

Abstract
Critically ill patients may require mechanical ventilatory support and short-term high-dose corticosteroid to treat some specific underlying disease processes. Diaphragm muscle inactivity induced by controlled mechanical ventilation produces dramatic alterations in diaphragm muscle structure and significant losses in function. Although the exact mechanisms responsible for losses in diaphragm muscle function are still unknown, recent studies have highlighted the importance of proteolysis and oxidative stress. In experimental animals, short-term strategies that maintain partial diaphragm muscle neuromechanical activation mitigate diaphragmatic force loss. In animal models, studies on the influence of combined controlled mechanical ventilation and short-term high-dose methylprednisolone have given inconsistent results in regard to the effects on diaphragm muscle function. In the critically ill patient, further research is needed to establish the prevalence and mechanisms of ventilator-induced diaphragm muscle dysfunction, and the possible interaction between mechanical ventilation and the administration of high-dose corticosteroid. Until then, in caring for these patients, it is imperative to allow partial activation of the diaphragm, and to administer the lowest dose of corticosteroid for the shortest duration possible.
Introduction
Through a complex integration of feedback signals, the respiratory center generates signal output to the diaphragm muscle leading to its rhythmic contractions. Some critically ill patients, including those with acute insults to the respiratory center, upper spinal cord, bilateral phrenic nerves or neuromuscular junction or those receiving neuromuscular paralysis - for instance, patients with acute respiratory distress syndrome [1] - must be supported with the application of controlled mechanical ventilation (CMV), where the ventilator takes full control of the act of breathing and the respiratory muscles do not contract. In addition to mechanical ventilation, some critically ill patients - such as victims of acute spinal cord injury [2], of lung transplant rejection [3], of hematologic malignancy [4] and of status asthmaticus [5] - may require administration of high doses of corticosteroids.
The extent to which CMV [6] or (short-term) high-dose corticosteroid administration [7] negatively impacts diaphragm muscle function has been demonstrated in experimental animals. In critically ill patients, however, the presence of confounding factors (for example, sepsis, malnutrition, hyperglycemia) makes it difficult to determine the extent to which diaphragm muscle dysfunction is attributable to disuse or high-dose corticosteroid alone, or in combination. The reported studies suggest that deleterious effects in the diaphragm occurred with both diaphragm muscle disuse [8] and possibly with the administration of high-dose corticosteroid [9], leading to difficulty weaning from mechanical ventilation.
The goal of the present article is to address two key issues: to identify the underlying mechanisms responsible for the loss of diaphragmatic function that occur as a result of CMV and acute high-doses of corticosteroids; and to determine the evidence of diaphragm muscle impairment in humans, and the potential approaches for protecting the diaphragm muscle.
Mechanisms of diaphragm muscle dysfunction with disuse
Several animal studies have demonstrated that CMV reduces the contractile function of previously healthy diaphragm muscle with intact neural outflow tract and neurotrophic influences, a condition referred to as ventilator-induced diaphragmatic dysfunction (VIDD) [6,10,11]. The impairment occurs fairly rapidly and is progressive. In rabbits, compared with a control, the diaphragmatic force-generating capacity declined by 25% after 24 hours of CMV, and by 44% after 72 hours of CMV [11]. In rats, the rate of diaphragmatic force loss was more profound than that in rabbits (46% after only 24 hours of CMV) [10]. The deleterious effects of CMV-induced diaphragmatic dysfunction are not exclusive to rodents [6,12,13]. It is plausible that the diaphragm's lack of constant rhythmic contractions makes it susceptible to functional derangement with inactivity, even when the inactivity is of short duration.
CMV induces diaphragm muscle inactivity via phrenic inhibition. Superimposed to the already inactive diaphragm from CMV application, the administration of cisatracurium - a benzylisoquinolinium nondepolarizing paralytic - does not exacerbate the force loss [14]. In contrast, rocuronium - an aminosteroid nondepolarizing paralytic - worsens diaphragmatic force loss [15]. Testelmans and colleagues postulated that this difference is related to rocuronium's corticosteroid molecular structure [15].
Studies assessing the mechanisms of CMV-induced diaphragm muscle dysfunction have attributed the dysfunction predominantly to increased proteolysis [16-18] with and without the requirement of oxidative stress [19,20]. Proteolysis is conducive to myofibrilar disruption and/or atrophy (reduced cross-sectional area) [21]. It should be noted that impairment in excitation-contraction coupling has not been investigated systematically. Impaired excitation-contraction coupling (that is, a decrease in sarcolemma resting membrane action potential and/or sarcoplasmic reticulum Ca2+ release capacity) leads to reduced force development [22].
Oxidative stress
Excessive oxidative stress results from a decrease in anti-oxidant buffering capacity and/or the overproduction of reactive oxygen species (ROS) [23]. CMV compromises anti-oxidant defenses [24,25]. CMV decreases the total anti-oxidant capacity and glutathione (a nonenzymatic antioxidant) concentrations [24,25]. The effects of CMV on enzymatic antioxidant (for example, glutathione peroxidase) are variable. For instance, in rats the glutathione-peroxidase activity decreases after 12 hours of CMV [25], while in piglets the activity remains unchanged after 3 days of CMV [24].
Overproduction of ROS can occur even following short periods of CMV. For instance, Zergeroglu and colleagues observed significant elevations in ROS levels after only 6 hours of CMV [26]. Importantly, the elevated ROS levels were associated with atrophy of all fiber types and diaphragmatic force loss after 12 to 18 hours of CMV [16,27]. The trigger for the increased oxidative stress remains unknown.
Oxidative stress pathways capable of producing ROS in skeletal muscle inactivity include nitric oxide synthase-generating, xanthine oxidase-generating, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-generating, and mitochondrial oxidant-generating pathways (Figure 1) [21].
Figure 1.

Oxidative stress pathways capable of producing reactive oxidant species. These pathways include nitric oxide synthase pathway, xanthine oxidase pathway, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathway, and mitochondrial oxidant-generating pathway. The mitochondrial oxidant-generating pathway is key to oxidative damage of diaphragm muscle inactivity. O2+ superoxide; NO+, nitric oxide. Adapted with permission from [21].
The nitric oxide synthase pathway does not seem to be involved in VIDD [28]. Conversely, Whidden and coworkers recently reported that the xanthine oxidase pathway contributes to the oxidative damage of diaphragm muscle [29]. This hypothesis was supported by the observation that administration of oxypurinol, a xanthine-oxidase inhibitor, partially attenuates diaphragmatic dysfunction after 12 hours and 18 hours of CMV [29]. Markers of protein and lipid peroxidation, protein carbonyls and 4-hydroxynoneal, respectively, are also suppressed with the administration of oxypurinol. While xanthine oxidase contributes to diaphragm muscle force loss, xanthine-oxidase inhibition does not attenuate CMV-induced diaphragm muscle atrophy [29], suggesting that other oxidative stress pathways may be involved in the atrophic process.
In addition to xanthine oxidase, McClung and colleagues demonstrated the role of the NADPH oxidase pathway in producing oxidative damage in the diaphragm [30]. In rats receiving 18 hours of CMV, apocynin (an inhibitor of NADPH oxidase) attenuated diaphragm muscle dysfunction, prevented atrophy of all myofiber types, and prevented CMV-induced reduction in glutathione. Furthermore, apocynin not only suppressed calpain-1 and caspase-3 activation, but in fact increased calpastatin, an endogenous calpain inhibitor. Among the oxidative stress pathways, however, the mitochondrial oxidant-generating pathway is key in the development of oxidative stress damage of the diaphragm with CMV [31]. Recently, Kavazis and colleagues demonstrated that mitochondriae are a major source of ROS production associated with mitochondrial oxidative damage and with mitochondrial respiratory dysfunction [31].
Consistent with the mitochondrial oxidant-generating pathway, an earlier study from the same laboratory demonstrated elevated intracellular oxidant production with CMV [25]. The latter was estimated from the intracellular increased emission of dichlorodihydrofluorescein dye, a chemical that fluoresces upon reaction with oxidative species [25]. The enhanced production of lipid and protein oxidation markers underscores the elevated oxidative stress [16]. Lipid oxidation may result in cellular membrane dysfunction (that is, decreased Ca2+ ATPase activity) and may delay Ca2+ removal from the cytosol, causing its accumulation within the cytosol itself [23]. The elevated Ca2+ concentration in the cytosol can activate calpain, the Ca2+-dependent proteases [21]. In fact, calpain-1 is an absolute requirement for oxidative stress-induced myofiber atrophy [32]. This contention was supported in experiments with hydrogen-peroxide-incubated myotube cell cultures. Hydrogen peroxide induced myotube atrophy. In contrast, calpain-1 RNA interference (gene knocked out) completely prevents the atrophy [32].
Protein oxidation preferentially targets myofibrillar proteins including myosin and actin [26]. The contractile proteins damaged by oxidation become susceptible to degradation by proteases [23]. Such protein degradation results in both decreased diaphragmatic force-generating capacity and diaphragm muscle atrophy. In mechanically ventilated animals, pretreatment with the antioxidant Trolox, a soluble vitamin E analog, preserves diaphragmatic force-generating capacity and prevents atrophy [27]. Trolox reduces production of protein carbonyls - oxidative byproducts of proteins [19] - but does not alter the suppressed antioxidant glutathione concentrations. The protective effect of the diaphragm by Trolox is achieved through a reduction in myofilament protein availability to degradation by the proteasome [19,23]. In addition to its antioxidant activity, Trolox has direct suppressive effects on calpain, caspase proteases, and 20S proteasome activity [19,21,27]. The 20S proteasome is the core structure of the 26S proteasome complex in the ubiquitin-proteasome pathway [33], and the unbound form can independently degrade oxidized proteins without requiring ubiquitin conjugation (Figure 2).
Figure 2.

The ubiquitin-proteasome pathway. The substrate proteins are designated for degradation by conjugation to ubiquitin in an ATP-dependent reaction. The ubiquitin-activating enzyme (E1) uses ATP to create a highly reactive thiolester form of ubiquitin, and then transfers it to a ubiquitin-carrier protein (E2). The subsequent transfer of the activated ubiquitin to the protein substrate requires a ubiquitin-protein ligase (E3). The E3 ligases muscle atrophy F-box (MAFbox) and muscle ring finger-1 (MuRF-1) have important roles in skeletal muscle atrophy. Once the ubiquitin conjugates are formed, they are transported to a proteolytic complex known as the 26S proteasome, consisting of two 19S regulators and the 20S core proteasome. The 19S regulators recognize and bind the ubiquitinated protein. Energy from ATP hydrolysis releases the ubiquitin chain and unfolds the substrate protein. The unfolded protein is fed into the 20S proteasome for degradation into small peptides and amino acids. The 20S proteasome can degrade oxidized protein without ubiquitination. Adapted with permission from [33].
Proteolytic systems
All of the major proteolytic systems are responsible for CMV-induced proteolysis; these include lysosomal proteases [18], calcium-dependent proteolysis or calpains [14], caspase-3 [34], and the ATP-dependent ubiquitin proteasome [17]. Lysosomal proteases are primarily responsible for proteolysis of extracellular proteins and cell surface receptors [35]. Calpain proteases are involved in the cleavage of cytoskeletal proteins (for example, titin, nebulin, desmin) that anchor contractile elements of myosin to actin [36]. Caspases (endoproteases responsible for the final execution of cell death) including caspase-3 proteases induce DNA fragmentation, induce myonuclear apoptosis, and cleave actomyosin complexes [34,37], whereas the ubiquitin-proteasome pathway degrades the myofilament actin and myosin [20].
Lysosomal proteases and calpains play a significant role in diaphragmatic dysfunction with inactivity [18]. Pretreatment with leupeptin - an inhibitor of lysosomal thiol proteases and calcium-activated proteases - completely prevents the CMV-induced reduction in diaphragmatic force and atrophy [18]. Likewise, caspase-3 vitally contributes in the detrimental effects of CMV on the diaphragm [34]. Treatment of animals with caspase inhibitor prevents myonuclei loss, DNA fragmentation, and myofiber atrophy [34].
The ubiquitin-proteasome pathway is responsible for most muscle protein degradation [33]. The ubiquitin-proteasome system, however, does not break down complexes of proteins contained in myofibrils. One or more other proteases are required in the initial process to release myofilament contractile proteins (that is, actin and myosin) for the ubiquitin-proteasome system to degrade those proteins [23]. The binding of ubiquitin to protein substrates requires ubiquitin-activating enzyme (E1), ubiquitin-carrier enzyme (E2), and ubiquitin ligases (E3) [33] (Figure 2). Two of the E3 ligases - the muscle atrophy F-box (MAFbox; atrogin-1, atrogenes) and muscle ring finger-1 (MuRF1) genes - are overexpressed in various models of skeletal muscle atrophy [38]. Similarly, MAFbox and MuRF1 are upregulated during CMV-induced diaphragm muscle inactivity [11,19].
An important upstream signaling pathway of atrogene expression is the insulin-like growth factor-1-phosphotidylinositol-3-kinase-protein kinase-B serine/threonine kinase (IGF-1/PI3K/Akt) pathway [39] (Figure 3). IGF-1/PI3K/Akt suppresses MAFbox by inactivating the expression of forkhead box-O and preventing its nuclear translocation. After 6 hours and 18 hours of CMV, diaphragmatic Akt activation decreased while both forkhead box-O nuclear translocation and MAFbox and MuRF1 expression increased [19]. IGF-1/PI3K/Akt signaling therefore seems to play an important role in regulating the E3 ligase in the ubiquitin-proteasome pathway.
Figure 3.

The insulin-like growth factor-1-phosphotidylinositol-3-kinase-protein kinase-B serine/threonine kinase-forkhead box-O pathway. (a) Increased insulin-like growth factor-1 (IGF-1) activates phosphotidylinositol-3-kinase (PI3K), leading to phosphorylation of protein kinase-B serine/threonine kinase (Akt) and forkhead box-O (Foxo). Phosphorylated Foxo is sequestered within the cytoplasm and prevents its nuclear translocation and atrogin-1 (muscle atrophy F-box (MAFbox)) activation. Phosphorylated Akt also activates mammalian target of rapamycin (mTOR) and p70Sk, resulting in increased protein synthesis. (b) Suppression of IGF-1 with controlled mechanical ventilation-induced diaphragm muscle inactivity deactivates Akt, leading to nuclear translocation of Foxo, which then activates atrogin-1 and other atrogenes resulting in increased proteolysis. Reprinted from Cell, 117, Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL, Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy, 14 Pages, Copyright (2004), with permission from Elsevier [39].
Influence of neuromechanical activation on diaphragmatic function
Maintaining diaphragm muscle activation with assist-control mechanical ventilation (AMV) represents an important strategy for maintaining diaphragm muscle function [40]. For instance, we have shown that 3 days of CMV produces a dramatic loss (~45%) in diaphragmatic function, as defined by the maximal isometric tension. In contrast, 3 days of AMV produces much smaller losses in diaphragmatic force-generating capacity, an approximately 15% loss in maximal isometric tension [40]. The minimal extent of diaphragmatic activation sufficient to preserve function is unclear. From our previous data [40], however, it appears that activation levels of 30% and above are associated with relatively small losses in diaphragm muscle function (Figure 4). The influence of minimal diaphragm muscle activation between 0% and 30% on maximal isometric tension remains unknown. It is also unclear whether AMV can preserve diaphragmatic force under prolonged mechanical ventilation >3 days. The decline in force with CMV was associated with an approximately threefold increase in MAFbox mRNA expression, while with AMV the expression did not differ significantly from controls [40].
Figure 4.
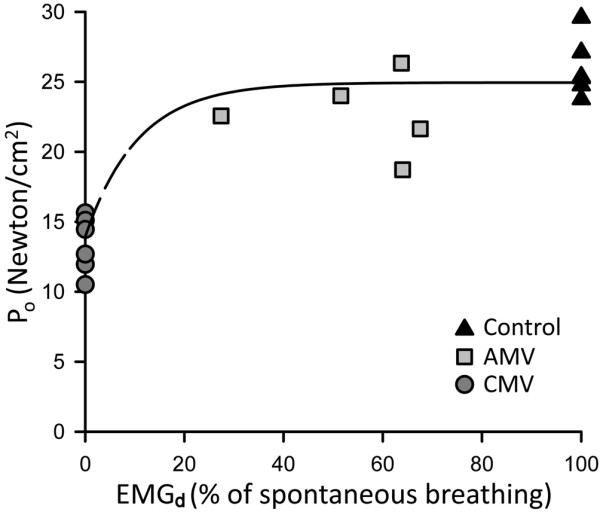
Monoexponential relationships between diaphragm muscle maximal tetanic force and its electrical activity. The maximal isometric tension (Po) is normalized for muscle cross-sectional area. The diaphragm muscle electrical activity (EMGd) during assist-control mechanical ventilation (AMV) was estimated by measuring the area subtended by the moving average EMGd curve and its baseline, and is expressed as a percentage of spontaneous breathing. Po is maintained almost identically to that of the control after 3 days of AMV with diaphragm muscle activation between 30% and 80% of spontaneous breathing. Whether diaphragm muscle activation between 0% and 30% is effective to maintain Po remains unknown. Data obtained from [41]: n = 6 for the control and controlled mechanical ventilation (CMV) groups; n = 5 for the AMV group.
In another study, when spontaneous breathing for 5 minutes or 60 minutes was interposed during 24 hours of CMV four times a day, the diaphragmatic force-generating capacity decreased by an average of 19% and decreased by 28% with continuous CMV, respectively [41]. Although the protective effects of a brief duration of diaphragm muscle activation on functional loss were modest (~9%), the activation prevents diaphragm muscle atrophy. Futier and colleagues recently demonstrated that maintaining diaphragmatic activation with pressure support ventilation for 18 hours did not augment proteolysis [42]. Protein carbonyls (markers of oxidative stress), however, were elevated to the same extent as with CMV. Unfortunately, measures of diaphragmatic function were not performed, and whether pressure support ventilation preserves diaphragmatic force therefore remains unknown [42].
Evidence of CMV-induced diaphragmatic dysfunction in humans and a potential approach to prevention
In critically ill patients it is extremely difficult to establish whether CMV is responsible for diaphragm muscle dysfunction and weaning failure, because multiple confounding factors (for example, sepsis, malnutrition, hyperglycemia) contribute to diaphragm muscle weakness and atrophy. Consistent with studies in animals, Levine and colleagues reported that diaphragm muscle atrophy also occurred fairly rapidly in brain-dead organ donors with CMV application of 18 to 69 hours, compared with control subjects who underwent lung surgery and received mechanical ventilation for 2 to 3 hours [8]. Diaphragm muscle atrophy involved both slow and fast fiber types, decreasing cross-sectional areas by 57% and 53%, respectively (Figure 5). The atrophy was associated with decreased antioxidant glutathione concentration (by 23%), increased active caspase-3 protease (by twofold), and elevated mRNA levels of MAFbox (by threefold) and MuRF1 (by sevenfold). Interestingly, a biopsy of the pectoralis major muscle did not show any fiber atrophy [8]. The study of Levine and colleagues lacked measurement of diaphragm muscle function, and was confined to brain-dead organ donors whose neural activation and possibly neurotrophic factors to the diaphragm were completely absent and thus were not typical of critically ill patients in the intensive care unit [8]. Nevertheless, the negative impact of diaphragm muscle disuse in critically ill patients cannot be ignored [43].
Figure 5.

Cross-sectional areas of diaphragm muscle. Cross-sections of diaphragm muscle from biopsy specimens of a representative organ donor subject ((a), (c), (e)) and from a control ((b), (d), (f)). (a) and (b) Muscle fibers in the organ donor subject are in general smaller than those in the control diaphragm. No inflammatory infiltrate or necrosis is seen. Stained with hematoxylin and eosin. (c) and (d) Stained with antibody specific for slow myosin, heavy chain. (e) and (f) Stained with antibody specific for fast myosin, heavy chain. In (c) to (f), fibers reacting with the antibody appear orange-red, whereas fibers not reacting with the antibody appear black; open circle, slow-twitch fibers; open square, fast-twitch fibers. In addition, all fibers in each section are outlined by an antibody reactive to laminin. Reproduced with permission from [8]. Copyright © 2008 Massachusetts Medical Society. All rights reserved.
In humans, the degree of diaphragm muscle activation that will preserve force remains unknown. In a prospective trial, critically ill patients who were predicted to receive mechanical ventilation for longer than 72 hours were randomized into controls (n = 13) and those receiving inspiratory muscle training (IMT) (n = 12) from the onset of mechanical ventilation [44]. A threshold load was used for the IMT by setting the ventilator pressure-triggering sensitivity at 10% or 20% of the initial maximum inspiratory pressure (PImax), whichever was tolerated, and was applied twice daily for 5 minutes. When the patient tolerated the initial load, the next training duration was increased by 5 minutes up to a maximum of 30 minutes. Afterwards, the load was increased by 10% increments until 40% of the initial PImax value was attained. Sedation and analgesia with intravenous midazolam and fentanyl, respectively, were administered. The IMT session was aborted according to specified criteria. Weaning with decreasing pressure support was initiated once the patient met the weaning criteria. The initial and final PImax values in the training group were similar to those of the control group (initial, -51 cmH2O vs. -48 cmH2O; final, -56 cmH2O vs. -55 cmH2O, respectively). The duration of mechanical ventilation or of the weaning trial was similar for both groups, with a trend toward a shorter duration for the IMT group compared with the control group (mean duration of mechanical ventilation, 8.6 days vs. 9.8 days; mean duration of weaning trial, 23 hours vs. 31 hours, respectively).
The lack of IMT benefits may be due to the small sample size [44]. It is also conceivable that the magnitude of the stimulus for IMT, as a percentage of the PImax, in the critically ill patients (that is, the threshold load applied, and/or session frequency and duration) was inadequate to elicit a physiological training effect. Measurements of PImax in the critically ill patients are challenging and highly dependent on patient volitional effort and the methods of measurement. Alternatively, in view of the complexity of the underlying mechanisms of CMV-induced diaphragmatic dysfunction, diaphragm muscle conditioning alone is inadequate, and pharmacological intervention may be required to mitigate diaphragm muscle weakness.
Mechanisms of the interactive effects of mechanical ventilation and short-term high-dose corticosteroid on diaphragm muscle dysfunction
Short-term high-dose corticosteroid has been administered for its anti-inflammatory and immunosuppressive effects in critically ill patients [9,45]; the treatment might be responsible for the development of acquired paresis in the critically ill patient, referred to as critical-illness myopathy [46]. Patients may receive both mechanical ventilation and short-term high-dose corticosteroid, yet the effects of acute high-dose corticosteroid alone or its interaction with mechanical ventilation is not well understood.
We recently studied the temporal relationship (1 to 3 days of 80 mg/kg/day intramuscularly) and dose-response effects (3 days of 80 mg/kg/day vs. 10 mg/kg/day intramuscularly) of methylprednisolone (MP) treatment in rabbits [7]. MP induced a progressive decline in diaphragmatic force by 19%, 24%, and 34% after 1 day, 2 days, and 3 days, respectively. The decline in diaphragmatic force correlated with the degree of abnormal myofibril volume density. Low-dose MP (10 mg/kg/day, but a high dose by clinical standards) decreased diaphragmatic force modestly, by 12%. The suppression of IGF-1 and upregulation of MAFbox mRNA were independent of the MP dose [7]. Both high-dose and low-dose MP decreased IGF-1 by 35%, and increased MAFbox mRNA by threefold [7]. Clearly, short-term high doses of MP in spontaneously breathing animals produced detrimental effects on the diaphragm. The combination of both CMV and high-dose MP is therefore expected to aggravate the decline in diaphragmatic force compared with either CMV or MP alone.
Interestingly, Maes and colleagues demonstrated in rats that 24 hours of combined CMV and high-dose MP (80 mg/kg/day intramuscularly) preserved the diaphragmatic force compared with CMV alone [47]. The mechanism by which MP prevented diaphragmatic force loss was via inhibition of calpain activity. Our preliminary data [48] in rabbits contrast with those of Maes and colleagues. After 2 days of combined MP (60 mg/kg/day intravenously) plus CMV, MP plus AMV, or MP plus continuous positive airway pressure, the diaphragmatic force decreased from that without MP by 10%, 16% and 18% from the average values of 16.1 Newton/cm2, 22.6 Newton/cm2, and 23.3 Newton/cm2 with CMV, AMV, and continuous positive airway pressure alone, respectively [48]. The diaphragmatic force with the combined CMV and MP approach was not significantly different from that with CMV alone. This suggests that both CMV and MP share common mechanisms for the decrease in diaphragmatic force. It is unclear whether the discrepancy between our preliminary results [48] and those of Maes and colleagues [47] is related to species differences or to the duration of MP treatment.
Evidence of methylprednisolone-induced diaphragmatic dysfunction in humans and a potential approach to prevention
As with CMV, the extent to which acute, high-dose MP could contribute to diaphragm muscle weakness in critically ill patients is difficult to determine. This difficulty stems from the many confounding factors in these patients, and from the lack of data on functional or structural alterations in humans. Indirect data, however, suggest that such interaction may occur in critically ill patients [46].
First, in a prospective study of critically ill patients receiving mechanical ventilation for longer than 7 days, De Jonghe and colleagues reported a strong association between the occurrence of neuromyopathy and the administration of corticosteroids [46]. Second, in patients with acute spinal cord injury, the administration of recommended high-dose MP for 48 hours resulted in paraspinal muscle necrosis and type II fiber atrophy in four out of five patients [9]. Three of the patients remained ventilator dependent at discharge from the Spinal Cord Injury Center despite the relatively low level of injury [9]. Finally, among 26 patients with chronic obstructive pulmonary disease who received mechanical ventilation and MP (240 mg/day), nine (35%) patients developed myopathy of the extremities - a condition associated with higher total doses of MP treatment (1,649 mg vs. 979 mg), with prolonged mechanical ventilation, and with prolonged hospital length of stay [45].
Whether a dose and duration of corticosteroids that confers beneficial anti-inflammatory effects and yet preserves diaphragm muscle integrity/function does exist remains unknown. More research is necessary to dissect the underlying mechanisms of the effects of corticosteroid on the diaphragm, particularly its interaction with mechanical ventilation. Because of the corticosteroid dose-response effects in both animal studies [7] and human studies [45], clinicians must carefully weigh the risks and benefits ratio, and must use the lowest corticosteroid dose for the shortest duration possible.
Future research
In laboratory animals the mechanisms responsible for VIDD have been the focus of intense investigation. Unfortunately, the triggering factor(s) for enhanced proteolysis in VIDD remain unknown. Similarly, the contribution of excitation-contraction coupling and the degree or duration of neuromechanical activation for preventing diaphragmatic force loss are unknown. Whether the benefits of AMV depend on the level of diaphragmatic activity or whether the benefits cease with time remains unclear. Diaphragm muscle conditioning using noninvasive phrenic nerve stimulation is a potential strategy for preventing VIDD that remains to be explored. In animal studies, treatment with specific inhibitors to the signaling cascade involved in proteolysis completely preserves diaphragm muscle function. Whether a similar strategy should be attempted in patients remains to be determined.
Abbreviations
Akt: protein kinase-B serine/threonine kinase; AMV: assist-control mechanical ventilation; CMV: controlled mechanical ventilation; IGF-1: insulin-like growth factor-1; IMT: inspiratory muscle training; MAFbox: muscle atrophy F-box; MP: methylprednisolone; MuRF1: muscle ring finger-1; NADPH: nicotinamide adenine dinucleotide phosphate; PI3K: phosphotidylinositol-3-kinase; PImax: maximal inspiratory pressure; ROS: reactive oxygen species; VIDD: ventilator-induced diaphragmatic dysfunction.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Catherine SH Sassoon, Email: csassoon@uci.edu.
Vincent J Caiozzo, Email: vjcaiozz@uci.edu.
Acknowledgements
The present work was supported by grants from the Department of Veterans Affairs Medical Research Service (to CSHS) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases AR-46856 (to VJC). We thank Ercheng Zhu, Ph.D. for generating the data presented in Figure 4.
References
- Gainnier M, Roch A, Forel JM, Thirion X, Arnal JM, Donati S, Papazian L. Effect of neuromuscular blocking agents on gas exchange in patients presenting with acute respiratory distress syndrome. Crit Care Med. 2004;32:113–119. doi: 10.1097/01.CCM.0000104114.72614.BC. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, Fehlings M, Herr DL, Hitchon PW, Marshall LF, Nockels RP, Pascale V, Perot PL Jr, Piepmeier J, Sonntag VK, Wagner F, Wilberger JE, Winn HR, Young W. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA. 1997;277:1597–1604. doi: 10.1001/jama.277.20.1597. [DOI] [PubMed] [Google Scholar]
- Nava S, Fracchia C, Callegari G, Ambrosino N, Barbarito N, Felicetti G. Weakness of respiratory and skeletal muscles after a short course of steroids in patients with acute lung rejection. Eur Respir J. 2002;20:497–499. doi: 10.1183/09031936.02.01732002. [DOI] [PubMed] [Google Scholar]
- Jagannath S. Treatment of myeloma in patients not eligible for transplantation. Curr Treat Options Oncol. 2005;6:241–253. doi: 10.1007/s11864-005-0007-0. [DOI] [PubMed] [Google Scholar]
- Kaplan PW, Rocha W, Sanders DB, D'Souza B, Spock A. Acute steroid-induced tetraplegia following status asthmaticus. Pediatrics. 1986;78:121–123. [PubMed] [Google Scholar]
- Sassoon CSH, Caiozzo VJ, Manka A, Sieck GC. Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol. 2002;92:2585–2595. doi: 10.1152/japplphysiol.01213.2001. [DOI] [PubMed] [Google Scholar]
- Sassoon CS, Zhu E, Pham HT, Nelson RS, Fang L, Baker MJ, Caiozzo VJ. Acute effects of high-dose methylprednisolone on diaphragm muscle function. Muscle Nerve. 2008;38:1161–1172. doi: 10.1002/mus.21048. [DOI] [PubMed] [Google Scholar]
- Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358:1327–1335. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- Qian T, Guo X, Levi AD, Vanni S, Shebert RT, Sipski ML. High-dose methylprednisolone may cause myopathy in acute spinal cord injury patients. Spinal Cord. 2005;43:199–203. doi: 10.1038/sj.sc.3101681. [DOI] [PubMed] [Google Scholar]
- Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol. 2002;92:1851–1858. doi: 10.1152/japplphysiol.00881.2001. [DOI] [PubMed] [Google Scholar]
- Zhu E, Sassoon CS, Nelson R, Pham HT, Zhu L, Baker MJ, Caiozzo VJ. Early effects of mechanical ventilation on isotonic contractile properties and MAF-box gene expression in the diaphragm. J Appl Physiol. 2005;99:747–756. doi: 10.1152/japplphysiol.00126.2005. [DOI] [PubMed] [Google Scholar]
- Radell PJ, Remahl S, Nichols DG, Eriksson LI. Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med. 2002;28:358–364. doi: 10.1007/s00134-002-1207-8. [DOI] [PubMed] [Google Scholar]
- Anzueto A, Peters JI, Tobin MJ, de los Santos R, Seidenfeld JJ, Moore G, Cox WJ, Coalson JJ. Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med. 1997;25:1187–1190. doi: 10.1097/00003246-199707000-00021. [DOI] [PubMed] [Google Scholar]
- Testelmans D, Maes K, Wouters P, Powers SK, Decramer M, Gayan-Ramirez G. Infusions of rocuronium and cisatracurium exert different effects on rat diaphragm function. Intensive Care Med. 2007;33:872–879. doi: 10.1007/s00134-007-0584-4. [DOI] [PubMed] [Google Scholar]
- Testelmans D, Maes K, Wouters P, Gosselin N, Deruisseau K, Powers S, Sciot R, Decramer M, Gayan-Ramirez G. Rocuronium exacerbates mechanical ventilation induced diaphragm dysfunction in rats. Crit Care Med. 2006;34:3018–3023. doi: 10.1097/01.CCM.0000245783.28478.AD. [DOI] [PubMed] [Google Scholar]
- Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166:1369–1374. doi: 10.1164/rccm.200202-088OC. [DOI] [PubMed] [Google Scholar]
- DeRuisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, Ordway GA, Powers SK. Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol. 2005;98:1314–1321. doi: 10.1152/japplphysiol.00993.2004. [DOI] [PubMed] [Google Scholar]
- Maes K, Testelmans D, Powers S, Decramer M, Gayan-Ramirez G. Leupeptin inhibits ventilator-induced diaphragm dysfunction in rats. Am J Respir Crit Care Med. 2007;175:1134–1138. doi: 10.1164/rccm.200609-1342OC. [DOI] [PubMed] [Google Scholar]
- McClung JM, Kavazis AN, Whidden MA, DeRuisseau KC, Falk DJ, Criswell DS, Powers SK. Antioxidant administration attenuates mechanical ventilation-induced rat diaphragm muscle atrophy independent of protein kinase B (PKB Akt) signalling. J Physiol. 2007;585:203–215. doi: 10.1113/jphysiol.2007.141119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung JM, Whidden MA, Kavazis AN, Falk DJ, Deruisseau KC, Powers SK. Redox regulation of diaphragm proteolysis during mechanical ventilation. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1608–R1617. doi: 10.1152/ajpregu.00044.2008. [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102:2389–2397. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- Clark BC, Fernhall B, Ploutz-Snyder LL. Adaptations in human neuromuscular function following prolonged unweighting: I. Skeletal muscle contractile properties and applied ischemia efficacy. J Appl Physiol. 2006;101:256–263. doi: 10.1152/japplphysiol.01402.2005. [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, DeRuisseau KC. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288:R337–R344. doi: 10.1152/ajpregu.00469.2004. [DOI] [PubMed] [Google Scholar]
- Jaber S, Sebbane M, Koechlin C, Hayot M, Capdevila X, Eledjam JJ, Prefaut C, Ramonatxo M, Matecki S. Effects of short vs. prolonged mechanical ventilation on antioxidant systems in piglet diaphragm. Intensive Care Med. 2005;31:1427–1433. doi: 10.1007/s00134-005-2694-1. [DOI] [PubMed] [Google Scholar]
- Falk DJ, Deruisseau KC, Van Gammeren DL, Deering MA, Kavazis AN, Powers SK. Mechanical ventilation promotes redox status alterations in the diaphragm. J Appl Physiol. 2006;101:1017–1024. doi: 10.1152/japplphysiol.00104.2006. [DOI] [PubMed] [Google Scholar]
- Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, DeRuisseau KC, Powers SK. Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol. 2003;95:1116–1124. doi: 10.1152/japplphysiol.00824.2002. [DOI] [PubMed] [Google Scholar]
- Betters JL, Criswell DS, Shanely RA, Van Gammeren D, Falk D, Deruisseau KC, Deering M, Yimlamai T, Powers SK. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med. 2004;170:1179–1184. doi: 10.1164/rccm.200407-939OC. [DOI] [PubMed] [Google Scholar]
- Van Gammeren D, Falk DJ, Deering MA, Deruisseau KC, Powers SK. Diaphragmatic nitric oxide synthase is not induced during mechanical ventilation. J Appl Physiol. 2007;102:157–162. doi: 10.1152/japplphysiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- Whidden MA, McClung JM, Falk DJ, Hudson MB, Smuder AJ, Nelson WB, Powers SK. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol. 2009;106:385–394. doi: 10.1152/japplphysiol.91106.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung JM, Van Gammeren D, Whidden MA, Falk DJ, Kavazis AN, Hudson MB, Gayan-Ramirez G, Decramer M, DeRuisseau KC, Powers SK. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit Care Med. 2009;37:1373–1379. doi: 10.1097/CCM.0b013e31819cef63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med. 2009;46:842–850. doi: 10.1016/j.freeradbiomed.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung JM, Judge AR, Talbert EE, Powers SK. Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. Am J Physiol Cell Physiol. 2009;296:C363–C371. doi: 10.1152/ajpcell.00497.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL, Elledge SJ, Harper JW. The cellular chamber of doom. Sci Am. 2001;284:68–73. doi: 10.1038/scientificamerican0101-68. [DOI] [PubMed] [Google Scholar]
- McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med. 2007;175:150–159. doi: 10.1164/rccm.200601-142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- Koh TJ, Tidball JG. Nitric oxide inhibits calpain-mediated proteolysis of talin in skeletal muscle cells. Am J Physiol Cell Physiol. 2000;279:C806–C812. doi: 10.1152/ajpcell.2000.279.3.C806. [DOI] [PubMed] [Google Scholar]
- Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–123. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/S0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoon CS, Zhu E, Caiozzo VJ. Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med. 2004;170:626–632. doi: 10.1164/rccm.200401-042OC. [DOI] [PubMed] [Google Scholar]
- Gayan-Ramirez G, Testelmans D, Maes K, Rácz GZ, Cadot P, Zádor E, Wuytack F, Decramer M. Intermittent spontaneous breathing protects the rat diaphragm from mechanical ventilation effects. Crit Care Med. 2005;33:2804–2809. doi: 10.1097/01.CCM.0000191250.32988.A3. [DOI] [PubMed] [Google Scholar]
- Futier E, Constantin JM, Combaret L, Mosoni L, Roszyk L, Sapin V, Attaix D, Jung B, Jaber S, Bazin JE. Pressure support ventilation attenuates ventilator-induced protein modifications in the diaphragm. Crit Care. 2008;12:R116. doi: 10.1186/cc7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck GC, Mantilla CB. Effects of mechanical ventilation on the diaphragm. N Engl J Med. 2008;358:1392–1393. doi: 10.1056/NEJMe0801226. [DOI] [PubMed] [Google Scholar]
- Caruso P, Denari SD, Ruiz SA, Bernal KG, Manfrin GM, Friedrich C, Deheinzelin D. Inspiratory muscle training is ineffective in mechanically ventilated critically ill patients. Clinics. 2005;60:479–484. doi: 10.1590/S1807-59322005000600009. [DOI] [PubMed] [Google Scholar]
- Amaya-Villar R, Garnacho-Montero J, Garcia-Garmendia JL, Madrazo-Osuna J, Garnacho-Montero MC, Luque R, Ortiz-Leyba C. Steroid-induced myopathy in patients intubated due to exacerbation of chronic obstructive pulmonary disease. Intensive Care Med. 2005;31:157–161. doi: 10.1007/s00134-004-2509-9. [DOI] [PubMed] [Google Scholar]
- De Jonghe B, Sharshar T, Lefaucheur JP, Authier FJ, Durand-Zaleski I, Boussarsar M, Cerf C, Renaud E, Mesrati F, Carlet J, Raphaël JC, Outin H, Bastuji-Garin S. Groupe de Réflexion et d'Etude des Neuromyopathies en Réanimation. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA. 2002;288:2859–2867. doi: 10.1001/jama.288.22.2859. [DOI] [PubMed] [Google Scholar]
- Maes K, Testelmans D, Cadot P, Deruisseau K, Powers SK, Decramer M, Gayan-Ramirez G. Effects of acute administration of corticosteroids during mechanical ventilation on rat diaphragm. Am J Respir Crit Care Med. 2008;178:1219–1226. doi: 10.1164/rccm.200702-296OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom L, Zhu E, Pham TH, Jiao G, Caiozzo VJ, Sassoon CSH. Effects of methylprednisolone on diaphragmatic contractile properties during mechanical ventilation. Proc Am Thoracic Soc. 2006;3:A137. [Google Scholar]