 ng/mL) for 24 h did not induce apoptosis compared to untreated cells in HCAECs (Figure S2).
Figure 1.
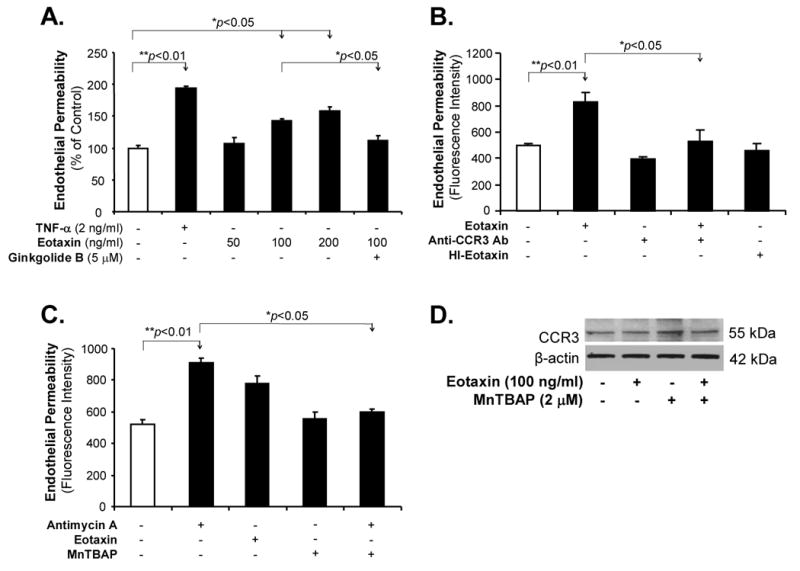
Effects of eotaxin, antimycin A, MnTBAP, ginkgolide B and CCR3 on endothelial monolayer permeability in HCAECs. Endothelial monolayer permeability was analyzed with a costar transwell system and a Texas Red-labeled dextran tracer. A, Concentration-dependent study. HCAECs were treated with increasing concentrations of eotaxin (50, 100 and 200 ng/mL), TNF-α (2 ng/mL) or with ginkgolide B (5 μM) for 24 h. n=3, *p<0.05 B, Effects of CCR3 receptor blocking and heat-inactivation (HI) of eotaxin on endothelial permeability. HCAECs were treated with eotaxin (100 ng/mL) or HI-eotaxin (100 gm/mL) with or without anti-CCR3 antibody 24 h. n=3, *p<0.05. C, Effect of antimycin A on endothelial permeability. HCAECs were treated with antimycin A (10 μg/mL) for 24 h in the presence or absence of antioxidant MnTBAP (2 μM). n=3, *p<0.05. D, Western blot analysis. HCAECs were treated with eotaxin (100 ng/mL) and/or MnTBAP (2 μM) for 24 h and CCR3 protein levels were determined by Western blot. Loading efficiency was determined by reprobing the blot with β-actin antibody. Experiments were repeated twice. Statistical comparison is indicated by the arrow head. Error bars represent SD.
CCR3 Receptors are Involved in Eotaxin-Induced Endothelial Permeability Increase
CC chemokines such as eotaxin acts predominantly via the CCR3 receptors, a member of the family of G-protein coupled receptors. Experiments were designed to determine whether CCR3 receptors are expressed on HCAECs and whether CCR3 receptors are involved in the effects of eotaxin on HCAECs. Western blotting analysis showed that CCR3 receptors were expressed in HCAECs and eotaxin treatment did not change CCR3 receptor expression levels compared with untreated cells (Figure 1D). To further determine the involvement of CCR3 receptors in eotaxin-mediated hyperpermeability, HCAECs were pretreated with anti-CCR3 antibody for 30 min followed by eotaxin treatment for another 24 h. Interestingly, anti-CCR3 antibody effectively reduced the basal endothelial permeability and eotaxin-induced permeability increase in HCAECs (n=3, p<0.05, Figure 1B). Immunohistological analysis showed that CCR3 was detected in human atherosclerotic arteries (Figure S3). These data demonstrate that CCR3 receptors play an important role in eotaxin-mediated endothelial permeability.
Eotaxin Decreases the Expression of Endothelial Tight Junction Proteins ZO-1, Claudin-1 and Occludin in HCAECs
To determine whether eotaxin could affect the expression of endothelial junction molecules especially the tight junction proteins at both mRNA and protein levels, HCAECs were treated with eotaxin for 24 h. Eotaxin treatment decreased the expression of tight junction proteins in a concentration-dependent manner. At 50, 100 and 200 ng/mL of eotaxin, ZO-1 mRNA was decreased by 8%, 25% (p<0.05) and 39% (p<0.05), respectively, compared with the control (n=3, Figure 2A); occludin mRNA was decreased by 19% (p<0.05), 33% (p<0.05) and 55% (p<0.05), respectively; and claudin-1 mRNA was decreased by 5%, 45% (p<0.05) and 48% (p<0.05), respectively, compared with the control. There were no significant differences of VE cadherin and JAM-1 mRNA levels after eotaxin treatment compared with the control. Western blot analysis showed a parallel decrease in the protein levels of ZO-1, occludin and claudin-1 in HCAECs when treated with eotaxin (100 ng/mL) compared with the control (Figure 2B). Commonly used antioxidant MnTBAP (2 μM), a cell permeable SOD mimetic, effectively blocked eotaxin-induced decrease in these molecules at the protein level. Consistent with the mRNA expression data, protein levels of VE cadherin and JAM-1 did not show any significant differences after eotaxin treatment (Figure 2B). To further confirm protein levels of tight junction proteins, flow cytometry analysis was used and showed that eotaxin treatment substantially decreased protein levels of ZO-1, occludin and claudin-1 in HCAECs (Figure 2C).
Figure 2.
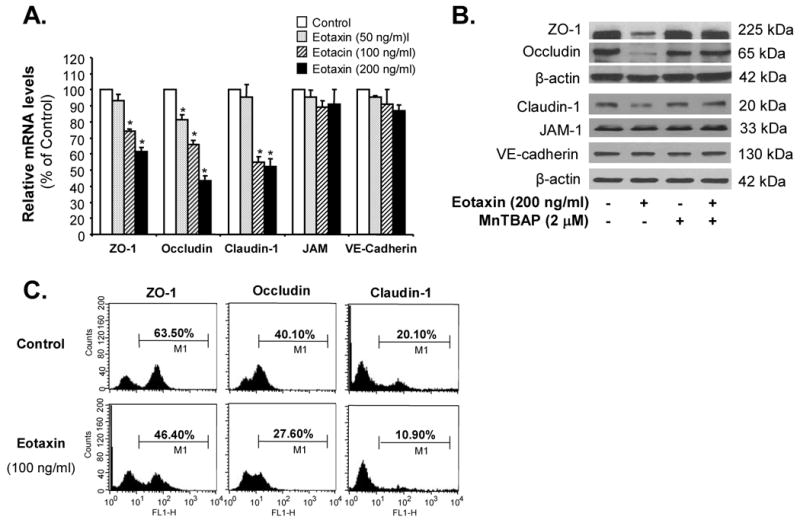
Effects of eotaxin on the expression of junctional molecules in HCAECs. HCAECs were treated with serial concentrations of eotaxin (50, 100 and 200 ng/mL) for 24 h. A, The mRNA levels of ZO-1, claudin-1, occludin, JAM-1 and VE cadherin were analyzed by real time PCR, and values were normalized to GAPDH [2(Ct GAPDH −Ct gene of interest)] in each sample. n=3 *p<0.05 (compared with controls). B, The protein levels of ZO-1, occludin, claudin-1, VE cadherin and JAM-1 were analyzed by Western blot analysis. Loading efficiency was determined by reprobing the blot with β-actin antibody. Eotaxin and antioxidant MnTBAP were used. Experiments were repeated twice. C, Histogram of flow cytometry analysis showing protein levels of ZO-1, claudin-1, and occludin under the control and eotaxin-treated conditions. HCAECs were treated with 100 ng/mL eotaxin for 24 h. Experiments were repeated twice.
We also performed a time course study of the expression of tight junction molecules ZO-1, occludin and claudin-1 in HCAECs. HCAECs were treated with eotaxin (100 ng/mL) and the mRNA levels of ZO-1, occludin and claudin-1 were determined at different time points (0, 45 min, 2 h, 6 h, 18 h and 24 h, respectively) by real time PCR. Results showed that mRNA levels of these tight junction molecules were significantly decreased by over 40% at or after 2 h of eotaxin treatment compared with untreated controls (n=3, p<0.05, Figure 3).
Figure 3.

Time course study of the effect of eotaxin on mRNA levels of tight junction molecules in HCAECs. Cells were treated with eotaxin (100 ng/mL) for different time points (0, 45 min, 2 h, 6 h, 18 h and 24 h) and the mRNA levels of ZO-1 (A), occludin (B) and claudin-1 (C) were determined by real-time PCR and values were normalized to GAPDH [2(Ct GAPDH −Ct gene of interest)] in each sample. n=3, *p<0.05 (compared with controls). Error bars represent SD.
Eotaxin Increases ROS Production in HCAECs
To determine whether oxidative stress could be involved in the action of eotaxin, ROS levels were analyzed by GSH assay. Superoxide can be converted to H2O2 by superoxide dismutase (SOD), and then H2O2 can be rapidly removed by catalase or peroxidases such as the GSH peroxidases, which use reduced GSH as the electron donor. Thus, cellular GSH levels are negatively correlated to ROS levels. Eotaxin (100 ng/mL) treatment of HCAECs for 45 min significantly reduced cellular GSH levels (n=3, p<0.05,Figure 4), while MnTBAP effectively blocked eotaxin-induced reduction in GSH levels in HCAECs. Two positive controls (antimycin A and TNF-α) also significantly reduced cellular GSH levels in HCAECs (n=3, p<0.05,Figure 4), while MnTBAP partially blocked the effect of antimycin A. These data demonstrate that eotaxin increases oxidative stress in HCAECs.
Figure 4.

Effects of eotaxin, antimycin A, TNF-α, and MnTBAP on the cellular glutathione (GSH) levels in HCAECs. Cells were treated with eotaxin (100 ng/mL), antimycin A (10 μg/mL), TNF-α (2 ng/mL) and/or MnTBAP (2 μM) for 45 min. Cellular GSH levels were determined by a GSH-Glo Glutathione assay kit. n=3. *p<005 (the comparison is indicated by the arrow head). Error bars represent SD.
Activation of p38, Stat3 and NF-κB is Involved in the Eotaxin-Induced Permeability Increase in HCAECs
To determine the signal transduction pathways involved in the action of eotaxin on HCAECs, the activation status of MAPK p38 was investigated using Western blot analysis because p38 is sensitive to oxidative stress. Treatment with eotaxin (100 ng/mL) substantially increased the phosphorylation of p38 at 45 min of the treatment (Figure 5A), but not at 120 min (Figure S4). Antimycin A treatment showed similar effects as eotaxin on p38 phosphorylation, while antioxidant MnTBAP effectively blocked eotaxin- or antimycin A-induced p38 phosphorylation (Figure 5A). To determine the functional significance of p38 activation, cells were pretreated with specific p38 inhibitor SB203580 followed by eotaxin treatment for 24 h and the monolayer permeability was analyzed using the Costar transwell system. Indeed, specific p38 inhibitor SB203580 (10 μM) completely blocked eotaxin-induced permeability increase in HCAECs (n=3, p<0.05, Figure 5B).
Figure 5.
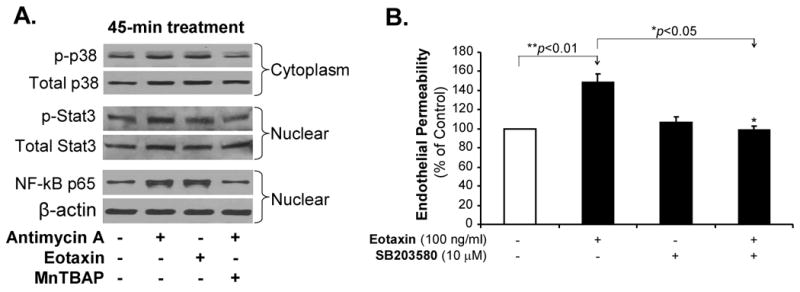
Effects of eotaxin, antimycin A and MnTBAP on the activation of p38, Stat3 and NFκB in HCAECs. A, Phosphorylation of p38 and Stat3 as well as nuclear translocation of NF-kB p65 in HCAECs. Cells were treated with either eotaxin (100 ng/mL) or antimycin A (10 μg/mL) for 45 min or pretreated with MnTBAP (2 μM) for 30 min followed by antimycin A treatment for 30 min. Cytoplasmic and nuclear extracts were isolated, respectively. Phosphorylated (p) and total p38 and Stat3 as well as NF-kB p65 proteins were detected by the Western blotting analysis. Loading efficiency was determined by reprobing the blot with β-actin antibody. The experiment was repeated twice. B, Endothelial permeability. HCAECs were treated with eotaxin (100 ng/mL) alone or pretreated with specific p38 inhibitor SB203580 followed by eotaxin treatment for 24 h. Endothelial permeability was assayed. n=3. *p<0.05 (the comparison is indicated by the arrow head). Error bars represent SD.
We further determined whether transcriptional factors such Stat3 and NF-κB could be involved in the eotaxin-induced signal pathways because these transcriptional factors are also sensitive to oxidative stress. Eotaxin or antimycin A treatment for 45 min substantially increased the phosphorylation of Stat3 and nuclear translocation of NF-κB in HCAECs, while antioxidant MnTBAP effectively blocked these effects (Figure 5A). Thus, MAPK p38 and transcriptional factors Stat3 and NF-κB may be involved in the eotaxin-induced signal transduction pathways though the oxidative stress mechanism.
Discussion
Eotaxin (CCL11) is an eosinophil-specific chemo attractant which has been found to be highly expressed at sites of vascular pathology.13 Eotaxin selectively attracts eosinophils by activating CCR3 receptors.22 However, it is not clear whether eotaxin could contribute to the progression of atherosclerosis. The present study, for the first time, reports three novel findings: 1). eotaxin increases the paracellular permeability in HCAECs; 2). eotaxin decreases the expression of tight junction proteins involved in the regulation of endothelial barrier functions; and 3). eotaxin may mediate its effects through oxidative stress and activation of the p38 MAPK, Stat3 and NF-κB signaling pathways.
In this study, we used a Costar transwell permeability model system to study the effect of eotaxin on paracellular permeability in HCAECs. This model has been used successfully in this laboratory to analyze the effects of ritanovir,20 lysophosphatidylcholine,21 stanniocalcin-1,17 and secretoneurin19 on endothelial permeability in vitro. We selected eotaxin concentrations ranging from 50 to 200 ng/mL based on the plasma eotaxin levels in patients with coronary artery disease.22 Our results show that eotaxin treatment for 24 h significantly increased endothelial permeability in HCAECs in a concentration-dependent manner. To rule out the possibility that the permeability increase is not due to the leakage through confluent HCAECs, monolayer was grown to confluence, stained with Calcein AM and checked under fluorescent microscope to make sure that the monolayer was confluent (data not shown). Heat-inactivated (HI) eotaxin did not show any effects on endothelial permeability indicating its specific effect, but not potential contamination of endotoxin. The endothelial apoptotic cascade is an important underlying mechanism of capillary leakage.23,24 In this study, we have shown that eotaxin did not affect apoptosis of HCAECs in the experimental condition.
Chemokines and chemokine receptors have emerged as important factors involved in the mobilization and function of leukocytes. Chemokine receptors are expressed on a wide range of leukocytes, as well as on endothelial cells, neurons, and possibly other cell types where they are involved in signaling events that can lead to eosinophil and mast cell degranulation, T cell activation, lymphocyte homing, chemotaxis, and mitogenic effects as well as hematopoiesis.25 In addition, overexpression of eotaxin and its receptor, CCR3, in human atherosclerosis have also been reported.11. In our study, we found that CCR3 receptor is constitutively expressed in HCAECs; while eotaxin treatment did not further increase the expression of CCR3 receptors. CCR3 expression was also observed in human atherosclerotic arteries. Pretreatment of HCAECs with anti-CCR3 antibody significantly reduced eotaxin-induced permeability increase. These findings suggest that CCR3 may play an important role in eotaxin-mediated endothelial permeability increase in HCAECs.
Intercellular junctional structures mediate adhesion and communication between adjoining endothelial cells and comprise of tight junction molecules including transmembrane proteins such as occludin, claudin and JAM-1 and intracellular proteins such as ZO-1 and cingulin as well as adherens junction proteins including transmembrane protein VE-cadherin and intracellular protein β-catenin. Tight junctions serve the major functional purpose of providing a “barrier” and a “fence” within the membrane by regulating paracellular permeability and maintaining cell polarity. In this study, we show that eotaxin significantly decreases the expression of tight junction proteins ZO-1, occludin and claudin-1 in a concentration-dependent manner at both mRNA and protein levels. However, eotaxin does not decrease the expression of VE-cadherin and JAM-1. These data indicate that down-regulation of tight junction proteins may be the key mechanism involved in the paracellular permeability increase in eotaxin-treated HCAECs.
ROS including superoxide anion, hydrogen peroxide, hydroxyl radical, and peroxynitrite play critical roles in cardiovascular disease.26 They could directly cause vascular damages, and could also act as signaling molecules for the gene expression in response to proinflammatory stimuli.27-29 ROS cause endothelial barrier dysfunction through alterations in the cytoskeleton and extracellular matrix.30 ROS are known to quench NO.30 NO synthesis inhibition can potentiate agonist-induced increases in vascular permeability or increase basal microvascular permeability via an alteration of endothelial actin cytoskeleton.31 In the present study, we showed a significant decrease of cellular GSH levels in eotaxin-treated cells, indicating that eotaxin may increase ROS production in HCAECs. Co-treatment of HCAECs with eotaxin and antioxidant MnTBAP substantially increase GSH levels, suggesting that MnTBAP may inhibit eotaxin-induced oxidative stress in HCAECs. To further elucidate the role of ROS in eotaxin-mediated permeability increase, we treated the cells with antimycin A, an inhibitor of electron transport in mitochondria and have been used as a ROS generator in biological systems. We found that antimycin A can induce endothelial permeability and ROS production in HCAECs. To further confirm the involvement of ROS in eotaxin-mediated hyper-permeability, HCAECs were treated with either antioxidant ginkgolide B32 or commonly used antioxidant MnTBAP, a cell permeable SOD mimetic, which effectively blocked the eotaxin-induced increase in vascular permeability and decrease in the expression of tight junction proteins, respectively. The natural antioxidant gingokolide B, a traditional Chinese herb from the plant Gingko Biloba, has less toxicity and side effects. Antioxidants are believed to counteract ROS and reduce the incidence of coronary artery disease.33 Thus, the current study suggest that eotaxin-induced oxidative stress may be one of the molecular mechanisms involved in the damage of endothelial barrier function and the use of antioxidants could be a novel strategy in the treatment of patients with high incidence of cardiovascular disease.
MAPKs play an important role in mediating cellular functions in response to many extracellular stimuli. There are three important MAPKs including extracellular signal regulated kinase (ERK1/2), c-Jun N-terminal kinase (JNK) and p38 in the cell. In the present study, we show that eotaxin can activate p38 in HCAECs. MAPK p38 activation was at 45 min of eotaxin treatment and there was no activation of p38 at 2 hours after eotaxin treatment. We previously reported that quick activation of MAPK p38 (within 5 to 10 min) was observed during lysophosphatidylcholine-induced increase of monolayer cell permeability in HCAECs.21 Sidestream cigarette smoke could also activate p38 within 60 min and induce endothelial permeability increase in human pulmonary endothelial cells.34 More importantly, specific p38 inhibitor SB203580 effectively blocked eotaxin-induced permeability increase in HCAECs. Blocking ROS generation also inhibited eotaxin-induced phosphorylation of p38, which indicates ROS acts as upstream effector of p38 under the stimulation of eotaxin. Possibly transient expression of p38 initiates the events leading to dysregulation of barrier function through the activation of downstream signal transduction pathways. Indeed, IL-6 can increase endothelial permeability through activation of transcriptional factor Stat3.35 Activation of transcription factor NF-κB has also been indicated for the TNF-α-permeability increase in HCAECs.18 We found activation of Stat3 and NF-κB under the stimulation of eotaxin in HCAEC. It is possible that Stat3 and NF-κB may regulate the expression of tight junction proteins. However, a direct link between Stat3 and NF-κB activation and repression of tight junction proteins has not been determined in this study.
In summary, the present study demonstrates that eotaxin can increase vascular permeability in HCAECs. The underlying molecular mechanisms may involve down-regulation of tight junction proteins, increase of oxidative stress and activation of MAPK p38, Stat3 and NF-κB. This study provides a new understanding of biological functions of eotaxin on the vascular system. Reducing oxidative stress or inhibiting p38 activation may be new strategies for inhibiting the detrimental effects of eotaxin on the vascular system, thereby preventing cardiovascular disease.
Supplementary Material
Acknowledgments
This work is partially supported by a NIH grant (R01HL083471 to C.C.) and by the Michael E. DeBakey Department of Surgery, Baylor College of Medicine. Authors would like to thank Dr. Shaoyu Yan for his technique assistance and data collection effort.
Footnotes
Disclosures: None.
References
- 1.Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778:794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Bonetti PO, Best PJ, Rodriguez-Porcel M, Holmes DR, Jr, Lerman LO, Lerman A. Endothelin type A receptor antagonism restores myocardial perfusion response to adenosine in experimental hypercholesterolemia. Atherosclerosis. 2003;168:367–373. doi: 10.1016/s0021-9150(03)00141-2. [DOI] [PubMed] [Google Scholar]
- 3.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2003;62:5381–5385. [PubMed] [Google Scholar]
- 4.Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol. 2008;23:243–249. doi: 10.1097/MOH.0b013e3282f97d86. [DOI] [PubMed] [Google Scholar]
- 5.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 6.Dejana E, Lampugnani MG, Martinez-Estrada O, Bazzoni G. The molecular organization of endothelial junctions and their functional role in vascular morphogenesis and permeability. Int J Dev Biol. 2000;44:743–748. [PubMed] [Google Scholar]
- 7.Bogatcheva NV, Garcia JG, Verin AD. Role of tyrosine kinase signaling in endothelial cell barrier regulation. Vasc Pharmacol. 2002;39:201–212. doi: 10.1016/s1537-1891(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 8.Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, Pardi R, Bender JR. Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol Heart Circ Physiol. 1997;273:H286–H294. doi: 10.1152/ajpheart.1997.273.1.H286. [DOI] [PubMed] [Google Scholar]
- 9.Ponath PD, Qin S, Ringler DJ, Clark-Lewis I, Wang J, Kassam N, Smith H, Shi X, Gonzalo JA, Newman W, Gutierrez-Ramos JC, Mackay CR. Cloning of the human eosinophil chemoattractant, eotaxin. Expression, receptor binding, and functional properties suggest a mechanism for the selective recruitment of eosinophils. J Clin Invest. 1996;97:604–612. doi: 10.1172/JCI118456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2:449–456. doi: 10.1038/nm0496-449. [DOI] [PubMed] [Google Scholar]
- 11.Haley KJ, Lilly CM, Yang JH, Feng Y, Kennedy SP, Turi TG, Thompson JF, Sukhova GH, Libby P, Lee RT. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation. 2000;102:2185–2189. doi: 10.1161/01.cir.102.18.2185. [DOI] [PubMed] [Google Scholar]
- 12.Kaehler J, Tuleweit A, Steven D, Krempl T, Haar A, Carstensen M, Koester R, Terres W, Meinertz T. Association between Eotaxin (CCL11), C-reactive protein, and antimicrobial antibodies in patients undergoing coronary angioplasty. J Invest Med. 2006;54:446–454. doi: 10.2310/6650.2006.06025. [DOI] [PubMed] [Google Scholar]
- 13.Farahi N, Cowburn AS, Upton PD, Deighton J, Sobolewski A, Gherardi E, Morrell NW, Chilvers ER. Eotaxin-1/CC chemokine ligand 11: a novel eosinophil survival factor secreted by human pulmonary artery endothelial cells. J Immunol. 2007;179:1264–1273. doi: 10.4049/jimmunol.179.2.1264. [DOI] [PubMed] [Google Scholar]
- 14.Economou E, Tousoulis D, Katinioti A, Stefanadis C, Trikas A, Pitsavos C, Tentolouris C, Toutouza MG, Toutouzas P. Chemokines in patients with ischaemic heart disease and the effect of coronary angioplasty. Int J Cardiol. 2001;80:55–60. doi: 10.1016/s0167-5273(01)00454-5. [DOI] [PubMed] [Google Scholar]
- 15.Zee RY, Cook NA, Cheng S, Erlich HA, Lindpaintner K, Lee RT, Ridker PM. Threonine for alanine substitution in the eotaxin (CCL11) gene and the risk of incident myocardial infarction. Atherosclerosis. 2004;175:91–94. doi: 10.1016/j.atherosclerosis.2004.01.042. [DOI] [PubMed] [Google Scholar]
- 16.Cheng SS, Lukacs NW, Kunkel SL. Eotaxin/CCL11 suppresses IL-8/CXCL8 secretion from human dermal microvascular endothelial cells. J Immunol. 2002;168:2887–2894. doi: 10.4049/jimmunol.168.6.2887. [DOI] [PubMed] [Google Scholar]
- 17.Kodali RB, Kim WJ, Galaria II, Miller C, Schecter AD, Lira SA, Taubman MB. CCL11 (Eotaxin) induces CCR3-dependent smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2004;7:1211–1216. doi: 10.1161/01.ATV.0000131654.90788.f5. [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Jamaluddin S, Yan S, Sheikh-Hamad D, Yao Q. Human stanniocalcin-1 blocks TNF-α-induced monolayer permeability in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:906–912. doi: 10.1161/ATVBAHA.108.163667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan S, Wang X, Chai H, Wang H, Yao Q, Chen C. Secretoneurin increases monolayer permeability in human coronary artery endothelial cells. Surgery. 2006;140:243–251. doi: 10.1016/j.surg.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Mu H, Chai H, Liao D, Yao Q, Chen C. Human immunodeficiency virus protease inhibitor ritonavir inhibits cholesterol efflux from human macrophage-derived foam cells. Am J Pathol. 2007;171:304–314. doi: 10.2353/ajpath.2007.060965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan S, Chai H, Wang H, Yang H, Nan B, Yao Q, Chen C. Effects of lysophosphatidylcholine on monolayer cell permeability of human coronary artery endothelial cells. Surgery. 2005;138:464–473. doi: 10.1016/j.surg.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 22.Emanuele E, Falcone C, D'Angelo A, Minoretti P, M P, Bertona M, Geroldi D. Association of plasma eotaxin levels with the presence and extent of angiographic coronary artery disease. Atherosclerosis. 2006;186:140–145. doi: 10.1016/j.atherosclerosis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Syrkina O, Jafari B, Hales CA, Quinn DA. Oxidant stress mediates inflammation and apoptosis in ventilator-induced lung injury. Respirology. 2008;3:333–340. doi: 10.1111/j.1440-1843.2008.01279.x. [DOI] [PubMed] [Google Scholar]
- 24.Vaschetto R, Kuiper JW, Chiang SR, Haitsma JJ, Juco JW, Uhlig S, Plötz FB, Della Corte F, Zhang H, Slutsky AS. Inhibition of poly(adenosine diphosphate-ribose) polymerase attenuates ventilator-induced lung injury. Anesthesiology. 2008;2:261–268. doi: 10.1097/01.anes.0000299434.86640.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hillyer P, Mordelet E, Flynn G, Male D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: discriminating the tissue-specific functions that affect leucocyte migration. Clin Exp Immunol. 2003;3:431–441. doi: 10.1111/j.1365-2249.2003.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Apel K, Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol. 2004;55:373–399. doi: 10.1146/annurev.arplant.55.031903.141701. [DOI] [PubMed] [Google Scholar]
- 27.Zhao RZ, Chen X, Yao Q, Chen C. TNF-alpha induces interleukin-8 and endothelin-1 expression in human endothelial cells with different redox pathways. Biochem Biophys Res Commun. 2005;327:985–992. doi: 10.1016/j.bbrc.2004.12.109. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 29.Berliner JA, Heinecke JW. The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med. 1996;20:707–727. doi: 10.1016/0891-5849(95)02173-6. [DOI] [PubMed] [Google Scholar]
- 30.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin AL, Thurston G, al Naemi H. Inhibition of nitric synthesis increases venular permeability and alters endothelial actin cytoskeleton. Am J Physiol. 1998;274:H1776–H1784. doi: 10.1152/ajpheart.1998.274.5.H1776. [DOI] [PubMed] [Google Scholar]
- 32.Xia SH, Fang DC. Pharmacological action and mechanisms of ginkgolide B. Chin Med J. 2007;120:922–928. [PubMed] [Google Scholar]
- 33.Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328:1450–1456. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- 34.Low B, Liang M, Fu J. p38 mitogen-activated protein kinase mediates sidestream cigarette smoke-induced endothelial permeability. J Pharmacol Sci. 2007;3:225–231. doi: 10.1254/jphs.fp0070385. [DOI] [PubMed] [Google Scholar]
- 35.Maruo N, Morita I, Shirao M, Murota S. IL-6 increases endothelial permeability in vitro. Endocrinology. 1992;131:710–714. doi: 10.1210/endo.131.2.1639018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.