

Abstract
We studied the effect on ureagenesis of a single dose of N-carbamylglutamate (NCG) in healthy young adults who received a constant infusion (300 min) of NaH13CO3. Isotope ratio-mass spectrometry was used to measure the appearance of label in [13C]urea. At 90 minutes after initiating the H13CO3− infusion each subject took a single dose of NCG (50 mg/kg). In 5/6 studies the administration of NCG increased the formation of [13C]urea. Treatment with NCG significantly diminished the concentration of blood alanine, but not that of glutamine or arginine. The blood glucose concentration was unaffected by NCG administration. No untoward side effects were observed. The data indicate that treatment with NCG stimulates ureagenesis and could be useful in clinical settings of acute hyperammonemia of various etiologies.
Keywords: N-Carbamyl-L-Glutamate, N-Acetyl-L-Glutamate Synthetase, Carbamyl-Phosphate Synthetase, Urea Cycle Defects, Stable Isotopes
Introduction
N-acetylglutamate (NAG) serves as an obligatory effector for the carbamyl phosphate synthase 1 (CPS1) (EC 6.3.4.16) reaction, the initial and rate-limiting first step of the urea cycle [1]. An absence of NAG, which can occur as a consequence of NAG synthase (NAGS) (EC 2.3.1.1) deficiency, compromises flux through CPS1, thereby impairing ureagenesis and causing hyperammonemia, neurological sequelae and even death [2–5]
N-carbamylglutmate (NCG) is a synthetic analog of NAG that activates CPS1 [6]. Treatment with NCG restores ureagenesis and normalizes blood ammonia concentrations in patients with NAGS deficiency [7–11]. The administration of NCG also improves ureagenesis in patients with inborn errors of metabolism such as propionic acidemia and methylmalonic acidemia [12, 13], in which a decreased mitochondrial concentration of NAG may limit ammonia flux through CPS1 [11, 14, 15]. NCG treatment might prove beneficial in other clinical settings that are characterized by a relative compromise of the CPS1 reaction. Examples include the hyperinsulinism and hyperammonemia syndrome (HIHA) [16], which involves a gain-of-function mutation in glutamate dehydrogenase (EC 1.4.1.4) that reduces the hepatic mitochondrial concentration of glutamate below the Km for NAGS [17]. Similarly, the level of NAG may be appreciably lowered in other hyperammonemic syndromes, including hepatic encephalopathy and valproate therapy [18, 19].
Indirect evidence indicates that, even when the CPS1 reaction is intact, only 35–60% of this mitochondrial enzyme is in an active form in the basal state [20]. The proportion of active CPS1, which is the most abundant mitochondrial matrix protein [21], depends in large measure on the mitochondrial concentration of NAG [20, 22, 23]. Molecules of CPS1 remain catalytically silent unless NAG binds to them [1]. Thus a saturating dose of NCG should activate unliganded CPS1, thereby increasing flux through this rate-limiting enzyme of the urea cycle.
We have tested this hypothesis by studying the effect of NCG on the rate of ureagenesis in healthy human volunteers. Our goal was to ascertain whether a single dose of NCG would augment ureagenesis as measured by the in vivo synthesis of [13C]urea during the intravenous administration of [13C]sodium bicarbonate. The rationale is that labeled bicarbonate is readily incorporated into [13C]urea at a rate that reflects flux through the urea cycle [24, 25] and that NCG would enhance [13C]urea synthesis by increasing flux through CPS1. Our data confirm that treatment with NCG has such a salutary effect and that this agent might therefore prove beneficial in the management of hyperammonemia secondary to a relative deficiency (or insufficiency) of NAG of diverse etiology.
1. Subjects and Methods
1.1 Subjects
We performed 6 studies in 5 healthy volunteers between the ages of 20–40 years. One individual (Subject 4) was studied on 2 occasions at an interval of 4 months. Subjects took no medications and were free of any evidence of major organ or systemic disease. None of the subjects had any medical or family history to suggest a urea cycle defect or other syndrome of protein intolerance. A negative pregnancy test was required of all female subjects before the study. The study was approved by the Institutional Review Boards at the Children’s Hospital of Philadelphia and Children’s National Medical Center. Informed consent was obtained from each individual before participation in the study.
1.2 Experimental Procedure
Subjects consumed a low protein (< 40 gm) meal at 6 PM on the evening prior to study. Thereafter, they consumed nothing but water until the morning of the study, which began at 8 AM. An indwelling catheter was placed in an antecubital vein for blood drawing. A separate intravenous line was placed on the dorsum of the contralateral hand for the purpose of isotope administration.
Baseline samples (5 ml) of heparinized blood were centrifuged to separate plasma, which was kept frozen (−70° C) until analyzed for H13CO3−, [13C]urea and plasma amino acids, as described below. In addition, a sample of expired breath was obtained by having the subject exhale into a plastic collection bag equipped with a one-way valve. The air was analyzed for 13CO2, as described below.
After obtaining the baseline specimens, a priming dose (0.083 mmol/kg) of a sterile solution of NaH13CO3 (98 atom % excess;) was infused intravenously over 10 minutes. This was immediately followed by a continuous infusion (0.089 mmol/kg/hr). Heparinzed blood samples were obtained at 10, 15, 30, 45, 60, 75, 90, 105, 130, 150, 170, 190, 210, 230, 250, 270 and 300 min during the infusion.
Ninety minutes after starting the infusion each subject received a single oral dose (50 mg/kg) of N-carbamylglutamate (Carbaglu, Orphan Europe). The dispersable tablets were dissolved in 60 ml of water. Subjects drank an additional 60 ml of water following administration of the drug.
1.3 Measurement of [13C]Urea and 13CO2
In a plastic tube, 40 µL of 60% perchloric acid and 0.5 mL of deionized water were added sequentially to 0.5 mL of plasma. The precipitated protein was removed by centrifugation. The tube was uncapped for 30 min to allow removal of most CO2. The supernatant was transferred to a new tube and the pH was adjusted to 6–7 with KOH. After removal of precipitated potassium perchlorate, the sample was applied to an AG-1 column (1 ml; Cl− form; 100–200 mesh) to remove traces of bicarbonate. The eluate was then combined with a 2 ml wash of 10 mmol/l HCl. The sample was dried in a glass tube under a stream of compressed air while heated to 80° C.
The dried sample was then left for 2 hours in an airtight container the bottom of which was lined with gauze soaked with 1M NaOH to remove any residual trace of CO2. The glass tube was then flushed with helium and capped with a rubber stopper. 400 µL of 0.5 mol/l pre-boiled and cooled (to remove any CO2) potassium phosphate solution, pH 6.0, containing urease (3 mg/0.4 ml) was injected through the stopper. After 60 min incubation at 37°C, 100 µL of 20% phosphoric acid (final concentration 0.4 mol/l) was injected through the rubber stopper. The atmosphere is then analyzed for 13CO2 by a Finnigan Delta Plus isotope ratio-mass spectrometer (Thermo Fisher Scientific, Waltham, MA). A commercial CO2 source (Airgas, Radnor, PA) was used as the standard.
The 13CO2 label in expired air was determined by using a gas-tight syring to transfer an aliquot of breath from the plastic collection device to the sampling tube of the mass spectrometer.
1.4 Amino Acid and Glucose Assay
Amino acids were measured in plasma with liquid chromatography of the o-pthalaldehyde derivatives [26]. Glucose was measured with the glucose oxidase assay [27].
1.5 Data Analysis
Label in 13C (Atom % Excess) is defined as APE = 100 * (Rs − Rb)/(100 + (Rs − Rb)), where Rs is the ratio (M+1/M) in the sample (either 13CO2 or [13C]urea) at any time after initiation of the infusion of NaH13CO3 and Rb is the same ratio at T0.
The rate of production of CO2 (QCO2) is QCO2 = d * (Id/I CO2 − 1), where d is the rate of infusion of NaH13CO3 and Id and ICO2 are respectively the 13C label in administered bicarbonate (98.0 atom % excess) and in plasma 13CO2 at steady-state.
The concentration of blood [13C]urea (C13C-Urea) = CUrea*IUrea/100, where CUrea is the concentration of blood urea (mmol/l) and IUrea is isotopic label (atom % excess) in blood urea at any given time following the initiation of the infusion of NaH13CO3.
The rate of urea synthesis was determined by performing linear regression analysis on C13C-Urea on time points from 10 to 150 minutes after starting the infusion of NaH13CO3.
Statistical analysis was by t-test for independent variables or, when appropriate, by paired t-test.
2. Results
2.1 Subjects of Study
In Table 1 is shown data for the 5 subjects. (Subject #4 was studied on 2 separate occasions). Each was a healthy young adult of normal weight and normal plasma ammonia (< 40 µmol/l) and urea (< 10 mmol/l).
TABLE 1.
Demographic data for the 5 subjects of study, including concentration of plasma ammonia and serum urea at baseline (0 min) and at termination (300 min) of the H13CO3− infusion.
| Subject | Sex | Age | Weight (kg) |
Baseline Plasma Ammonia (µmol/l) |
Final Plasma Ammonia (µmol/l) |
Baseline Serum Urea (mmol/l) |
Final Serum Urea (mmol/l) |
|---|---|---|---|---|---|---|---|
| 1 | M | 33 | 67 | 36 | 16 | 4.1 | 3.9 |
| 2 | M | 25 | 77 | 27 | 27 | 4.0 | 4.0 |
| 3 | F | 36 | 70 | 1 | 1 | 4.3 | 4.3 |
| 4A* | M | 38 | 75 | 35 | 54 | 10.8 | 10.2 |
| 4B* | M | 38 | 75 | 21 | 28 | 5.8 | 5.7 |
| 5 | F | 39 | 62 | 1 | 9 | 2.6 | 3.1 |
Subject 4 was studied twice
2.1 13CO2 in Breath and [13C]urea in Blood
Fig 1 illustrates label in breath 13CO2 for the 6 studies. Data are expressed as atom % excess relative to the baseline (T0) value to 13C label was attained by 10 minutes. The horizontal line denotes the mean value. The range of steady-state 13CO2 enrichment was 0.5–1.1 atom % excess with a mean of 0.713 ± 0.213 (SD). At a constant infusion rate of 0.089 mmol/kg/hr, this corresponds to a rate of carbon dioxide production of 21.7 ± 5.4 mol/70 kg/day, a result in good agreement with published values [28]. Administration of NCG did not affect bicarbonate turnover (Fig.1). Patients did not demonstrate acidemia or alkalemia post-treatment (data not shown).
Figure 1.
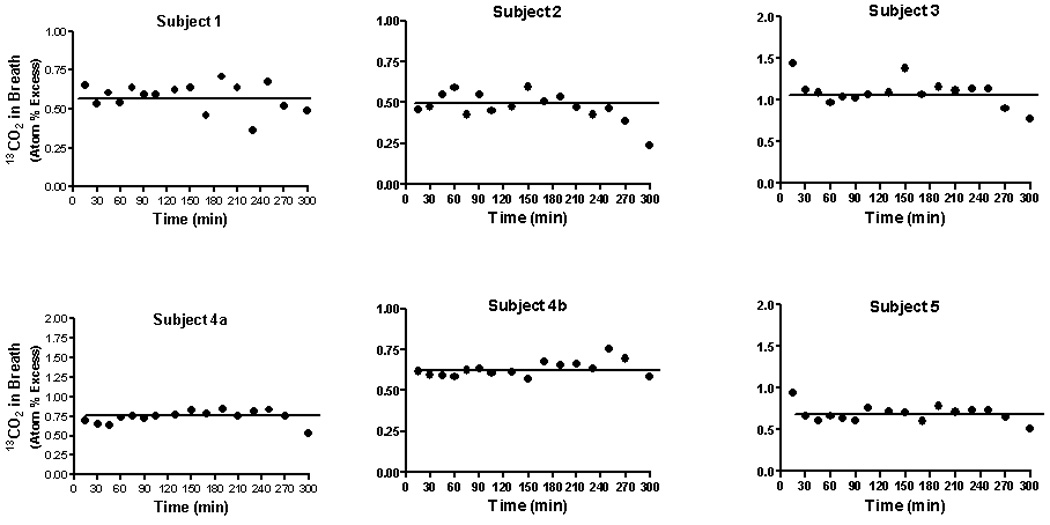
Isotopic abundance (atom % excess) in 13CO2 in expired air in healthy adults who received an infusion of NaH13CO3 (0.089 mmol/kg/hr; 98 atom % excess) for a period of 300 minutes. The solid horizontal line denotes the mean isotopic abundance from 10 to 300 minutes. During the initial 10 minutes of the experiment each subject also received a priming dose (0.083 mmol/kg) of the labeled bicarbonate. Breath was taken at the times indicated by having each individual exhale into a plastic collection device fitted with a one-way valve. At 90 minutes each individual received an oral dose of NCG (100 mg/kg).
The appearance of [13C]urea in blood is shown in Figure 2. As indicated above (Methods) these values correspond to the product of isotopic abundance (atom % excess) and the concentration of blood urea (µmol/liter). Isotopic enrichment above baseline was evident by 10 minutes and increased steadily thereafter. In each study the incremental pattern of 13C labeling was linear until 130 minutes (Fig. 2, solid circles) with a mean rate of increase of blood [13C]urea of 0.021 ± 0.011 µmol/min/l.
Fig. 2.
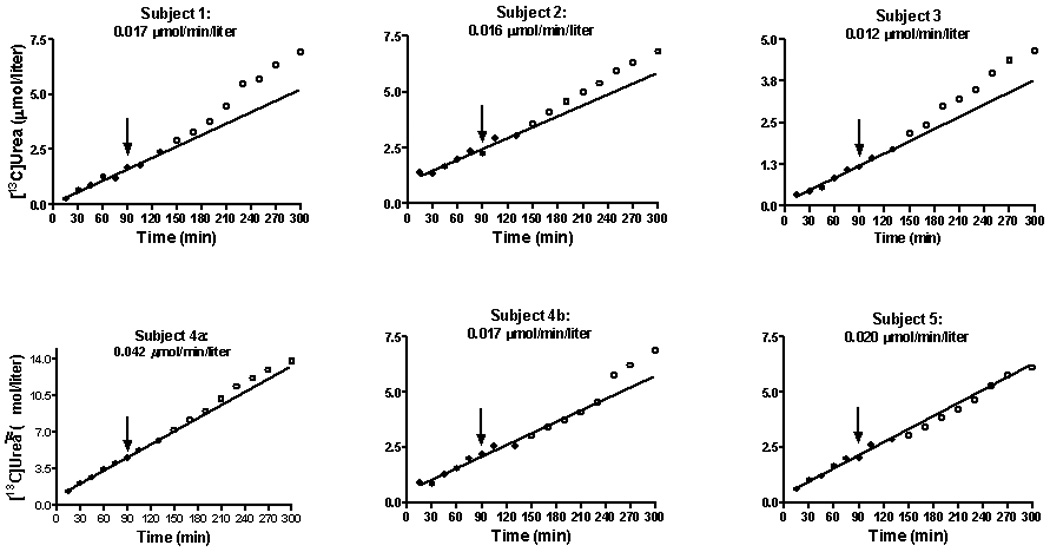
The concentration of [13C]urea in plasma during the constant infusion of NaH13CO3 (Fig. 1) in healthy adults. The line represents linear regression analysis from 10 to 150 minutes. At 90 minutes each subject received an oral dose (50 mg/kg) of NCG. The closed circles correspond to the period during which the linear regression was performed. The open circles are points from 150 until 300 minutes and correspond to the post-treatment values. The time from 10 until 150 minutes is included in the regression because this is a period of drug absorption. The number at the top of each graph(µmol/min/liter) correspond to the rate of [13C]urea appearance in blood. Calculation was done as explained above (Methods).
Each subject received a single dose of NCG at 90 minutes (Fig. 2, arrow). In 5 of the 6 studies we noted an upward inflection in the slope of the curve of ureagenesis. In the first 4 studies this increase in ureagenesis was manifest at 130 minutes, or within 60 minutes of drug administration. In study 4b (a repeat study of subject 4) the increase was evident by 250 minutes. In Subject 5 no drug effect was noted.
Table 2 describes the observed vs expected concentration of blood [13C]urea at either 270 or 300 minutes. The expected values were calculated from the linear regression analysis that is represented in Fig. 2. In 5 of the 6 studies, the expected concentration was appreciably lower than that actually noted. In several instances the observed values were as much as 20–30% greater than the predicted result. In one instance (Subject 5) there was a close correlation between the concentration we measured and that we anticipated.
TABLE 2.
Observed and expected plasma [13C]Urea (µmol/L) at 270 min and 300 min. Also shown is the ratio (Variance Obs/Exp) of the observed/expected value. The expected values are calculated from the linear regression analysis, as shown in Fig. 2.
| Subject | Observed (270 min) |
Expected (270 min) |
Variance Obs/Exp: 270 min |
Observed (300 min) |
Expected (300 min) |
Variance Obs/Exp: 300 min |
|---|---|---|---|---|---|---|
| 1 | 6.340 | 4.662 | 1.360 | 6.907 | 5.172 | 1.335 |
| 2 | 6.330 | 5.330 | 1.188 | 6.800 | 5.810 | 1.170 |
| 3 | 4.380 | 3.422 | 1.280 | 4.660 | 3.791 | 1.229 |
| 4A | 12.980 | 12.172 | 1.066 | 13.820 | 13.432 | 1.029 |
| 4B | 6.198 | 5.127 | 1.209 | 6.860 | 5.637 | 1.217 |
| 5 | 5.747 | 5.770 | 0.996 | 6.092 | 6.370 | 0.956 |
Treatment with NCG augmented the appearance of [13C]urea with H13CO3− as precursor, but it did not consistently change the final (300 minute) concentration of blood ammonia or blood urea (Table 1).
2.2 Blood Amino Acid Levels
Plasma amino acid concentrations before-and-after administration of NCG are shown in Fig. 3. The data for each time point are expressed as a fraction (%) of the mean baseline (0–90 min) value. Significant changes post-treatment were observed for alanine, which declined from a baseline level of 100 ± 3.8% (221.9 ± 11.8 nmol/ml) to a post-treatment (105–300 min) value of 92.0 ± 4.9% (205.5 ± 16.5 nmol/ml) (p < .001). Similarly, blood isoleucine concentration was 100 ± 1.9% at baseline (59.8 ± 1.7 nmol/ml) and 92.0 ± 4.9% after treatment with a single dose of NCG (53.3 ± 2.3 nmol/ml). Of note is the observation that administration of NCG did not significantly affect the blood glutamine concentration (100 ± 3.7% at baseline vs 99.0 ± 5.1% post-treatment; 328.6 ± 26.6 nmol/ml vs 321.3 ± 15.0 nmol/ml). Similarly, treatment did not alter the blood arginine level (100 ± 9.3% vs 96.0 ± 5.2%; 101.2 ± 10.9 nmol/ml vs 96.8 ± 7.0 nmol/ml).
Fig. 3.
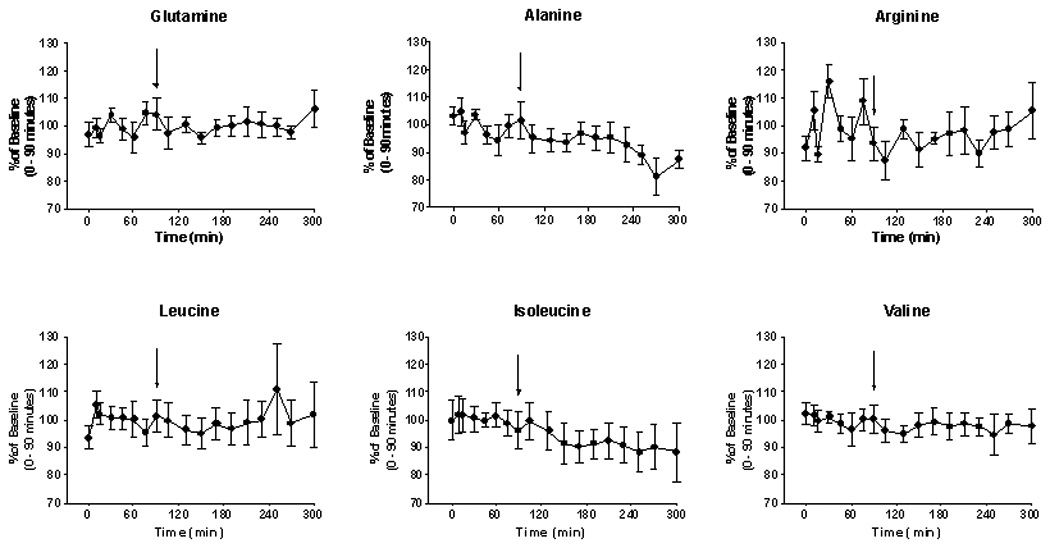
Plasma amino acid levels (mean ± SEM) for the 6 studies in healthy volunteers. The data are expressed as a percentage of the baseline T0) value in order to facilitate comparison. The arrow denotes an oral dose (50 mg/kg) of NCG, taken in an aqueous solution. As noted (Results), significant (p < .05) changes in concentration post-NCG were observed with regard to the concentration of alanine and of isoleucine.
2.3 Effect of N-Carbamylglutamate on Glucose Concentration in Blood
Ureagenesis and gluconeogenesis are inter-related processes since the conversion of amino acid N to urea may be linked to the conversion of a portion of amino acid carbon to glucose. We therefore followed the blood glucose concentration before-and-after treatment with NCG. As shown in Fig. 4, the blood glucose concentration (5.8 ± 0.2 mmol/l) remained constant during the period of experimentation. Administration of NCG at 90 minutes did not have a discernible effect on this parameter.
Fig. 4.
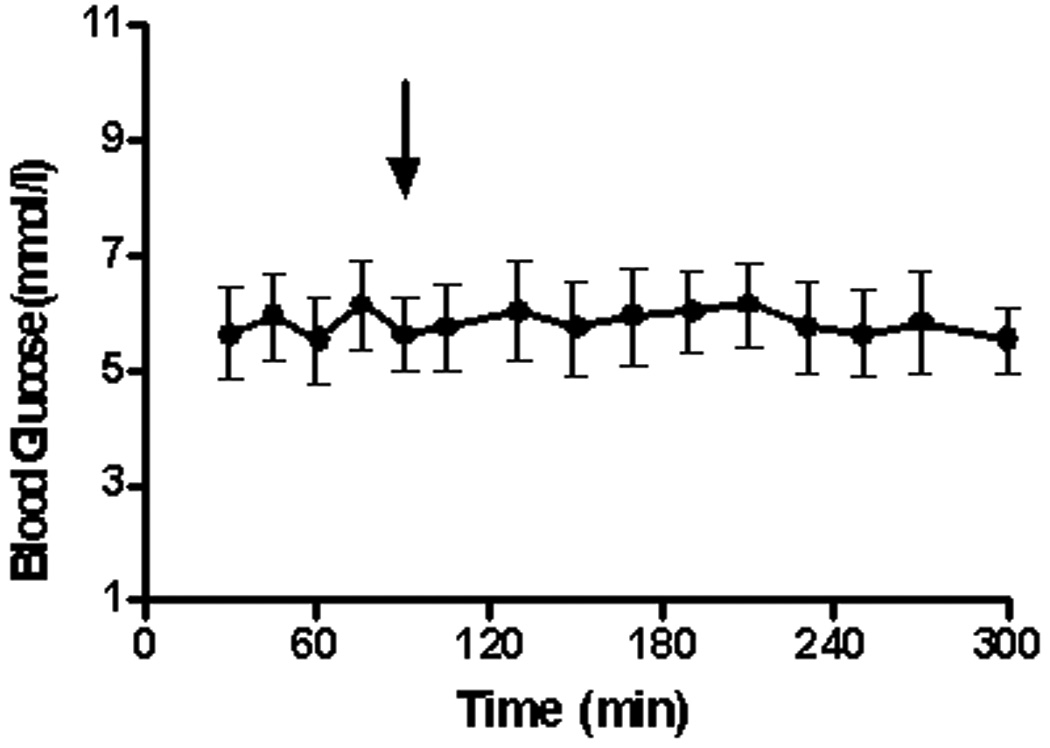
Blood glucose concentration (mmol/l; mean ± SEM) for the 6 studies. The arrow denotes the time (90 min) of administration of NCG (50 mg/kg).
3. Discussion
The urea cycle prevents the accumulation of ammonia, a potent neurotoxin. Vivid evidence of the importance of ureagenesis is the fact that CPS1, the initial and rate-limiting step of the urea cycle, constitutes almost 20% of hepatic mitochondrial protein [21]. Notwithstanding the abundance of this enzyme, the conversion of ammonia into carbamyl phosphate is almost nil unless CPS1 is allosterically activated by NAG [9].
Our interest in NCG derives from the fact that this compound can dramatically stimulate ureagenesis in patients with a probable deficiency of NAG. Examples include individuals with NAGS deficiency and those with propionic and methylmalonic acidemia [7, 9–11, 14, 15]. In the latter instance, a partial NAG deficiency may result from reduced concentration of acetyl-CoA or of free coenzyme A. In our prior investigations we deployed stable isotope methodologies ([1-13C]acetate and 15NH3) to demonstrate the restoration of a normal rate of ureagenesis when patients with DNA-documented NAGS deficiency were treated for 3 days with NCG [5, 11].
In the current study we have utilized a constant infusion method to assess the transfer of carbon from NaH13CO3 to [13C]urea, this parameter corresponding to the rate of ureagenesis. We monitored both the basal rate of urea synthesis and the response to treatment with NCG. The labeled H13CO3− articulates with the intra-mitochondrial pool to become [13C]carbamylphosphate in the CPS1 reaction. The latter species ultimately is converted to [13C]urea as it traverses the remaining steps of the urea cycle.
There is no single, “correct” tool with which to gauge the competency of ureagenesis or the response of the urea cycle to a putative therapeutic agent. An advantage of the constant infusion method is that it avoids the problem of variable gastrointestinal absorption of an orally administered tracer such as [1-13C]acetate. Indeed, we found, using a priming dose/constant infusion paradigm (Fig. 1), that we rapidly (< 10 min) achieved a steady-state with regard to label in 13CO2. Furthermore, we demonstrated (Fig. 2) that our method could detect within 10–15 minutes the appearance of this label in [13C]urea. Our study encompassed a period of 300 minutes, but our data (Fig. 2) suggest that we could shorten the experiment. This is an important consideration in studying children with defects of ureagenesis, including youngsters with urea cycle defects, organic acidemias, liver disease and those receiving anti-epileptic drugs such as valproate. In addition, we found that linear regression analysis describes the initial rate of labeling in [13C]urea (Fig. 2). The simplicity of the technique affords a useful tool with which to test the acute effect of a therapeutic agent like NCG.
For 5/6 studies the rate of rise of plasma [13C]urea was found to be within a narrow range (0.012–0.020 µmol/min/liter; Fig. 2). An exception was Subject 4a, in whom the increase was 0.042 µmol/min/liter (Fig. 2). For the group as a whole the mean was 0.021 ± 0.011 µmol/min/liter). A rate of total body urea synthesis can be estimated by normalizing this value to the plateau label in 13CO2, which had a mean level of 0.713 atom % excess (Fig. 1). This yields 2.95 µmol/min/liter. If we assume urea distribution in controls to be ~ 60% of body weight [29], this equates to 106 µmol/hr/kg, which is similar to the value reported by Lee et al (132 µmol/hr/kg) [30]. and to the value we previously reported (105 µmol/kg/hr) [31].
The sensitivity of the isotopic approach is documented by the finding (Fig. 2) that 13C label increased even though the concentration in blood of urea or ammonia did not change in a consistent manner when comparing the baseline and terminal (300 min) values (Table 1). Of course, the constancy in the level of these metabolites may reflect the fact that the current series of studies was done in healthy subjects. In our prior investigation of patients with NAGS deficiency we did note a frank increase of the blood urea concentration after treatment with NCG for 3 days [7, 11].
Similarly, we did not find an acute diminution of the blood glutamine level after administration of an agent that promoted ureagenesis (Fig. 3), but we did observe a fall of blood glutamine concentration in our patients with NAGS deficiency [11]. The current study showed a significant drop of the blood alanine level, which by 270 and 300 minutes declined by about 15% from the baseline value (Fig. 3). Administration of NCG may have augmented hepatic conversion of alanine N to urea, a well-known phenomenon [32,33]. Alanine is a major gluconeogenic precursor [34], and it is possible that alanine carbon was incorporated into glucose in these post-absorptive subjects.
Of interest is the rapidity of the hepatic response to NCG. In most studies a clear upward inflection in the rate of [13C]urea production became apparent by ~ 60 minutes after drug administration (Fig. 2), probably corresponding to the time necessary for this agent to be absorbed from the gut and to be transported to hepatic mitochondria and there to become bound to CPS1 . Indirect evidence suggests that 35–60% of CPS 1 exists in the active form in the basal state [20]. Hence, our data imply that a single large dose of NCG swiftly activates unbound enzyme. Intestinal absorption of the drug may have been sluggish in the one individual (Subject 5) who seemed refractory to drug treatment. Future dose-response studies should help to clarify this point.
The results of this study, together with our prior investigations, may provide a rationale for the use of NCG to treat hyperammonemia in conditions other than NAGS deficiency or organic acidemias. Hyperammonemia consequent to hepatic failure might be such an application [19]. In both clinical settings patients typically are treated with low-protein diets, which in themselves tend to down-regulate NAGS activity [35]. Similarly, it is possible that a subset of patients with mutations of CPS1 could benefit from treatment, especially if the mutation affected the binding of NAG. It also may be that binding of NCG to CPS1 could help to stabilize an otherwise labile enzyme, as may occur with regard to the binding of pyridoxine to cystathionine synthase (EC 4.2.1.22) [36] or reduced biopterin to phenylalanine hydroxylase (EC 1.5.1.34) [37].
Indeed, treatment with NCG could prove a useful therapeutic adjunct in other urea cycle defects. Treatment of patients with citrullinemia or argininosuccinic aciduria usually entails administration of a low-protein diet as well as high doses of arginine, since this amino acid furnishes the carbon “backbone” by which waste nitrogen is excreted into the urine as citrulline or argininosuccinate. The low-protein diet, although therapeutically necessary to minimize NH3 production, also might attenuate flux through CPS1. Concurrent administration of NCG, which augments the activity of CPS1, might thereby facilitate the synthesis of a vehicle for elimination of excess nitrogen.
NCG therapy even might prove useful in ornithine transcarbamylase (OTC) (EC 2.1.3.3) deficiency, the most common of the urea cycle defects [38–40]. If the mutant OTC had poor affinity for carbamyl phosphate, then augmenting the pool of the latter intermediate ought to be of clinical value. Similarly, if the binding of carbamyl phosphate to OTC stabilized an otherwise labile enzyme, then treatment with NCG might confer a beneficial effect. Finally, increasing hepatic production of carbamyl phosphate should favor synthesis of orotic acid [41,42]. The latter metabolite is rapidly excreted in the urine, thereby providing an auxiliary route for waste N elimination. The amount of orotic acid produced is low relative to urea synthesis, but in patients with a partial deficiency of OTC a very fine line may exist between metabolic competency and decompensation. In these situations, just enough N might be excreted as orotate to improve overall function.
Acknowledgements
This work was supported by NIH grants HD058567, HD26979, DK53761, RR00240, RR019453, NS054900,. We thank the CTRC nursing staff at the Children’s Hospital of Philadelphia for their expert support.
Abbreviations
- APE
atom % excess
- CPS1
carbamyl phosphate synthase 1
- NAG
N-acetylglutamate
- NAGS
N-acetylglutamate synthase
- NCG
N-carbamylglutamate
- HIHA
hyperinsulinism/hyperammonemia syndrome
Contributor Information
Marc Yudkoff, Email: yudkoff@email.chop.edu.
Nicholas Ah Mew, Email: nahmew@cnmc.org.
Irma Payan, Email: payan@email.chop.edu.
Yevgeny Daikhin, Email: daikhin@email.chop.edu.
Ilana Nissim, Email: nissimi@email.chop.edu.
Itzhak Nissim, Email: nissim@email.chop.edu.
Mendel Tuchman, Email: mtuchman@cnmc.org.
References
- 1.Hall LM, Metzenberg RL, Cohen PP. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230:1013–1021. [PubMed] [Google Scholar]
- 2.Bachmann C, Colombo JP, Jaggi K. N-acetylglutamate synthetase (NAGS) deficiency: diagnosis, clinical observations and treatment. Adv Exp Med Biol. 1982;153:39–45. doi: 10.1007/978-1-4757-6903-6_6. [DOI] [PubMed] [Google Scholar]
- 3.Schubiger G, Bachmann C, Barben P, Colombo JP, Tonz O, Schupbach D. N-acetylglutamate synthetase deficiency: diagnosis, management and follow-up of a rare disorder of ammonia detoxication. Eur J Pediatr. 1991;150:353–356. doi: 10.1007/BF01955939. [DOI] [PubMed] [Google Scholar]
- 4.Caldovic L, Morizono H, Panglao MG, Cheng SF, Packman S, Tuchman M. Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Hum Genet. 2003;112:364–368. doi: 10.1007/s00439-003-0909-5. [DOI] [PubMed] [Google Scholar]
- 5.Caldovic L, Morizono H, Panglao MG, Lopez GY, Shi D, Summar ML, Tuchman M. Late onset N-acetylglutamate synthase deficiency caused by hypomorphic alleles. Hum Mutat. 2005;25:293–298. doi: 10.1002/humu.20146. [DOI] [PubMed] [Google Scholar]
- 6.Grisolia S, Cohen PP. The catalytic role of carbamyl glutamate in citrulline biosynthesis. J Biol Chem. 1952;198:561–571. [PubMed] [Google Scholar]
- 7.Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, Tuchman M. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr. 2004;145:552–554. doi: 10.1016/j.jpeds.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 8.Morris AA, Richmond SW, Oddie SJ, Pourfarzam M, Worthington V, Leonard JV. N-acetylglutamate synthetase deficiency: favourable experience with carbamylglutamate. J Inherit Metab Dis. 1998;21:867–868. doi: 10.1023/a:1005478904186. [DOI] [PubMed] [Google Scholar]
- 9.Guffon N, Vianey-Saban C, Bourgeois J, Rabier D, Colombo JP, Guibaud P. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J Inherit Metab Dis. 1995;18:61–65. doi: 10.1007/BF00711374. [DOI] [PubMed] [Google Scholar]
- 10.Hinnie J, Colombo JP, Wermuth B, Dryburgh FJ. N-Acetylglutamate synthetase deficiency responding to carbamylglutamate. J Inherit Metab Dis. 1997;20:839–840. doi: 10.1023/a:1005344507536. [DOI] [PubMed] [Google Scholar]
- 11.Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Korson M, Burton B, Yudkoff M. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64:213–217. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shafai T, Sweetman L, Weyler W, Goodman SI, Fennessey PV, Nyhan WL. Propionic acidemia with severe hyperammonemia and defective glycine metabolism. J Pediatr. 1978;92:84–86. doi: 10.1016/s0022-3476(78)80081-x. [DOI] [PubMed] [Google Scholar]
- 13.Cathelineau L, Briand P, Ogier H, Charpentier C, Coude FX, Saudubray JM. Occurrence of hyperammonemia in the course of 17 cases of methylmalonic acidemia. J Pediatr. 1981;99:279–280. doi: 10.1016/s0022-3476(81)80478-7. [DOI] [PubMed] [Google Scholar]
- 14.Gebhardt B, Dittrich S, Parbel S, Vlaho S, Matsika O, Bohles H. N-carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemia. J Inherit Metab Dis. 2005;28:241–244. doi: 10.1007/s10545-005-5260-7. [DOI] [PubMed] [Google Scholar]
- 15.Gebhardt B, Vlaho S, Fischer D, Sewell A, Bohles H. N-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduria. Mol Genet Metab. 2003;79:303–304. doi: 10.1016/s1096-7192(03)00095-7. [DOI] [PubMed] [Google Scholar]
- 16.Yorifuji T, Muroi J, Uematsu A, Hiramatsu H, Momoi T. Hyperinsulinism-hyperammonemia syndrome caused by mutant glutamate dehydrogenase accompanied by novel enzyme kinetics. Hum Genet. 1999;104:476–479. doi: 10.1007/s004390050990. [DOI] [PubMed] [Google Scholar]
- 17.Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol Genet Metab. 2004;81 Suppl 1:S45–S51. doi: 10.1016/j.ymgme.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Coude FX, Rabier D, Cathelineau L, Grimber G, Parvy P, Kamoun P. A mechanism for valproate-induced hyperammonemia. Adv Exp Med Biol. 1982;153:153–161. doi: 10.1007/978-1-4757-6903-6_21. [DOI] [PubMed] [Google Scholar]
- 19.Kim WH, Park H, Yun C, Cho H, Kim S, Paik WK, Jeon SH, Lee JH. Mixture of N-carbamoyl-L-glutamate plus L-arginine can protect rats with liver cirrhosis from acute ammonia intoxication. J Hepatol. 2001;35:719–725. doi: 10.1016/s0168-8278(01)00199-4. [DOI] [PubMed] [Google Scholar]
- 20.Cheung CW, Raijman L. The regulation of carbamyl phosphate synthetase (ammonia) in rat liver mitochondria. Effects of acetylglutamate concentration and ATP translocation. J Biol Chem. 1980;255:5051–5057. [PubMed] [Google Scholar]
- 21.Cohen NS, Cheung CW, Kyan FS, Jones EE, Raijman L. Mitochondrial carbamyl phosphate and citrulline synthesis at high matrix acetylglutamate. J Biol Chem. 1982;257:6898–6907. [PubMed] [Google Scholar]
- 22.Shigesada K, Aoyagi K, Tatibana M. Role of acetylglutamate in ureotelism. Variations in acetylglutamate level and its possible significance in control of urea synthesis in mammalian liver. Eur J Biochem. 1978;85:385–391. doi: 10.1111/j.1432-1033.1978.tb12250.x. [DOI] [PubMed] [Google Scholar]
- 23.Meijer AJ, Lof C, Ramos IC, Verhoeven AJ. Control of ureogenesis. Eur J Biochem. 1985;148:189–196. doi: 10.1111/j.1432-1033.1985.tb08824.x. [DOI] [PubMed] [Google Scholar]
- 24.Jahoor F, Wolfe RR. Reassessment of primed constant-infusion tracer method to measure urea kinetics. Am J Physiol. 1987;252:E557–E564. doi: 10.1152/ajpendo.1987.252.4.E557. [DOI] [PubMed] [Google Scholar]
- 25.Matthews DE, Downey RS. Measurement of urea kinetics in humans: a validation of stable isotope tracer methods. Am J Physiol. 1984;246:E519–E527. doi: 10.1152/ajpendo.1984.246.6.E519. [DOI] [PubMed] [Google Scholar]
- 26.Jones BN, Gilligan JP. o-Phthaldialdehyde precolumn derivatization and reversed-phase high-performance liquid chromatography of polypeptide hydrolysates and physiological fluids. J Chromatogr. 1983;266:471–482. doi: 10.1016/s0021-9673(01)90918-5. [DOI] [PubMed] [Google Scholar]
- 27.Marks V. An improved glucose-oxidase method for determining blood, C.S.F. and urine glucose levels. Clin Chim Acta. 1959;4:395–400. doi: 10.1016/0009-8981(59)90110-x. [DOI] [PubMed] [Google Scholar]
- 28.el-Khoury AE, Sanchez M, Fukagawa NK, Gleason RE, Young VR. Similar 24-h pattern and rate of carbon dioxide production, by indirect calorimetry vs. stable isotope dilution, in healthy adults under standardized metabolic conditions. J Nutr. 1994;124:1615–1627. doi: 10.1093/jn/124.9.1615. [DOI] [PubMed] [Google Scholar]
- 29.Schoeller DA, van Santen E, Peterson DW, Dietz W, Jaspan J, Klein PD. Total body water measurement in humans with 18O and 2H labeled water. Amer J Clin Nutr. 1980;33:2686–2693. doi: 10.1093/ajcn/33.12.2686. [DOI] [PubMed] [Google Scholar]
- 30.Lee B, Yu H, Jahoor F, O’Brien W, Beaudet AL, Reeds P. In vivo urea cycle flux distinguishes and correlates with phenotypic severity in disorders of the urea cycle. Proc Natl Acad Sci USA. 2000;97:8021–8026. doi: 10.1073/pnas.140082197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yudkoff M, Daikhin Y, Nissim I, Jawad A, Wilson J, Batshaw M. In Vivo Nitrogen Metabolism in Ornithine Transcarbamylase Deficiency. J. Clin. Invest. 1996;98:2167–2173. doi: 10.1172/JCI119023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brosnan JT, Brosnan ME, Yudkoff M, Nissim I, Daikhin Y, Lazarow A, Horyn O, Nissim I. Alanine metabolism in the perfused rat liver. Studies with (15)N. J Biol Chem. 2001;276:31876–31882. doi: 10.1074/jbc.M103890200. [DOI] [PubMed] [Google Scholar]
- 33.Yang D, Hazey JW, David F, Singh J, Rivchum R, Streem JM, Halperin ML, Brunengraber H. Integrative physiology of splanchnic glutamine and ammonium metabolism. Am J Physiol Endocrinol Metab. 2000;278:E469–E476. doi: 10.1152/ajpendo.2000.278.3.E469. [DOI] [PubMed] [Google Scholar]
- 34.Felig P. The glucose-alanine cycle. Metabolism. 1973;22:179–207. doi: 10.1016/0026-0495(73)90269-2. [DOI] [PubMed] [Google Scholar]
- 35.Schimke RT. Differential effects of fasting and protein-free diets on levels of urea cycle enzymes in rat liver. J Biol Chem. 1962;237:1921–1924. [PubMed] [Google Scholar]
- 36.Fowler B. Recent advances in the mechanism of pyridoxine-responsive disorders. J Inherit Metab Dis. 1985;8 Suppl 1:76–83. doi: 10.1007/BF01800664. [DOI] [PubMed] [Google Scholar]
- 37.Matalon R, KochK R, Michals-Matalon K, Moseley K, Surendran S, Tyring S, Erlandsen H, Gamez A, Stevens RC, Romstad A, Moller LB, Guttler F. Biopterin responsive phenylalanine hydroxylase deficiency. Genet Med. 2004;6:27–32. doi: 10.1097/01.gim.0000108840.17922.a7. [DOI] [PubMed] [Google Scholar]
- 38.Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, McCarter RJ, Krischer JP, Batshaw ML. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–170. [PubMed] [Google Scholar]
- 40.Nagata N, Matsuda I, Oyanagi K. Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet. 1991;39:228–229. doi: 10.1002/ajmg.1320390226. [DOI] [PubMed] [Google Scholar]
- 41.Natale PJ, Tremblay GC. Studies on the availability of intramitochondrial carbamoylphosphate for utilization in extramitochondrial reactions in rat liver. Arch Biochem Biophys. 1974;162:357–368. doi: 10.1016/0003-9861(74)90193-3. [DOI] [PubMed] [Google Scholar]
- 42.Alonso E, Rubio V. Orotic aciduria due to arginine deprivation: changes in the levels of carbamoyl phosphate and of other urea cycle intermediates in mouse liver. J Nutr. 1989;119:1188–1195. doi: 10.1093/jn/119.8.1188. [DOI] [PubMed] [Google Scholar]