

Abstract
Sepsis, despite recent therapeutic progress, still carries unacceptably high mortality rates. The adrenergic system, a key modulator of organ function and cardiovascular homeostasis, could be an interesting new therapeutic target for septic shock. β-Adrenergic regulation of the immune function in sepsis is complex and is time dependent. However, β2 activation as well as β1 blockade seems to downregulate proinflammatory response by modulating the cytokine production profile. β1 blockade improves cardiovascular homeostasis in septic animals, by lowering myocardial oxygen consumption without altering organ perfusion, and perhaps by restoring normal cardiovascular variability. β-Blockers could also be of interest in the systemic catabolic response to sepsis, as they oppose epinephrine which is known to promote hyperglycemia, lipid and protein catabolism. The role of β-blockers in coagulation is less clear cut. They could have a favorable role in the septic pro-coagulant state, as β1 blockade may reduce platelet aggregation and normalize the depressed fibrinolytic status induced by adre-nergic stimulation. Therefore, β1 blockade as well as β2 activation improves sepsis-induced immune, cardiovascular and coagulation dysfunctions. β2 blocking, however, seems beneficial in the metabolic field. Enough evidence has been accumulated in the literature to propose β- adrenergic modulation, β1 blockade and β2 activation in particular, as new promising therapeutic targets for septic dyshomeostasis, modulating favorably immune, cardiovascular, metabolic and coagulation systems.
Introduction
Sepsis still places a burden on the healthcare system, with an annual increase in incidence of about 9% and a mortality of about 25% and up to 60% when shock is present [1,2]. Uncontrolled systemic inflammatory response is the hallmark of sepsis and contributes to the development of organ dysfunction and shock [3]. The exact mechanisms of cardiovascular failure following severe infection, however, remain poorly elucidated. The adrenergic system is a key modulator of organ function and cardiovascular homeostasis. These receptors are widely distributed in the body, including in circulating immune cells, vessels, the heart, airways, lungs, adipose tissues, skeletal muscles, and brain. Furthermore, β-adrenergic modulation is a frequent therapeutic intervention in the intensive care setting [4] - addressing the issue of its consequences in sepsis.
The present review summarizes current knowledge on the effects of β-adrenergic agonists and antagonists on immune, cardiac, metabolic and hemostasis functions during sepsis. A comprehensive understanding of this complex regulation system will enable the clinician to better apprehend the impact of β-stimulants and β-blockers in septic patients.
β-Adrenergic receptor and signaling cascade
The β-adrenergic receptor is a G-protein-coupled seven-transmembrane domain receptor. There are three receptor subtypes: β1, β2 and β3. β1-receptors and β2-receptors are widely distributed, but β1-receptors predominate in the heart and β2-receptors are mainly found in smooth muscles such as vessels and bronchus. Table 1 presents details of the β-adrenergic system. Mixed β1β2-agonists include epinephrine and isoproterenol, selective β1-agonists include dobutamine, norepinephrine and dopamine, and selective β2-agonists include salbutamol, terbutaline and dopexamine.
Table 1.
The β-adrenergic system
| Target | Sympathetic receptor | |
|---|---|---|
| Heart | Cardiac muscle | β1β2 increases |
| Heart rate | β1β2 increases | |
| Blood vessels | Vascular smooth muscle | β2 relaxes |
| Smaller coronary arteries | β2 dilates | |
| Hepatic artery | β2 dilates | |
| Arteries to skeletal muscle | β2 dilates | |
| Veins | β2 dilates | |
| Respiratory system | Smooth muscles of bronchioles | β2 relaxes |
| Digestive system | Smooth muscle of gastrointestinal tract | β2 relaxes |
| Sphincters of gastrointestinal tract | β2 contracts | |
| Kidney | β1 enhances renin secretion | |
| Liver | β2 increases glycogenolysis, gluconeogenesis | |
| Adipose cells | β1β3 stimulates lipolysis | |
| Urinary system | Detrusor | β2 relaxes |
| Sphincter | β2 relaxes | |
| Reproductive system | Uterus | β2 relaxes |
| Nervous system | Ciliary muscles | β2 relaxes |
Upon activation by specific agonists, activated Gs proteins increase intracystosolic cAMP via an adenylate cyclase-dependent pathway [5]. cAMP activates protein kinase A, which in turn phosphorylates numerous targets in the cell such as transmembrane channels, and modulates nucleus transcription via the Ras, Raf, MEK and ERK pathways [6]. The β-receptor itself can be phosphorylated by protein kinase A, inducing its uncoupling from the G protein (acute response) and its internalization (chronic response) - the whole process leading to a downregulation of β-adrenergic signaling.
β-Adrenergic-mediated immune modulation
Immune response to sepsis
By definition, sepsis corresponds to a syndrome of systemic inflammatory response triggered by invading pathogens [7].
In bone marrow tissues, sepsis is associated with a shift in the myelopoietic production towards the monocyte lineage, at the expense of the granulocytic lineage [8]. Activated mononuclear cells release a broad variety of proinflammatory cytokines, including IL-1, IL-6, TNFα, IL-12, IL-15 and IL-18, as well as the so-called late mediators, high mobility group box 1 and macrophage migration inhibitory factor [3]. Generally, the synthesis of proinflammatory cytokines is mediated by NFκ B. In parallel, a physiologic counter-inflammatory response is initiated with the release of IL-10, IL-1-receptor antagonist and soluble TNFα-receptor among various anti-inflammatory mediators. Mononuclear cells are subsequently reprogrammed, allowing the inflammation to be turned off. In addition, following the initial hyperinflammatory response, immune cell apoptosis occurs, taking part in the secondary impairment of immune function. Apoptosis concerns mainly B lymphocytes and CD4+ cells, as well as dendritic cells and epithelial cells. It appears that, apart from direct immune cell stock depletion, apoptotic bodies induce macrophage anergy and favor anti-inflammatory cytokine secretion [9].
Sepsis is therefore characterized by a balance between pro-inflammatory signals and anti-inflammatory signals to immuno-effector cells [10]. Excessive systemic inflammation may favor the development of organ failure, and excess anti-inflammatory mediators may compromise the local response to infection. This issue has not yet been elucidated, and the ambivalence of immune response in sepsis reflects the difficulty of finding therapeutic targets for immunomodulation.
β-Adrenergic system and immune modulation
The β-adrenergic system is a well-known powerful modulator of the immune system [11]. Lymphoid organs such as the spleen, thymus, lymph nodes and bone marrow are predominantly innervated by the sympathetic system. The majority of lymphoid cells express β-adrenergic receptors on their surface, with the exception of T-helper type 2 (Th2) cells. The density of cell surface receptors varies with cell type, natural killer cells having the highest density. The efficiency of receptor coupling with adenylate cyclase also differs among immune cells, with natural killer cells and monocytes being the most responsive cells.
In bone marrow, monocytic production appears to be under the influence of sympathetic activation via β2-receptors. Indeed, monocytes have an increased sensitivity to epinephrine. Upon adrenergic stimulation, monocytes differentiate into mature macrophages [12] that are functionally different in their cytokine response [13].
Immune cell apoptosis is at least partly mediated by catecholamines, via α-adrenergic and β-adrenergic pathways. Nonspecific and specific β2 blockade induce splenocyte apoptosis [14]. Epinephrine also exerts apoptosis, however, suggesting that β blockade creates an imbalance towards the α-adrenergic pathway that is proapoptotic [15].
Catecholamines, via a β2-mediated pathway [16], down-regulate the synthesis of proinflammatory cytokines such as TNFα, IL-6 and IL-1 [17-19], and upregulate synthesis of anti inflammatory cytokines (for example, IL-10) [18,19]. The pattern of cytokine production in sepsis is dependent on the CD4+ T-helper type 1 (Th1) and Th2 balance [20] (Figure 1). T-helper cells coordinate the adaptative immune response towards cellular or humoral response by secreting different subsets of cytokines. Subclass Th1 cells promote cellular immunity by secreting IFNγ, IL-2 and transforming growth factor beta, by activating macrophages and natural killer cells, and by producing inflammatory mediators. Subclass Th2 cells promote humoral response and synthesize primarily IL-4 and IL-10. Th2 response therefore inhibits macrophage activation, T-cell proliferation and proinflammatory cytokine production [11]. Th1 cells express β2-adrenergic receptors on their surface, whereas Th2 cells do not [21]. Sepsis-induced β2-adrenergic stimulation therefore selectively inhibits Th1 function and favors the Th2 response.
Figure 1.
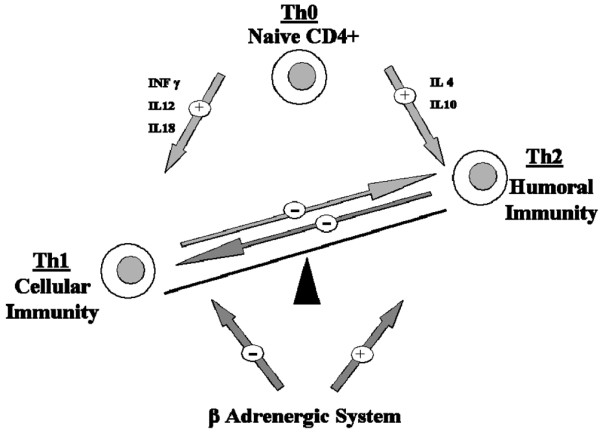
T-helper type 1 and T-helper type 2 balance and the adrenergic system. Naive CD4+, T-helper type 0 (Th0) cells are bipotential and are precursors of T-helper type 1 (Th1) cells and T-helper type 2 (Th2) cells. IL-12, produced by antigen-presenting cells, is the major inducer of Th1 differentiation. Th1 and Th2 responses are mutually inhibitory. IL-12 and IFNγ therefore inhibit Th2 cell activity, while IL-4 and IL-10 inhibit the Th1 response. The stimulation of β-adrenergic receptors potently inhibits the production of IL-12 by antigen-presenting cells, and thus inhibits the development of Th1 cells while promoting Th2 cells.
The immunosuppressive effects of catecholamines may be attenuated in prolonged septic shock. Indeed, epinephrine did not alter TNFα or IL-10 in patients in septic shock [22]. β-Adrenergic regulation of the immune function in sepsis is therefore complex and is time dependent. Nonselective β blockade by propranolol inhibited the mobilization and activation of natural killer cells [23]. Two studies in septic mice showed that propranolol upregulated Th1-mediated IFNγ production and downregulated the Th2-mediated IL-6 synthesis [15,24]. In these experiments, however, propranolol-treated animals had a greater mortality rate. In another study on severely burnt children, propranolol administration significantly decreased serum TNF and IL-1β concentrations, and did not increase mortality [25]. Interestingly, in septic rats, selective β1 blocking by esmolol decreased circulating TNFα and IL-1β concentrations [26]. Similarly, landiolol, another selective β1-blocker, also decreased circulating levels of TNFα, IL-6, and high mobility group box 1 [27]. While IL-10 production was not altered by β blockade [28], it was increased by β-agonists [18,19]. The mechanisms by which β1-adrenoceptor blockade modulates cytokine production remain unknown.
Counteracting the adrenergic storm of sepsis, β-blockers modulate cytokine profile production. β2-blockers seem to induce a proinflammatory profile, whereas β1 blockade has the opposite effect. The numerous studies conducted, however, show conflicting results on sepsis outcome. This inconsistency reflects the difficulty to apprehend the beneficial and harmful effects of the modulation of both proinflammatory and anti-inflammatory processes.
β-Adrenergic-mediated cardiovascular modulation
Physiopathology of myocardial dysfunction in sepsis
Myocardial depression, defined as diminution of the left ventricular ejection fraction, occurs in about 50% of patients with septic shock [29]. The depression is characterized by both left and right ventricular dysfunction. Systolic dysfunction occurs early in shock with lower ejection fraction and acute ventricle dilatation. Interestingly, patients who do not dilate their ventricles have worse prognosis than those with acute and reversible dilated cardiomyopathy. Diastolic dysfunction may also be altered, with slower ventricular filling on echocardiography and altered relaxation. Whether diastolic dysfunction carries a poor prognosis in sepsis is still unclear. Of note, in spite of the myocardial depression, following adequate fluid resuscitation, cardiac output remains high until death or recovery [30]. The mechanisms of myocardial depression are multiple [31], including microvascular abnormalities, autonomic dysregulation, metabolic changes, mitochondrial dysfunction and cardiomyocyte apoptosis [32].
Beside excessive adrenergic nervous system activation, patients with septic shock receive a substantial amount of exogenous catecholamines. This adrenergic storm is thought to be detrimental to cardiac function, as it is in chronic heart failure [33]. Studies have shown that intracystosolic cAMP levels are attenuated in sepsis after β-adrenergic stimulation [34], leading to decreased myocardial performance. Sepsis downregulates β-adrenoceptors by phosphorylation and internalization, reducing the density of receptors on the cell surface [35,36]. Uncoupling of β-adrenoceptors alters transduction pathways [37] via decreased expression of Gs proteins and increased inhibitory activity of Gi proteins [38]. The sustained adrenergic stimulation per se may trigger cytokine production by cardiomyocytes [39].
From another viewpoint, physiological oscillations in heart rate and blood pressure are directly correlated to the activity of the autonomic nervous system. Bedside analysis of heart rate variability may help in investigating the vagal to sympathetic balance [40]. In critically ill patients, the loss of heart rate variability may contribute to the progression of organ dysfunction [41] and is associated with increased risk of death [42-44]. Sepsis is often characterized by altered cardiovascular variability, and particularly by impaired sympathetic control on heart and vessel tone [45-47]. The loss in sympathetic modulation of the cardiovascular system preceded shock in both experimental sepsis and clinical sepsis [47,48].
β-Adrenergic modulation and sepsis-induced myocardial dysfunction
The first studies of adrenergic modulation in septic shock were published in the late 1960s [49,50]. These studies have evidenced that septic shock induced excessive β-adrenergic stimulation, altering splanchnic and pulmonary circulation. Infusion of propranolol then improved the arterial pressure and pH, and eventually improved survival.
Following the same line of evidence, excessive β1 stimulation by dobutamine increased the mortality rate in critically ill patients [51]. On the other hand, treatment with isoproterenol, a nonselective β-stimulant, in β1-adrenoceptor knockout mice prevented apoptosis in the myocardium [52]. These findings suggest that β 2-adrenergic stimulation protects myocardial function during sepsis. In septic rats, esmolol, a selective β1-antagonist, improved myocardial function and oxygen consumption [26]. There was also evidence of improved relaxation and an increase of end-diastolic volume. In patients with sepsis, esmolol lowered the heart rate and cardiac output without altering whole-body oxygen consumption, suggesting an increase in tissue oxygen extraction [53]. Organ perfusion also remained unaltered. In another study, oral metoprolol induced a significant increase in arterial pH and decreased arterial lactate concentrations [54]. In 22.5% of the metoprolol-treated patients, however, vasopressor therapy had to be increased.
On the sympathovagal modulation level, specific β1 blockade by metoprolol and atenolol may restore heart rate variability in conditions such as coronary artery disease [55] or chronic heart failure [56,57]. Whether selective β1-antagonist may restore normal cardiovascular oscillations in sepsis deserves to be investigated.
Evidence for a beneficial effect of β blockade is far more clear cut than for immune modulation. Indeed, β1 blockade seems beneficial to myocardial function in sepsis, improving its diastolic function, oxygen extraction and consumption. β2 stimulation should be respected as some evidence shows a protective action. Selective β1 blockade could also maintain global cardiovascular homeostasis by restoring adequate sympathovagal balance.
Metabolic effects of β-adrenergic modulation
Sepsis-associated metabolic dysfunction
Sepsis is associated with a systemic adaptative catabolic response [58] that is characterized by increased resting energy expenditure, extensive protein and fat catabolism, negative nitrogen balance, hyperglycemia, and progressive loss of lean body mass. Although this response to stress might be adaptative in the early stage, when sustained it may cause malnutrition and immunosuppression and may promote organ dysfunction [59] and death [60].
Counteracting hyperglycemia may favorably impact morbimortality [61,62]. Indeed, increased extracellular glucose impairs the host response to infection, increases oxidative stress, and favors procoagulant factors, sympathetic hyperactivity, and the proinflammatory response [63]. Hyperglycemia is multifactorial, and may result from enhanced counter-regulatory hormones glucagon, cortisol and catecholamines that promote hepatic glycogenolysis and gluconeogenesis, as well as peripheral insulin resistance [64].
Skeletal muscle protein loss is multifactorial, caused by an imbalance between an increased rate of muscle protein degradation and increased synthesis of proteins such as cytokines [65]. The decrease in basal muscle protein synthesis seems partly correlated by the availability of a branched-chain amino acid, leucine [66]. An increased leucine concentration stimulates muscle protein synthesis [67]. Sepsis is associated with muscle leucine resistance, via a mechanism not yet clearly understood.
At the cellular level, sepsis is characterized by impaired cellular respiration. Numerous studies have shown in septic shock that the tissue partial pressure of oxygen is normal or high, and cells are unable to utilize oxygen. This cytopathic hypoxia is a direct consequence of mitochondrial dysfunction [68], which is at least partly related to excessive release of nitric oxide following overexpression of inducible nitric oxide synthase [69].
Adrenergic modulation of sepsis-associated metabolic dysfunction
The increase in protein and lipid catabolism and hyperglycemia are both partly mediated via β2-adrenergic signaling [70,71]. Epinephrine induces insulin resistance [72] and enhances glucose hepatic production [73,74], but norepi-nephrine or dobutamine does not [75]. Epinephrine-induced gluconeogenesis increases hepatic oxygen consumption. As epinephrine also decreases hepatosplanchnic blood flow, relative splanchnic ischemia may develop [76,77]. Norepinephrine, on the contrary, increases hepatic blood flow and does not promote gluconeogenesis due to its lack of β2 activity. Dobutamine seems to increase blood flow but also increases splanchnic oxygen consumption [78,79]. Two studies conducted on healthy volunteers showed that epinephrine, although elevating the global metabolic rate, did not increase muscle proteolysis with regard to circulating leucine levels [77,80]. Basal metabolic rates of septic patients are far from those of healthy volunteers, however, and these results cannot be directly extrapolated.
In experimental sepsis, nonspecific β blockade by propranolol reduced the plasma glucose concentration via a decrease in endogenous glucose production [81,82]. Propranolol improved the nitrogen balance, suggesting reduced muscle proteolysis [83]. A key trial by Herndon and colleagues showed that propranolol treatment in severely burnt children, a condition associated with hypercatabolism and severe muscle wasting, attenuated the resting energy expenditure and reversed muscle protein catabolism [84]. By contrast, these effects were not seen with β1-selective blockers [53].
There is little information on the effects of β-adrenergic modulation on sepsis-associated cytopathic dysoxia. Nevertheless, in chronic heart failure and myocardial infarction, carvedilol exerted cardiac mitochondrial protection via inhibition of mitochondrial permeability transition [85,86].
In sepsis-induced metabolic disorder, β2 antagonization is beneficial - lowering gluconeogenesis, hyperglycemia, proteolysis and resting energy expenditure. β1-blockers seem to lack any activity on metabolism modulation.
β-Adrenergic modulation and the coagulation system
β-Adrenergic modulation of platelet function
Platelets play a complex role in sepsis - they contribute to thrombus formation and release mediators of neutrophils and macrophages, and ensure vascular tone and endothelial integrity [87]. Sepsis is characterized by thrombocytopenia and altered platelet function. Studies in patients with sepsis showed variably reduced [88] or increased [89] platelet aggregability, and showed increased β-thromboglobulin, a marker of activated platelets [90].
Platelets express on their surface adrenergic receptors, which can modulate their functions [91]. α2-Adrenoceptor stimulation enhances aggregability via increased intra-cystosolic calcium and decreased cAMP, whereas β2-adrenoceptors reduce aggregability by stimulating cytosolic cAMP. A vast majority of in vivo and in vitro studies showed that epinephrine activated platelet aggregation via the α2-mediated pathway [91,92]. The effects of β blockade are more controversial. Indeed, β2 inhibition may enhance platelet activation by unopposed α2 stimulation. Chronic β-adrenergic blockade, however, could transregulate the α-adrenergic signal by reducing the density of α2-adrenoceptors on the cell surface and by impairing the signaling cascade [93]. In patients with ischemic heart disease, propranolol reduced platelet aggregation. Interestingly, in hypertensive patients, β1 blockade also reduced platelet aggregation, although the underlying mechanisms remain unclear [94].
These results were obtained in healthy volunteers and in patients with hypertension or ischemic heart disease, not in sepsis patients. The effects of catecholamines on in vivo septic platelet activation deserve to be investigated.
β-Adrenergic modulation of coagulation in sepsis
Sepsis induces an imbalance between exaggerated coagulation activation and inhibition of fibrinolysis. Plasma tissue factor and von Willebrand factor levels are increased, activating coagulation in conjunction with activated factor VII. Amplification by the coagulation cascade leads to thrombin formation, and then to fibrin formation [95]. Thrombi formation leads to endothelial damage, exposing more tissue factor and accentuating the coagulation activation. Endotoxins induce endothelial cell apoptosis by macrophage activation, further damaging the endothelium [96].
Sepsis also downregulates the physiologic anticoagulant proteins, tissue factor pathway inhibitor, antithrombin and activated protein C. Indeed, tissue factor pathway inhibitor activity is impaired as a result of glycosaminoglycan down-regulation. Circulating antithrombin levels are decreased by impaired synthesis and increased consumption. Liver protein C synthesis and activation are impaired. Finally, as fibrin clots are generated by the coagulation process, the fibrinolytic system degrades fibrin by its key enzyme, plasmin. Plasmin formation from plasminogen is mainly activated by tissue plasminogen activator and inhibited by plasminogen activator inhibitor 1. In response to an increased circulating TNFα and IL-1β concentration, plasminogen activator inhibitor 1 production is enhanced, leading to impaired fibrinolysis. The net result in sepsis is a procoagulant state, where coagulation is activated and amplified, and physiologic anticoagulation and fibrinolytic mechanisms are impaired.
To the extreme, disseminated intravascular coagulation occurs, promoting microcirculation alteration - which is one of the key factors of multiple organ failure. Furthermore, as inflammation interacts with coagulation, complement activation by sepsis leads to products such as C3a, C4a and C5a, which may promote procoagulant activity [97]. C5a induces tissue factor on endothelial cells, C5b stimulates prothrombinase activity, and C4b affects the protein S system. Antagonization of C5a by specific antibodies in septic rats reduced procoagulant activity and prevented fibrinolysis impairment [98].
Epinephrine increases the factor VIII concentration [92]. This effect of epinephrine remains unaltered by selective β1-antagonists [99], suggesting a β2-adrenoceptor-mediated mechanism. In addition, salbutamol and not norepinephrine reproduced epinephrine hemostatic effects. Epinephrine also increased von Willebrand factor concentrations, an effect abolished by nonselective β blockade [100]. Adrenergic agonist did not affect tissue factor concentrations, but epinephrine might increase the procoagulant activity by promoting P-selectin expression on the platelet surface [101]. There is no information on adrenergic modulation of tissue factor pathway inhibitor, antithrombin or activated protein C.
Adrenergic stimulation has been known to increase fibrinolytic activity since the 1960s [102], due to stimulated tissue plasminogen activator release by specific β2-adrenoceptor activation. β1 activation, on the other hand, did not modify tissue plasminogen activator or plasminogen activator inhibitor 1 levels [103] - but may decrease fibrinolysis via reduction of prostacyclin synthesis by endothelial cells [104]. Accordingly, nonselective β-blockers reduced tissue plasminogen activator concentrations and decreased fibrinolysis, but β1-blockers did not. It is thought that β1-blockers could normalize the depressed fibrinolytic status induced by β1-adrenergic stimulation [103].
Complement modulation by β blockade or β activation has not been investigated. Hepatic complement clearance seems to be influenced by the adrenergic system, however, as propranolol restores Kupffer cell clearance function in vitro, hence reducing circulating activated complement fractions [105].
The sepsis-induced procoagulant state is therefore at least partly mediated by the adrenergic system. β1 and β2 pathways, however, seem to have opposite effects. β2 blocking could be detrimental by counteracting a β2-induced decrease in platelet activation and improved fibrinolytic activity. β1 blockade could therefore be beneficial for the fibrinolytic status.
Conclusion
The β-adrenergic system has a wide range of effects in various organ systems. Tight regulation of β1-adrenergic and β2-adrenergic receptors may contribute to restore immune, metabolic, cardiovascular and coagulation homeostasis. Modulating the β-adrenergic system, and in particular β blockade, is therefore an exciting new therapeutic approach. The use of hypotensive drugs such as β-blockers in severe sepsis and septic shock, however, raises justified safety issues. Manipulation of the β-adrenergic system in septic shock should be carefully tested in various animal models of sepsis prior to considering its evaluation in patients.
Abbreviations
IFN: interferon; IL: interleukin; NF: nuclear factor; Th1: T-helper type 1; Th2: T-helper type 2; TNF: tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Etienne de Montmollin, Email: edemontmollin@gmail.com.
Jerome Aboab, Email: jerome.aboab@rpc.aphp.fr.
Arnaud Mansart, Email: arnaud.mansart@rpc.aphp.fr.
Djillali Annane, Email: djillali.annane@rpc.aphp.fr.
References
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med. 2003;168:165–172. doi: 10.1164/rccm.2201087. [DOI] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Oberbeck R, Kobbe P. Beta-adrenergic antagonists: indications and potential immunomodulatory side effects in the critically ill. Curr Med Chem. 2009;16:1082–1090. doi: 10.2174/092986709787581770. [DOI] [PubMed] [Google Scholar]
- Wallukat G. The beta-adrenergic receptors. Herz. 2002;27:683–690. doi: 10.1007/s00059-002-2434-z. [DOI] [PubMed] [Google Scholar]
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth. 2002;88:101–123. doi: 10.1093/bja/88.1.101. [DOI] [PubMed] [Google Scholar]
- American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–874. [PubMed] [Google Scholar]
- Santangelo S, Gamelli RL, Shankar R. Myeloid commitment shifts toward monocytopoiesis after thermal injury and sepsis. Ann Surg. 2001;233:97–106. doi: 10.1097/00000658-200101000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro da Silva F, Nizet V. Cell death during sepsis: integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis. 2009;14:509–521. doi: 10.1007/s10495-009-0320-3. [DOI] [PubMed] [Google Scholar]
- Pinsky MR. Dysregulation of the immune response in severe sepsis. Am J Med Sci. 2004;328:220–229. doi: 10.1097/00000441-200410000-00005. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve - an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- Muthu K, Deng J, Romano F, He LK, Gamelli R, Shankar R, Jones SB. Thermal injury and sepsis modulates beta-adrenergic receptors and cAMP responses in monocyte-committed bone marrow cells. J Neuroimmunol. 2005;165:129–138. doi: 10.1016/j.jneuroim.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Cohen MJ, Shankar R, Stevenson J, Fernandez R, Gamelli RL, Jones SB. Bone marrow norepinephrine mediates development of functionally different macrophages after thermal injury and sepsis. Ann Surg. 2004;240:132–141. doi: 10.1097/01.sla.0000130724.84914.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberbeck R. Catecholamines: physiological immunomodulators during health and illness. Curr Med Chem. 2006;13:1979–1989. doi: 10.2174/092986706777584997. [DOI] [PubMed] [Google Scholar]
- Oberbeck R, Schmitz D, Wilsenack K, Schuler M, Pehle B, Schedlowski M, Exton MS. Adrenergic modulation of survival and cellular immune functions during polymicrobial sepsis. Neuroimmunomodulation. 2004;11:214–223. doi: 10.1159/000078439. [DOI] [PubMed] [Google Scholar]
- Sekut L, Champion BR, Page K, Menius JA Jr, Connolly KM. Anti-inflammatory activity of salmeterol: down-regulation of cytokine production. Clin Exp Immunol. 1995;99:461–466. doi: 10.1111/j.1365-2249.1995.tb05573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severn A, Rapson NT, Hunter CA, Liew FY. Regulation of tumor necrosis factor production by adrenaline and beta-adrenergic agonists. J Immunol. 1992;148:3441–3445. [PubMed] [Google Scholar]
- Muthu K, Deng J, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of cytokine release in bone marrow progenitor-derived macrophage following polymicrobial sepsis. J Neuroimmunol. 2005;158:50–57. doi: 10.1016/j.jneuroim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Deng J, Muthu K, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of splenic macrophage cytokine release in polymicrobial sepsis. Am J Physiol Cell Physiol. 2004;287:C730–C736. doi: 10.1152/ajpcell.00562.2003. [DOI] [PubMed] [Google Scholar]
- Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol. 1997;158:4200–4210. [PubMed] [Google Scholar]
- Ramer-Quinn DS, Baker RA, Sanders VM. Activated T helper 1 and T helper 2 cells differentially express the beta-2-adrenergic receptor: a mechanism for selective modulation of T helper 1 cell cytokine production. J Immunol. 1997;159:4857–4867. [PubMed] [Google Scholar]
- Bergmann M, Gornikiewicz A, Sautner T, Waldmann E, Weber T, Mittlbock M, Roth E, Fugger R. Attenuation of catecholamine-induced immunosuppression in whole blood from patients with sepsis. Shock. 1999;12:421–427. doi: 10.1097/00024382-199912000-00002. [DOI] [PubMed] [Google Scholar]
- Schedlowski M, Hosch W, Oberbeck R, Benschop RJ, Jacobs R, Raab HR, Schmidt RE. Catecholamines modulate human NK cell circulation and function via spleen-independent beta 2-adrenergic mechanisms. J Immunol. 1996;156:93–99. [PubMed] [Google Scholar]
- Schmitz D, Wilsenack K, Lendemanns S, Schedlowski M, Oberbeck R. β-Adrenergic blockade during systemic inflammation: impact on cellular immune functions and survival in a murine model of sepsis. Resuscitation. 2007;72:286–294. doi: 10.1016/j.resuscitation.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Jeschke MG, Norbury WB, Finnerty CC, Branski LK, Herndon DN. Propranolol does not increase inflammation, sepsis, or infectious episodes in severely burned children. J Trauma. 2007;62:676–681. doi: 10.1097/TA.0b013e318031afd3. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Morisaki H, Serita R, Yamamoto M, Kotake Y, Ishizaka A, Takeda J. Infusion of the beta-adrenergic blocker esmolol attenuates myocardial dysfunction in septic rats. Crit Care Med. 2005;33:2294–2301. doi: 10.1097/01.CCM.0000182796.11329.3B. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Iwasaka H, Maeda H, Noguchi T. Landiolol, an ultra-short-acting beta1-adrenoceptor antagonist, has protective effects in an LPS-induced systemic inflammation model. Shock. 2009;31:515–520. doi: 10.1097/SHK.0b013e3181863689. [DOI] [PubMed] [Google Scholar]
- Boost KA, Flondor M, Hofstetter C, Platacis I, Stegewerth K, Hoegl S, Nguyen T, Muhl H, Zwissler B. The beta-adrenoceptor antagonist propranolol counteracts anti-inflammatory effects of isoflurane in rat endotoxemia. Acta Anaesthesiol Scand. 2007;51:900–908. doi: 10.1111/j.1399-6576.2007.01363.x. [DOI] [PubMed] [Google Scholar]
- Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, Damske BA, Parrillo JE. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- Court O, Kumar A, Parrillo JE. Clinical review: Myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–508. doi: 10.1186/cc1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–1608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- Feldman AM, Bristow MR. The beta-adrenergic pathway in the failing human heart: implications for inotropic therapy. Cardiology. 1990;77(Suppl 1):1–32. doi: 10.1159/000174645. [DOI] [PubMed] [Google Scholar]
- Romanosky AJ, Giaimo ME, Shepherd RE, Burns AH. The effect of in vivo endotoxin on myocardial function in vitro. Circ Shock. 1986;19:1–12. [PubMed] [Google Scholar]
- Bristow MR, Hershberger RE, Port JD, Gilbert EM, Sandoval A, Rasmussen R, Cates AE, Feldman AM. Beta-adrenergic pathways in nonfailing and failing human ventricular myocardium. Circulation. 1990;82(2 Suppl):I12–I25. [PubMed] [Google Scholar]
- Tang C, Yang J, Wu LL, Dong LW, Liu MS. Phosphorylation of beta-adrenergic receptor leads to its redistribution in rat heart during sepsis. Am J Physiol. 1998;274(4 Pt 2):R1078–R1086. doi: 10.1152/ajpregu.1998.274.4.R1078. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Hattori Y, Akaishi Y, Suzuki Y, Kemmotsu O, Gando S. Impairment of cardiac beta-adrenoceptor cellular signaling by decreased expression of G(s alpha) in septic rabbits. Anesthesiology. 2000;93:1465–1473. doi: 10.1097/00000542-200012000-00019. [DOI] [PubMed] [Google Scholar]
- Brodde OE, Michel MC, Zerkowski HR. Signal transduction mechanisms controlling cardiac contractility and their alterations in chronic heart failure. Cardiovasc Res. 1995;30:570–584. [PubMed] [Google Scholar]
- Murray DR, Prabhu SD, Chandrasekar B. Chronic beta-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation. 2000;101:2338–2341. doi: 10.1161/01.cir.101.20.2338. [DOI] [PubMed] [Google Scholar]
- Heart rate variability. Standards of measurement, physiological interpretation, and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Eur Heart J. 1996;17:354–381. [PubMed] [Google Scholar]
- Pontet J, Contreras P, Curbelo A, Medina J, Noveri S, Bentancourt S, Migliaro ER. Heart rate variability as early marker of multiple organ dysfunction syndrome in septic patients. J Crit Care. 2003;18:156–163. doi: 10.1016/j.jcrc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Yien HW, Hseu SS, Lee LC, Kuo TB, Lee TY, Chan SH. Spectral analysis of systemic arterial pressure and heart rate signals as a prognostic tool for the prediction of patient outcome in the intensive care unit. Crit Care Med. 1997;25:258–266. doi: 10.1097/00003246-199702000-00011. [DOI] [PubMed] [Google Scholar]
- Gang Y, Malik M. Heart rate variability in critical care medicine. Curr Opin Crit Care. 2002;8:371–375. doi: 10.1097/00075198-200210000-00002. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Muller-Werdan U, Hoffmann T, Francis DP, Piepoli MF, Rauchhaus M, Prondzinsky R, Loppnow H, Buerke M, Hoyer D, Werdan K. Autonomic dysfunction predicts mortality in patients with multiple organ dysfunction syndrome of different age groups. Crit Care Med. 2005;33:1994–2002. doi: 10.1097/01.CCM.0000178181.91250.99. [DOI] [PubMed] [Google Scholar]
- Korach M, Sharshar T, Jarrin I, Fouillot JP, Raphael JC, Gajdos P, Annane D. Cardiac variability in critically ill adults: influence of sepsis. Crit Care Med. 2001;29:1380–1385. doi: 10.1097/00003246-200107000-00013. [DOI] [PubMed] [Google Scholar]
- Godin PJ, Fleisher LA, Eidsath A, Vandivier RW, Preas HL, Banks SM, Buchman TG, Suffredini AF. Experimental human endotoxemia increases cardiac regularity: results from a prospective, randomized, crossover trial. Crit Care Med. 1996;24:1117–1124. doi: 10.1097/00003246-199607000-00009. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Kempski MH, Stair D, Tipton RB, DeKing D, DeLong DJ, DeAsla R, Cox C, Lund N, Woolf PD. Autonomic modulation of heart rate variability during endotoxin shock in rabbits. Crit Care Med. 1995;23:1694–1702. doi: 10.1097/00003246-199510000-00014. [DOI] [PubMed] [Google Scholar]
- Annane D, Trabold F, Sharshar T, Jarrin I, Blanc AS, Raphael JC, Gajdos P. Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med. 1999;160:458–465. doi: 10.1164/ajrccm.160.2.9810073. [DOI] [PubMed] [Google Scholar]
- Berk JL, Hagen JF, Beyer WH, Gerber MJ, Dochat GR. The treatment of endotoxin shock by beta adrenergic blockade. Ann Surg. 1969;169:74–81. doi: 10.1097/00000658-196901000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk JL, Hagen JF, Maly G, Koo R. The treatment of shock with beta adrenergic blockade. Arch Surg. 1972;104:46–51. doi: 10.1001/archsurg.1972.04180010040011. [DOI] [PubMed] [Google Scholar]
- Hayes MA, Timmins AC, Yau EH, Palazzo M, Hinds CJ, Watson D. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717–1722. doi: 10.1056/NEJM199406163302404. [DOI] [PubMed] [Google Scholar]
- Patterson AJ, Zhu W, Chow A, Agrawal R, Kosek J, Xiao RP, Kobilka B. Protecting the myocardium: a role for the beta2 adrenergic receptor in the heart. Crit Care Med. 2004;32:1041–1048. doi: 10.1097/01.CCM.0000120049.43113.90. [DOI] [PubMed] [Google Scholar]
- Gore DC, Wolfe RR. Hemodynamic and metabolic effects of selective beta1 adrenergic blockade during sepsis. Surgery. 2006;139:686–694. doi: 10.1016/j.surg.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Schmittinger CA, Dunser MW, Haller M, Ulmer H, Luckner G, Torgersen C, Jochberger S, Hasibeder WR. Combined milrinone and enteral metoprolol therapy in patients with septic myocardial depression. Crit Care. 2008;12:R99. doi: 10.1186/cc6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemela MJ, Airaksinen KE, Huikuri HV. Effect of beta-blockade on heart rate variability in patients with coronary artery disease. J Am Coll Cardiol. 1994;23:1370–1377. doi: 10.1016/0735-1097(94)90379-4. [DOI] [PubMed] [Google Scholar]
- Aronson D, Burger AJ. Effect of beta-blockade on heart rate variability in decompensated heart failure. Int J Cardiol. 2001;79:31–39. doi: 10.1016/S0167-5273(01)00401-6. [DOI] [PubMed] [Google Scholar]
- Lin LY, Lin JL, Du CC, Lai LP, Tseng YZ, Huang SK. Reversal of deteriorated fractal behavior of heart rate variability by beta-blocker therapy in patients with advanced congestive heart failure. J Cardiovasc Electrophysiol. 2001;12:26–32. doi: 10.1046/j.1540-8167.2001.00026.x. [DOI] [PubMed] [Google Scholar]
- Trager K, DeBacker D, Radermacher P. Metabolic alterations in sepsis and vasoactive drug-related metabolic effects. Curr Opin Crit Care. 2003;9:271–278. doi: 10.1097/00075198-200308000-00004. [DOI] [PubMed] [Google Scholar]
- Chiolero R, Revelly JP, Tappy L. Energy metabolism in sepsis and injury. Nutrition. 1997;13(9 Suppl):45S–51S. doi: 10.1016/S0899-9007(97)00205-0. [DOI] [PubMed] [Google Scholar]
- Hasselgren PO. Catabolic response to stress and injury: implications for regulation. World J Surg. 2000;24:1452–1459. doi: 10.1007/s002680010262. [DOI] [PubMed] [Google Scholar]
- Berghe G van den, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- Berghe G Van den, Wouters PJ, Bouillon R, Weekers F, Verwaest C, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P. Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Crit Care Med. 2003;31:359–366. doi: 10.1097/01.CCM.0000045568.12881.10. [DOI] [PubMed] [Google Scholar]
- Brierre S, Kumari R, Deboisblanc BP. The endocrine system during sepsis. Am J Med Sci. 2004;328:238–247. doi: 10.1097/00000441-200410000-00007. [DOI] [PubMed] [Google Scholar]
- Chambrier C, Laville M, Rhzioual Berrada K, Odeon M, Bouletreau P, Beylot M. Insulin sensitivity of glucose and fat metabolism in severe sepsis. Clin Sci (Lond) 2000;99:321–328. doi: 10.1042/CS20000017. [DOI] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab. 2007;293:E453–E459. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Jefferson LS. Signaling pathways and molecular mechanisms through which branched-chain amino acids mediate translational control of protein synthesis. J Nutr. 2006;136(1 Suppl):227S–231S. doi: 10.1093/jn/136.1.227S. [DOI] [PubMed] [Google Scholar]
- Lang CH, Frost RA. Endotoxin disrupts the leucine-signaling pathway involving phosphorylation of mTOR, 4E-BP1, and S6K1 in skeletal muscle. J Cell Physiol. 2005;203:144–155. doi: 10.1002/jcp.20207. [DOI] [PubMed] [Google Scholar]
- Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:219–237. doi: 10.1016/S0749-0704(05)70161-5. [DOI] [PubMed] [Google Scholar]
- Fink MP. Bench-to-bedside review: Cytopathic hypoxia. Crit Care. 2002;6:491–499. doi: 10.1186/cc1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GW, Doxey JC, Walter DS, Reid JL. The role of alpha- and beta-adrenoceptor subtypes in mediating the effects of catecholamines on fasting glucose and insulin concentrations in the rat. Br J Pharmacol. 1990;100:699–704. doi: 10.1111/j.1476-5381.1990.tb14078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner CA, Kendall MJ. Metabolic effects of beta 2-agonists. J Clin Pharm Ther. 1992;17:155–164. doi: 10.1111/j.1365-2710.1992.tb01285.x. [DOI] [PubMed] [Google Scholar]
- Lang CH. Sepsis-induced insulin resistance in rats is mediated by a beta-adrenergic mechanism. Am J Physiol. 1992;263(4 Pt 1):E703–E711. doi: 10.1152/ajpendo.1992.263.4.E703. [DOI] [PubMed] [Google Scholar]
- McGuinness OP, Shau V, Benson EM, Lewis M, Snowden RT, Greene JE, Neal DW, Cherrington AD. Role of epinephrine and norepinephrine in the metabolic response to stress hormone infusion in the conscious dog. Am J Physiol. 1997;273(4 Pt 1):E674–E681. doi: 10.1152/ajpendo.1997.273.4.E674. [DOI] [PubMed] [Google Scholar]
- Trager K, Radermacher P, Debacker D, Vogt J, Jakob S, Ensinger H. Metabolic effects of vasoactive agents. Curr Opin Anaesthesiol. 2001;14:157–163. doi: 10.1097/00001503-200104000-00006. [DOI] [PubMed] [Google Scholar]
- Levy B, Mansart A, Bollaert PE, Franck P, Mallie JP. Effects of epinephrine and norepinephrine on hemodynamics, oxidative metabolism, and organ energetics in endotoxemic rats. Intensive Care Med. 2003;29:292–300. doi: 10.1007/s00134-002-1611-0. [DOI] [PubMed] [Google Scholar]
- Dahn MS, Lange P, Lobdell K, Hans B, Jacobs LA, Mitchell RA. Splanchnic and total body oxygen consumption differences in septic and injured patients. Surgery. 1987;101:69–80. [PubMed] [Google Scholar]
- Ensinger H, Trager K, Geisser W, Anhaupl T, Ahnefeld FW, Vogt J, Georgieff M. Glucose and urea production and leucine, ketoisocaproate and alanine fluxes at supraphysiological plasma adrenaline concentrations in volunteers. Intensive Care Med. 1994;20:113–118. doi: 10.1007/BF01707665. [DOI] [PubMed] [Google Scholar]
- De Backer D, Creteur J, Noordally O, Smail N, Gulbis B, Vincent JL. Does hepatosplanchnic VO2/DO2 dependency exist in critically ill septic patients? Am J Respir Crit Care Med. 1998;157(4 Pt 1):1219–1225. doi: 10.1164/ajrccm.157.4.9705075. [DOI] [PubMed] [Google Scholar]
- Asfar P, De Backer D, Meier-Hellmann A, Radermacher P, Sakka SG. Clinical review: influence of vasoactive and other therapies on intestinal and hepatic circulations in patients with septic shock. Crit Care. 2004;8:170–179. doi: 10.1186/cc2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DE, Pesola G, Campbell RG. Effect of epinephrine on amino acid and energy metabolism in humans. Am J Physiol. 1990;258(6 Pt 1):E948–E956. doi: 10.1152/ajpendo.1990.258.6.E948. [DOI] [PubMed] [Google Scholar]
- Norbury WB, Jeschke MG, Herndon DN. Metabolism modulators in sepsis: propranolol. Crit Care Med. 2007;35(9 Suppl):S616–S620. doi: 10.1097/01.CCM.0000278599.30298.80. [DOI] [PubMed] [Google Scholar]
- Hargrove DM, Bagby GJ, Lang CH, Spitzer JJ. Adrenergic blockade prevents endotoxin-induced increases in glucose metabolism. Am J Physiol. 1988;255(5 Pt 1):E629–E635. doi: 10.1152/ajpendo.1988.255.5.E629. [DOI] [PubMed] [Google Scholar]
- Dickerson RN, Fried RC, Bailey PM, Stein TP, Mullen JL, Buzby GP. Effect of propranolol on nitrogen and energy metabolism in sepsis. J Surg Res. 1990;48:38–41. doi: 10.1016/0022-4804(90)90142-O. [DOI] [PubMed] [Google Scholar]
- Herndon DN, Hart DW, Wolf SE, Chinkes DL, Wolfe RR. Reversal of catabolism by beta-blockade after severe burns. N Engl J Med. 2001;345:1223–1229. doi: 10.1056/NEJMoa010342. [DOI] [PubMed] [Google Scholar]
- Ichihara S, Yamada Y, Ichihara G, Kanazawa H, Hashimoto K, Kato Y, Matsushita A, Oikawa S, Yokota M, Iwase M. Attenuation of oxidative stress and cardiac dysfunction by bisoprolol in an animal model of dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;350:105–113. doi: 10.1016/j.bbrc.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Caruana JA, Whalen DA Jr, Maben WC, Chen HH. Beta-adrenergic blockade increases the hepatic extraction of glucose in sepsis. Arch Surg. 1986;121:319–323. doi: 10.1001/archsurg.1986.01400030073013. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Yagushi A, Pradier O. Platelet function in sepsis. Crit Care Med. 2002;30(5 Suppl):S313–S317. doi: 10.1097/00003246-200205001-00022. [DOI] [PubMed] [Google Scholar]
- Boldt J, Menges T, Wollbruck M, Sonneborn S, Hempelmann G. Platelet function in critically ill patients. Chest. 1994;106:899–903. doi: 10.1378/chest.106.3.899. [DOI] [PubMed] [Google Scholar]
- Gawaz M, Dickfeld T, Bogner C, Fateh-Moghadam S, Neumann FJ. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. 1997;23:379–385. doi: 10.1007/s001340050344. [DOI] [PubMed] [Google Scholar]
- Mavrommatis AC, Theodoridis T, Orfanidou A, Roussos C, Christopoulou-Kokkinou V, Zakynthinos S. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit Care Med. 2000;28:451–457. doi: 10.1097/00003246-200002000-00027. [DOI] [PubMed] [Google Scholar]
- Hjemdahl P, Larsson PT, Wallen NH. Effects of stress and beta-blockade on platelet function. Circulation. 1991;84(6 Suppl):VI44–VI61. [PubMed] [Google Scholar]
- von Kanel R, Dimsdale JE. Effects of sympathetic activation by adrenergic infusions on hemostasis in vivo. Eur J Haematol. 2000;65:357–369. doi: 10.1034/j.1600-0609.2000.065006357.x. [DOI] [PubMed] [Google Scholar]
- Schwencke C, Schmeisser A, Weinbrenner C, Braun-Dullaeus RC, Marquetant R, Strasser RH. Transregulation of the alpha2-adrenergic signal transduction pathway by chronic beta-blockade: a novel mechanism for decreased platelet aggregation in patients. J Cardiovasc Pharmacol. 2005;45:253–259. doi: 10.1097/01.fjc.0000154372.03531.e1. [DOI] [PubMed] [Google Scholar]
- Markel A, Brook JG, Levy Y, Aviram M, Youdim MB. Increased platelet adhesion and aggregation in hypertensive patients: effect of atenolol. Br J Clin Pharmacol. 1983;16:663–668. doi: 10.1111/j.1365-2125.1983.tb02238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med. 2004;30:1032–1040. doi: 10.1007/s00134-004-2291-8. [DOI] [PubMed] [Google Scholar]
- Schouten M, Wiersinga WJ, Levi M, Poll T van der. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83:536–545. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- Laudes IJ, Chu JC, Sikranth S, Huber-Lang M, Guo RF, Riedemann N, Sarma JV, Schmaier AH, Ward PA. Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am J Pathol. 2002;160:1867–1875. doi: 10.1016/S0002-9440(10)61133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gader AM, Da Costa J, Cash JD. The effect propranolol, alprenolol and practolol on the vibrinolytic and factor VIII responses to adrenaline and salbutamol in man. Thromb Res. 1974;4:25–33. doi: 10.1016/0049-3848(74)90201-1. [DOI] [PubMed] [Google Scholar]
- von Kanel R, Dimsdale JE, Adler KA, Dillon E, Perez CJ, Mills PJ. Effects of nonspecific beta-adrenergic stimulation and blockade on blood coagulation in hypertension. J Appl Physiol. 2003;94:1455–1459. doi: 10.1152/japplphysiol.00892.2002. [DOI] [PubMed] [Google Scholar]
- Osterud B. Cellular interactions in tissue factor expression by blood monocytes. Blood Coagul Fibrinolysis. 1995;6(Suppl 1):S20–S25. doi: 10.1097/00001721-199506001-00004. [DOI] [PubMed] [Google Scholar]
- Ogston D, Mc DG, Fullerton HW. The influence of anxiety in tests of blood coagulability and fibrinolytic activity. Lancet. 1962;2(7255):521–523. doi: 10.1016/S0140-6736(62)90398-7. [DOI] [PubMed] [Google Scholar]
- Teger-Nilsson AC, Larsson PT, Hjemdahl P, Olsson G. Fibrinogen and plasminogen activator inhibitor-1 levels in hypertension and coronary heart disease. Potential effects of beta-blockade. Circulation. 1991;84(6 Suppl):VI72–VI77. [PubMed] [Google Scholar]
- Adler B, Gimbrone MA Jr, Schafer AI, Handin RI. Prostacyclin and beta-adrenergic catecholamines inhibit arachidonate release and PGI2 synthesis by vascular endothelium. Blood. 1981;58:514–517. [PubMed] [Google Scholar]
- Loegering DJ, Commins LM. Effect of beta-receptor stimulation on Kupffer cell complement receptor clearance function. Circ Shock. 1988;25:325–332. [PubMed] [Google Scholar]