

Abstract
Rationale: Impaired endothelium-dependent vasodilation has been documented in patients with sleep apnea. This impairment may result in blood flow dysregulation during apnea-induced fluctuations in arterial blood gases.
Objectives: To test the hypothesis that hypoxic and hypercapnic vasodilation in the forearm and cerebral circulation are impaired in patients with sleep apnea.
Methods: We exposed 20 patients with moderate to severe sleep apnea and 20 control subjects, to isocapnic hypoxia and hyperoxic hypercapnia. A subset of 14 patients was restudied after treatment with continuous positive airway pressure.
Measurements and Main Results: Cerebral flow velocity (transcranial Doppler), forearm blood flow (venous occlusion plethysmography), arterial pressure (automated sphygmomanometry), oxygen saturation (pulse oximetry), ventilation (pneumotachograph), and end-tidal oxygen and carbon dioxide tensions (expired gas analysis) were measured during three levels of hypoxia and two levels of hypercapnia. Cerebral vasodilator responses to hypoxia (−0.65 ± 0.44 vs. −1.02 ± 0.72 [mean ± SD] units/% saturation; P = 0.03) and hypercapnia (2.01 ± 0.88 vs. 2.57 ± 0.89 units/mm Hg; P = 0.03) were smaller in patients versus control subjects. Hypoxic vasodilation in the forearm was also attenuated (−0.05 ± 0.09 vs. −0.10 ± 0.09 unit/% saturation; P = 0.04). Hypercapnia did not elicit forearm vasodilation in either group. Twelve weeks of continuous positive airway pressure treatment enhanced hypoxic vasodilation in the cerebral circulation (−0.83 ± 0.32 vs. −0.46 ± 0.29 units/% saturation; P = 0.01) and forearm (−0.19 ± 0.15 vs. −0.02 ± 0.08 units/% saturation; P = 0.003), and hypercapnic vasodilation in the brain showed a trend toward improvement (2.24 ± 0.78 vs. 1.76 ± 0.64 units/mm Hg; P = 0.06).
Conclusions: Vasodilator responses to chemical stimuli in the cerebral circulation and the forearm are impaired in many patients with obstructive sleep apnea. Some of these impairments can be improved with continuous positive airway pressure.
Keywords: hypoxia, sleep, vasodilation, cerebral vascular circulation, regional blood flow
AT A GLANCE COMMENTARY
Scientific Knowledge on the Subject
Impaired hypoxic vasodilation in the cerebral circulation has been demonstrated in patients with obstructive sleep apnea (OSA).
What This Study Adds to the Field
Our study demonstrates that hypercapnic vasodilation in the cerebral circulation and hypoxic vasodilation in the forearm are impaired in patients with OSA. Furthermore, this study shows that such impairments are ameliorated by CPAP treatment.
Obstructive sleep apnea (OSA) is associated with hypertension and other forms of cardiovascular disease (1). The mechanisms underlying these relationships are not fully understood; however, one hypothesis is that vascular dysfunction is an important link between the conditions (2). Several impairments in vascular regulation are present in patients with OSA, including increased sympathetic nerve activity (3, 4) and diminished reactive hyperemia (5). Observations of decreased flow-mediated (6, 7) and acetylcholine-induced (8–10) vasodilation suggest that OSA impairs function of the vascular endothelium.
Patients with OSA demonstrate impaired vasodilator responses to acute hypoxia in the forearm (11) and the cerebral circulation (12). Because hypoxic vasodilation minimizes the changes in tissue Po2 during episodes of hypoxemia, these findings raise the possibility that, over time, OSA results in increasingly more severe tissue hypoxia during acute apneic events.
Some (13), but not all (14, 15), previous investigators have reported attenuated cerebral vasodilator responses to hypercapnia in patients with OSA. Cerebrovascular CO2 reactivity minimizes changes in brain Pco2 during fluctuations in arterial Pco2; therefore, reductions in reactivity could exacerbate breathing instability during sleep by exaggerating the accumulation and also the washout of CO2 from central chemoreceptors during fluctuations in ventilation. Thus, impaired vascular responses to hypercapnia and hypoxia are potential contributors to both the causes and the consequences of OSA.
The purpose of this study was to compare forearm and cerebral blood flow responses to exposures to hypoxia and hypercapnia during wakefulness in patients with OSA and control (CON) subjects. Also, we evaluated the effects of elimination of sleep-disordered breathing on vascular responses to the two stimuli by repeating these measurements in a subset of patients after 6 and 12 weeks of treatment with nasal continuous positive airway pressure (CPAP). We hypothesized that, compared with CON subjects, patients with OSA would have impaired vasodilator responses to both stimuli in both vascular beds, and that these impairments would be ameliorated by CPAP treatment. Some of the results of these studies have been previously reported in the form of an abstract (16).
METHODS
Subjects
Participants were 20 males and females with OSA and 20 CON subjects (Table 1). Patients were recruited on the basis of nocturnal polysomnography (apnea–hypopnea index, ≥25 events per hour of sleep). Control subjects were recruited from the community and screened by nocturnal oximetry (Pulsox 3i; Minolta, Osaka, Japan). Exclusion criteria were as follows: age less than 18 or greater than 50 years, tobacco use, diabetes mellitus, pulmonary disease, cardiovascular disease other than hypertension, and use of α- and β-adrenergic blocking medications. Five patients with OSA took antihypertensive agents, which were not withheld before testing. Medication use remained stable throughout the study. All subjects provided written, informed consent. Procedures were approved by the University of Wisconsin Health Sciences (Madison, WI) Institutional Review Board.
TABLE 1.
SUBJECT CHARACTERISTICS
| Patients with OSA (n = 20) | CON Subjects (n = 20) | |
|---|---|---|
| Age | 36.1 ± 7.5 | 34.1 ± 5.9 |
| Sex, males:females | 18:2 | 17:3 |
| BMI, kg/m2 | 37.1 ± 7.2 | 31.0 ± 5.8 |
| AHI, events/h | 68.9 ± 29.9 | 0.5 ± 0.9 |
| Time < 90% SaO2, % | 18.1 ± 10.3 | 0.1 ± 0.2 |
| Nadir SaO2, % |
74.8 ± 7.6 |
90.9 ± 2.7 |
Definition of abbreviations: AHI = apnea–hypopnea index; BMI = body mass index; CON = control; OSA = obstructive sleep apnea; SaO2 = arterial oxygen saturation.
BMI was higher in patients with OSA versus CON subjects (P < 0.00 by t test). By design, there were large between-group differences in the measures of sleep-disordered breathing.
Measurements
Cerebral blood flow velocity (CFV) was measured by transcranial Doppler ultrasound (Neurovision 500M; Multigon, Yonkers, NY) as previously described (17). Forearm blood flow (FBF) was measured by venous occlusion plethysmography (18) (EC6; Hokanson, Bellevue, WA). We obtained heart rate (HR) from the electrocardiogram, arterial oxygen saturation (SaO2) by pulse oximetry (Biox 3740; Ohmeda, Madison, WI), and blood pressure by automated sphygmomanometry (Dinamap 1846SX/P; Critikon, Tampa, FL). Subjects breathed through a mouthpiece with the nose occluded. Airflow was measured with a heated pneumotachograph (#5719; Hans Rudolph, Kansas City, MO), tidal volume and breathing frequency were calculated, and end-tidal oxygen pressure (PetO2) and carbon dioxide pressure (PetCO2) were measured by expired air analysis (S-3A/I and CD-3A; Ametek, Pittsburgh, PA).
Protocol
Graded isocapnic hypoxia and hyperoxic hypercapnia were presented in random order during wakefulness. For the hypoxia trial, 5 minutes of stable room air breathing was followed by 5 minutes with SaO2 held constant at 90, 85, and 80%. End-tidal CO2 was maintained at the eupneic level. For the hypercapnia trial, 5 minutes of baseline breathing was followed by 5 minutes with PetCO2 held constant at +5 mm Hg and at +10 mm Hg above eupnea. The hypercapnia trial was conducted with a hyperoxic background (FiO2, 0.40).
CPAP Treatment
CPAP pressures were prescribed on the basis of titration studies. Compliance was assessed every 6 weeks, using time-at-pressure monitors built into the devices. In-home oximetry was performed at Weeks 4, 6, and 12 to verify the effectiveness of CPAP.
Data Analysis
All signals were analyzed with custom-written software. Beat-by-beat values for CFV were determined as the velocity–time integrals. Mean arterial pressure (MAP) was calculated as 1/3 pulse pressure + diastolic pressure. Cerebrovascular and forearm vascular conductance (CVC and FVC) were calculated as CFV × 100/MAP and FBF × 100/MAP, respectively.
Statistics
Subject characteristics and baseline values were compared by unpaired t tests. To assess vascular responsiveness to hypoxia and hypercapnia, our primary outcome measures, we computed the slopes of the relationships between cardiorespiratory variables and SaO2 and PetCO2. We used SaO2 as the denominator of hypoxia response slopes because it is more linearly related to CFV than is PetO2. Between-group comparisons of slopes were made by unpaired t tests. We did not correct for multiple comparisons because there were only two primary outcome measures (CVC and FVC), and because the comparisons were preplanned. To assess the effect of CPAP on vascular responsiveness, we used one-way repeated-measures analyses of variance followed by Fisher's protected least significant difference tests. P values less than 0.05 were considered significant. All analyses (19) were performed with SAS statistical software version 9.1 (SAS Institute Inc., Cary, NC). In the text and tables, reported values are means ± SD. In the figures, reported values are means ± SE. Additional details on methods are provided in the online supplement.
RESULTS
Comparison of Subject Characteristics and Baseline Cardiorespiratory Variables
Patients with OSA and CON subjects were of similar age; however, the body mass index (BMI) was higher in patients with OSA. The apnea–hypopnea index (AHI) and percentage of time at less than 90% SaO2 were higher and minimal SaO2 was lower in patients with OSA versus CON subjects (Table 1). Baseline cardiorespiratory variables are shown in Table 2. HR and FBF were higher in patients with OSA versus CON subjects, whereas other baseline values were comparable in the two groups.
TABLE 2.
BASELINE VALUES RECORDED DURING 5 MINUTES OF EUPNEIC BREATHING BEFORE THE START OF THE INITIAL INTERVENTION (HYPOXIA OR HYPERCAPNIA)
| Patients with OSA (n = 20) | CON Subjects (n = 20) | P Value | |
|---|---|---|---|
| MAP, mm Hg | 87 ± 9 | 86 ± 8 | 0.38 |
| HR, bpm | 76 ± 10* | 65 ± 7 | 0.0002 |
| CFV, cm/s | 55.9 ± 10.0 | 56.7 ± 15.7 | 0.42 |
| FBF, ml/100 ml/min | 6.7 ± 3.5* | 4.0 ± 1.4 | 0.0015 |
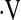 e, L/min e, L/min |
8.5 ± 2.7 | 7.8 ± 1.6 | 0.17 |
| PetCO2, mm Hg |
40.6 ± 3.9 |
39.7 ± 4.3 |
0.23 |
Definition of abbreviations: bpm = beats per minute; CFV = cerebral flow velocity; FBF = forearm blood flow; HR = heart rate; MAP = mean arterial pressure, PetCO2 = end-tidal carbon dioxide tension; 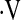 e = ventilation.
e = ventilation.
HR and FBF values were higher in patients with OSA versus CON subjects. Statistically significant values are in boldface type.
P < 0.05, patients with OSA versus CON subjects by t test.
Cardiorespiratory Responses to Isocapnic Hypoxia
The HR responses to graded hypoxia, as judged by the slopes of the stimulus–response relationship between SaO2 and HR, were blunted in OSA versus CON. In contrast, MAP and 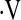 e responses were comparable in the two groups (Table 3). Group mean values for SaO2 and vascular conductance at each level of hypoxia are shown in Figure 1. Vascular responsiveness to hypoxia was reduced in patients with OSA versus CON subjects, both in the cerebral circulation and the forearm (Figures 1 and 2, and Table 3). Although the mean values for vascular responsiveness were significantly different in the two groups, there was substantial overlap in the responses of individual subjects (Figure 2). Mean values for all cardiorespiratory variables at each level of graded hypoxia are shown in the online supplement (see Table E1).
e responses were comparable in the two groups (Table 3). Group mean values for SaO2 and vascular conductance at each level of hypoxia are shown in Figure 1. Vascular responsiveness to hypoxia was reduced in patients with OSA versus CON subjects, both in the cerebral circulation and the forearm (Figures 1 and 2, and Table 3). Although the mean values for vascular responsiveness were significantly different in the two groups, there was substantial overlap in the responses of individual subjects (Figure 2). Mean values for all cardiorespiratory variables at each level of graded hypoxia are shown in the online supplement (see Table E1).
TABLE 3.
STIMULUS–RESPONSE SLOPES FOR CARDIORESPIRATORY VARIABLES DURING HYPOXIA AND HYPERCAPNIA IN PATIENTS WITH OBSTRUCTIVE SLEEP APNEA AND CONTROL SUBJECTS
| Patients with OSA (n = 20) | CON Subjects (n = 20) | P Value | |
|---|---|---|---|
| Hypoxia response slopes | |||
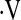 e, L/min/% SaO2 e, L/min/% SaO2
|
−0.51 ± 0.34 | −0.47 ± 0.32 | 0.34 |
| MAP, mm Hg/% SaO2 | −0.15 ± 0.53 | −0.14 ± 0.28 | 0.48 |
| HR, bpm/% SaO2 | −0.67 ± 0.30 | −0.94 ± 0.44 | 0.01 |
| CVC, units/% SaO2 | −0.65 ± 0.44 | −1.02 ± 0.72 | 0.03 |
| FVC, units/% SaO2 | −0.05 ± 0.09 | −0.10 ± 0.09 | 0.04 |
| Hypercapnia response slopes | |||
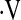 e, L/min/mm Hg e, L/min/mm Hg |
2.21 ± 1.29 | 2.02 ± 1.48 | 0.34 |
| MAP, mm Hg/mm Hg | 0.72 ± 1.00 | 0.59 ± 0.55 | 0.31 |
| HR, bpm/mm Hg | 0.80 ± 0.68 | 0.80 ± 0.49 | 0.50 |
| CVC, units/mm Hg | 2.01 ± 0.88 | 2.57 ± 0.89 | 0.03 |
| FVC, units/mm Hg |
−0.02 ± 0.18 |
0.02 ± 0.12 |
0.19 |
Definition of abbreviations: bpm = beats per minute; CVC = cerebrovascular conductance; FVC = forearm vascular conductance; HR = heart rate; MAP = mean arterial pressure; SaO2 = arterial oxygen saturation; 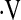 e = ventilation.
e = ventilation.
HR, CVC, and FVC responses to hypoxia and CVC responses to hypercapnia were blunted in patients with OSA versus CON subjects. Statistically significant values are in boldface type.
Figure 1.

Group mean values ± SE and individual subject vascular responses to isocapnic hypoxia in the brain (top) and forearm (bottom) in patients with obstructive sleep apnea (OSA) and control subjects (CON). In OSA, hypoxic vasodilation in both vascular beds was attenuated (P = 0.03 and 0.04).
Figure 2.

Individual subject values for the slope representing the stimulus–response relationship between arterial oxygen saturation (SaO2) and vascular conductance in the brain (left) and forearm (right). Note that although the means were significantly different, there was considerable overlap between the patient and control groups. *P < 0.05, obstructive sleep apnea (OSA) versus control subjects (CON).
Moderate to good associations were observed between the slopes of the SaO2:CVC relationship and AHI and percentage of sleep time spent with SaO2 less than 90% (Table 4). In contrast, the correlations between cerebrovascular responsiveness to hypoxia and age and BMI were only fair and were not statistically significant. Likewise, the correlations between FVC and AHI, percentage of time spent with SaO2 less than 90%, age, and BMI were poor to fair and were not statistically significant.
TABLE 4.
CORRELATION COEFFICIENTS AND CORRESPONDING P VALUES FOR ASSOCIATIONS BETWEEN VASCULAR RESPONSE SLOPES AND PATIENT CHARACTERISTICS
| AHI | Time < 90% SaO2 | BMI | Age | |
|---|---|---|---|---|
| SaO2:CVC slope | 0.70 | 0.55 | 0.32 | 0.35 |
| P Values | 0.005 | 0.04 | 0.26 | 0.22 |
| SaO2:FVC slope | 0.38 | 0.27 | 0.07 | 0.08 |
| P Values | 0.18 | 0.35 | 0.81 | 0.79 |
| PetCO2:CVC slope | −0.32 | −0.28 | −0.43 | 0.64 |
| P Values | 0.26 | 0.33 | 0.12 | 0.01 |
| PetCO2:FVC slope | 0.13 | −0.16 | −0.20 | 0.35 |
| P Values |
0.66 |
0.58 |
0.49 |
0.22 |
Definition of abbreviations: AHI = apnea–hypopnea index; BMI = body mass index; bpm = beats per minute; CON = control; CVC = cerebrovascular conductance; FVC = forearm vascular conductance; HR = heart rate; MAP = mean arterial pressure; OSA = obstructive sleep apnea; SaO2 = arterial oxygen saturation; 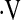 e = ventilation.
e = ventilation.
CVC responses to hypoxia were correlated with measures of OSA severity, whereas CVC responses to hypercapnia were correlated with age. Statistically significant values are in boldface type.
Cardiorespiratory Responses to Graded Hyperoxic Hypercapnia
The increases in HR, MAP, and 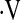 e elicited by graded hypercapnia were comparable in OSA and CON (Table 3). Group mean values for PetCO2 and vascular conductance at each level of hypercapnia are shown in Figure 3. Vascular responsiveness to hypercapnia was reduced in OSA versus CON in the cerebral circulation, whereas there was no between-group difference in forearm vascular responsiveness to hypercapnia (Figures 3 and 4, and Table 3). As in our observations of hypoxic vascular reactivity, there was substantial overlap in the responses of individual subjects in the two groups (Figure 4). Mean values for all cardiorespiratory variables at each level of graded hypercapnia are shown in the online supplement (see Table E2).
e elicited by graded hypercapnia were comparable in OSA and CON (Table 3). Group mean values for PetCO2 and vascular conductance at each level of hypercapnia are shown in Figure 3. Vascular responsiveness to hypercapnia was reduced in OSA versus CON in the cerebral circulation, whereas there was no between-group difference in forearm vascular responsiveness to hypercapnia (Figures 3 and 4, and Table 3). As in our observations of hypoxic vascular reactivity, there was substantial overlap in the responses of individual subjects in the two groups (Figure 4). Mean values for all cardiorespiratory variables at each level of graded hypercapnia are shown in the online supplement (see Table E2).
Figure 3.

Group mean values ± SE and individual subject vascular responses to hyperoxic hypercapnia in the brain (top) and forearm (bottom) in patients with obstructive sleep apnea (OSA) and control subjects (CON). Hypercapnic vasodilation in the cerebral circulation was attenuated in OSA versus CON (P = 0.03), whereas in the forearm, the slopes were comparable (hypercapnia did not cause vasodilation in either group).
Figure 4.

Individual subject values for the slope representing the stimulus–response relationship between PetCO2 and vascular conductance in the brain (left) and forearm (right). Note that although the mean values for cerebrovascular responsiveness were significantly different, there was considerable overlap between the patient and control groups. In the forearm, the slopes were comparable in the two groups. *P < 0.05, OSA versus CON.
The slope of the CVC:CO2 relationship was not correlated with measures of OSA severity; however, cerebrovascular responsiveness to CO2 did show a moderate to good correlation with age (Table 4). Forearm vascular responsiveness to hypercapnia was not correlated with measures of OSA severity, BMI, or age.
Effects of CPAP Treatment on Cardiorespiratory Responses to Hypoxia and Hypercapnia
The mean CPAP usage over the 12-week observation period was 5.1 ± 1.4 hours per night. Overnight oximetry performed at 4, 8, and 12 weeks of treatment revealed minimal SaO2 values of 90 ± 1, 91 ± 3, and 92 ± 2, respectively. At the same time points, the number of desaturation events were 0.8 ± 0.5, 0.7 ± 0.8, and 0.5 ± 0.6 per hour of sleep. Over the 12 weeks of CPAP treatment, there was no change in body weight (111.9 ± 26.4, 113.1 ± 26.0, and 112.2 ± 26.0 kg; P > 0.05) or in baseline values for any of the variables of interest (Table 5).
TABLE 5.
BASELINE VALUES RECORDED DURING 5 MINUTES OF EUPNEIC BREATHING BEFORE THE INITIAL INTERVENTION (HYPOXIA OR HYPERCAPNIA) AT THE PRE–CONTINUOUS POSITIVE AIRWAY PRESSURE VISIT AND 6 AND 12 WEEKS AFTER INITIATION OF TREATMENT IN PATIENTS WITH OBSTRUCTIVE SLEEP APNEA
| Pre-CPAP | 6 wk of CPAP | 12 wk of CPAP | P Value (ANOVA) | |
|---|---|---|---|---|
| MAP, mm Hg | 89 ± 9 | 87 ± 8 | 87 ± 11 | 0.78 |
| HR, bpm | 76 ± 10 | 80 ± 14 | 77 ± 13 | 0.34 |
| CFV, cm/s | 54.6 ± 10.5 | 58.7 ± 11.0 | 60.9 ± 12.0 | 0.11 |
| FBF, ml/100 ml/min | 6.6 ± 3.9 | 6.7 ± 3.4 | 6.9 ± 2.8 | 0.87 |
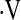 e, L/min e, L/min |
8.4 ± 2.4 | 8.9 ± 2.4 | 9.2 ± 1.7 | 0.20 |
| PetCO2, mm Hg |
40.9 ± 3.9 |
41.3 ± 3.6 |
42.0 ± 3.0 |
0.12 |
Definition of abbreviations: ANOVA = analysis of variance; bpm = beats per minute; CFV = cerebral flow velocity; CPAP = continuous positive airway pressure; FBF = forearm blood flow; HR = heart rate; MAP = mean arterial pressure, PetCO2 = end-tidal carbon dioxide tension; 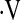 e = ventilation.
e = ventilation.
Patients with obstructive sleep apnea: n = 14.
There were no significant differences in any of these baseline variables over time.
The MAP response to hypoxia was significantly reduced with CPAP treatment, whereas the 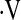 e and HR responses to hypoxia and the MAP,
e and HR responses to hypoxia and the MAP, 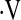 e, and HR responses to hypercapnia remained stable over time (Table 6). There was a substantial amount of variability among subjects in terms of CPAP-related changes in vascular responsiveness. The amount of improvement in vascular responsiveness in the two vascular beds was not correlated with hours of CPAP usage, at least over the rather constricted range we observed (4–7 h per night). In contrast, the amount of change in cerebrovascular responses to hypoxia and hypercapnia was positively correlated with the amount of impairment present before CPAP treatment (r = −0.65 and r = −0.66, respectively; both P = 0.01). The same was true for improvement in forearm vascular responses to hypoxia and hypercapnia (r = −0.70 and r = −0.72, respectively; P = 0.005 and 0.003). When slopes of only those subjects with clearly abnormal baseline responses (slopes less than the 95% confidence intervals around the control group means; n = 12) were compared over time, statistically significant CPAP-associated increases in hypoxic vasodilation were observed in the cerebral circulation and the forearm (Figure 5 and Table 6). Increases in hypercapnic vasodilation in the cerebral circulation approached statistical significance (P = 0.06), whereas there was no change over time in forearm vascular responses to hypercapnia (Figure 5 and Table 6). In the two subjects eliminated from these analyses, the severity of OSA was relatively mild in terms of frequency of events (25 and 41 events/h) and percentage of time at less than 90% SaO2 (0.1 and 6.4%).
e, and HR responses to hypercapnia remained stable over time (Table 6). There was a substantial amount of variability among subjects in terms of CPAP-related changes in vascular responsiveness. The amount of improvement in vascular responsiveness in the two vascular beds was not correlated with hours of CPAP usage, at least over the rather constricted range we observed (4–7 h per night). In contrast, the amount of change in cerebrovascular responses to hypoxia and hypercapnia was positively correlated with the amount of impairment present before CPAP treatment (r = −0.65 and r = −0.66, respectively; both P = 0.01). The same was true for improvement in forearm vascular responses to hypoxia and hypercapnia (r = −0.70 and r = −0.72, respectively; P = 0.005 and 0.003). When slopes of only those subjects with clearly abnormal baseline responses (slopes less than the 95% confidence intervals around the control group means; n = 12) were compared over time, statistically significant CPAP-associated increases in hypoxic vasodilation were observed in the cerebral circulation and the forearm (Figure 5 and Table 6). Increases in hypercapnic vasodilation in the cerebral circulation approached statistical significance (P = 0.06), whereas there was no change over time in forearm vascular responses to hypercapnia (Figure 5 and Table 6). In the two subjects eliminated from these analyses, the severity of OSA was relatively mild in terms of frequency of events (25 and 41 events/h) and percentage of time at less than 90% SaO2 (0.1 and 6.4%).
TABLE 6.
STIMULUS–RESPONSE SLOPES FOR CARDIORESPIRATORY VARIABLES DURING HYPOXIA AND HYPERCAPNIA BEFORE CONTINUOUS POSITIVE AIRWAY PRESSURE TREATMENT AND AFTER 6 AND 12 WEEKS OF TREATMENT IN PATIENTS WITH OBSTRUCTIVE SLEEP APNEA
| Pre-CPAP | 6 wk | 12 wk | P Values (ANOVA) | |
|---|---|---|---|---|
| Hypoxia response slopes | ||||
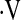 e, L/min/% SaO2 e, L/min/% SaO2
|
−0.50 ± 0.34 | −0.43 ± 0.28 | −0.43 ± 0.26 | 0.68 |
| MAP, mm Hg/% SaO2 | −0.21 ± 0.38 | 0.12 ± 0.37* | −0.06 ± 0.32 | 0.04 |
| HR, bpm/% SaO2 | −0.70 ± 0.29 | −0.55 ± 0.37 | −0.75 ± 0.32 | 0.18 |
| CVC, units/% SaO2 | −0.46 ± 0.29 | −0.88 ± 0.47* | −0.83 ± 0.32* | 0.01 |
| FVC, units/% SaO2 | −0.02 ± 0.08 | −0.12 ± 0.14* | −0.19 ± 0.15* | 0.003 |
| Hypercapnia response slopes | ||||
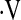 e, L/min/mm Hg e, L/min/mm Hg |
2.28 ± 1.32 | 2.23 ± 1.14 | 2.40 ± 1.22 | 0.73 |
| MAP, mm Hg/mm Hg | 0.52 ± 0.68 | 0.52 ± 0.72 | 0.58 ± 0.76 | 0.95 |
| HR, bpm/% mm Hg | 0.81 ± 0.54 | 0.62 ± 0.39 | 0.74 ± 0.45 | 0.44 |
| CVC, units/mm Hg | 1.76 ± 0.64 | 2.20 ± 0.78 | 2.24 ± 0.78 | 0.06 |
| FVC, units/mm Hg |
−0.05 ± 0.12 |
−0.05 ± 0.27 |
0.12 ± 0.30 |
0.21 |
Definition of abbreviations: See Table 3.
The MAP responses to hypoxia decreased over time, whereas the CVC and FVC responses increased. No change was observed in any of the responses to hypercapnia. Statistically significant values are in boldface type.
P < 0.05 versus baseline by Fisher's protected least significant difference post hoc test. There were no significant difference between 6 and 12 weeks of treatment.
Figure 5.

Effects of continuous positive airway pressure (CPAP) on hypoxic and hypercapnic vasodilation in the brain and forearm (means ± SE; n = 12). CPAP elicited improvements in hypoxic vasodilation in both vascular beds that was evident after 6 weeks of treatment. No statistically significant changes in hypercapnic vasodilation were observed; however, there was a trend toward improvement in hypercapnic cerebral vasodilation (P = 0.06). *P < 0.05 versus pre-CPAP. Note that the negative slopes for hypoxic vasodilation have been changed to positive slopes to aid in visual comparison.
DISCUSSION
We found that hypoxic vasodilation in the brain and the forearm was attenuated versus age-matched control subjects in most, but not all, of the patients with OSA studied. These impairments in vascular regulation, which correlated with measures of OSA severity, were ameliorated by CPAP treatment. Hypercapnic vasodilation in the cerebral circulation was also blunted in most of the patients with OSA; however, it was not correlated with measures of OSA severity, and CPAP-related improvement in this response was not statistically significant. We interpret these findings to mean that moderate to severe OSA can impair basic vasoregulatory mechanisms in multiple vascular beds. Impairments in vasodilatory responses to hypoxia are reversible when sleep-disordered breathing is eliminated with CPAP, whereas impairments in hypercapnic vasodilation seem to be somewhat less amenable to treatment.
Methodological Considerations
Our conclusions regarding cerebrovascular responses are predicated on the assumption that Doppler measurements of flow velocity are reflective of volume flow, an assumption that is satisfied only when the cross-sectional area of the artery remains constant. We did not measure diameter; however, previous investigators have shown that middle cerebral artery diameter varies by no more than 4% during changes in arterial pressure, CO2 tension (20), or gravitational stress (21). In addition, velocity and flow through the middle cerebral artery are highly correlated (22).
In our subjects, average BMI was higher in OSA versus CON; therefore, it is possible that obesity confounded our between-group comparisons of cardiorespiratory responses to hypoxia and hypercapnia (23). Nevertheless, we believe it unlikely that obesity was a primary determinant of blunted vascular responsiveness in OSA, because CPAP treatment improved vascular responses to hypoxia even though body weight did not change.
We considered the possibility that hypoxic forearm vasodilation may have been diminished in OSA secondary to higher baseline blood flows (i.e., a “ceiling” effect). We believe this is unlikely because forearm vasodilation during hypoxia was greater after versus before CPAP treatment even though baseline values remained the same. The reason for higher FBF in OSA versus CON is unknown; however, it may be caused by chronic exposure to hypoxia, as has been reported in patients with chronic obstructive pulmonary disease (24, 25). However, this explanation is unlikely to account for all of the between-group difference in FBF, because the difference persisted after intermittent hypoxia was eliminated by CPAP treatment. We speculate that the higher FBF values we observed in patients with OSA are related to obesity and the increased contribution of adipose tissue flow to total forearm flow (26). High baseline FBF in our subjects is consistent with several previous reports of increased brachial artery diameter in patients with OSA versus CON subjects (7, 9) and it highlights the need for caution in interpreting reports of impaired flow-mediated dilation that are based on percentage of baseline diameter.
Comparison with Previous Findings
Our observations of OSA-related decrements in hypoxic vasodilation in the cerebral and forearm circulations confirm findings of previous investigators (11, 12). The deficit in cerebral vasodilation we observed (−35% relative to CON subjects) is similar to that reported by Foster and colleagues (12). Likewise, the CPAP effects are similar, even though the observation period was twice as long in our study. Our data indicate that CPAP-associated improvement in hypoxic vasodilation occurs within 6 weeks of the initiation of treatment; with little, if any, additional improvement after 12 weeks.
The deficit in forearm hypoxic vasodilation we observed (−50% relative to CON subjects) is smaller than that reported by Remsburg and colleagues (11), who observed a complete lack of forearm vasodilation during hypoxic exposure in OSA. We believe the most likely reason for this discrepancy is a difference in the duration of the experimental intervention. We used 5-minute steady state exposures to graded hypoxia, whereas the previous investigators used a 3- to 4-minute ramp rebreathing protocol (11). Our findings are relatively consistent with those of previous investigators who used flow-mediated dilation in the forearm to assess vascular function (6). Despite the fact that Ip and colleagues (6) measured responses to vascular occlusion in conduit vessels and we measured responses to hypoxia in resistance vessels, the OSA-related reductions in reactivity are similar (−37 and −50% relative to CON subjects, respectively). In contrast, the previously reported increases in flow-mediated dilation after CPAP treatment (6) are much smaller than the increases in hypoxic vasodilation we observed (+47 vs. +89%), possibly because of a shorter treatment period and/or less severe impairment at the onset of treatment.
We found that hypercapnic cerebral vasodilation was reduced by 22% in patients with OSA versus CON subjects. This finding is consistent with previous observations of blunted cerebral vasodilation during rebreathing (13) and also during breath-holding (27), responses that are caused mainly by hypercapnia (17). Because hypercapnic vasodilation is an endothelium-dependent process, we speculate that altered CO2 responsiveness is one manifestation of OSA-induced endothelial dysfunction (6, 8–10). In contrast to our finding of blunted cerebrovascular response to CO2 in OSA versus CON, previous investigators found no between-group difference in this variable (15). Features of our study that may account for this discrepancy include increased sample size, multiple levels of the stimulus, decreased mean age and AHI in our subjects, and inclusion of female subjects. Interestingly, Foster and colleagues, who found that hypercapnic vasodilation in the cerebral circulation was not different in patients with OSA versus CON subjects, observed a CPAP-related enhancement in cerebral responsiveness to hypercapnia (15). In our study, there was some indication that CPAP therapy improved hypercapnic vasodilation in the cerebral circulation; however, this change failed to reach statistical significance (P = 0.06).
We are not aware that attenuated HR responses to hypoxia, as we observed in our patients with OSA, have been reported previously. This difference was not secondary to accentuated responses in the CON subjects, because their mean values are nearly identical to those reported previously (28). The cause of the OSA-related attenuation in the HR response to hypoxia, which persisted during 12 weeks of CPAP treatment, is unknown; however, it may reflect augmented carotid chemoreflex control of parasympathetic outflow to the sinus node (29).
In contrast to the OSA-related decrements in heart rate responses to hypoxia and vascular responses to hypoxia and hypercapnia we observed, there were no between-group differences in the ventilatory responses to the two stimuli. There is considerable disagreement among previous reports of the effects of OSA on hypoxic and hypercapnic ventilatory responses. Some investigators report augmented hypoxic ventilatory responses in patients with OSA versus CON subjects (30, 31), whereas others report similar responses in the two groups (32). Hypercapnic ventilatory responses are reported to be increased (32) or the same (15, 30) in patients with OSA versus CON subjects. We speculate that much of the variability in the present and previous findings is attributable to performance of these studies during wakefulness, when nonchemoreceptor, behavioral inputs have a substantial influence on respiratory output.
Mechanisms Governing Vascular Responses to Hypoxia and Hypercapnia
Vasomotor tone is determined by the net effect of neural and hormonal influences, mechanical effects of pressure and flow, and locally produced chemicals. Hypoxic exposure elicits increases in sympathetic outflow and vasoconstrictor hormones that are opposed by locally mediated vasodilation. In the cerebral and skeletal muscle circulations, hypoxic vasodilation is caused primarily by prostacyclin released from the endothelium (33, 34); however, it is evident that multiple, partially redundant mediators (including nitric oxide and adenosine) are operative, depending on the severity of hypoxia (34). Hypercapnia also elicits increases in sympathetic outflow that are opposed by local vasodilation. In the cerebral circulation, sympathetic vasoconstrictor effects are limited by the sparsity of α-adrenergic receptors and the blood–brain barrier limits access to circulating hormones (35). Hypercapnic vasodilation in the brain is a complex, endothelium-dependent process: nitric oxide, prostacyclin, and cytochrome P-450 metabolites have all been implicated (36–38). In the peripheral circulation, local hypercapnic vasodilation appears to be overwhelmed by increases in sympathetic activity, as evidenced by the present and previous findings (39, 40).
Beneficial Effects of CPAP on Cardiovascular Regulation
Several potential mechanisms for the beneficial effects of CPAP on hypoxic vasodilation can be postulated. Sympathetic vasoconstrictor outflow (4) and circulating angiotensin II (41) are reduced after elimination of sleep-disordered breathing with CPAP. Both effects may be caused by reversal of the previously described OSA-related augmentation in carotid chemoreflex control of sympathetic outflow (30). In addition, endothelium-dependent vasodilation is improved after CPAP treatment (10). Previous reports suggest that CPAP increases circulating metabolites of nitric oxide (42) and decreases production of superoxide ion by neutrophils (43). If similar CPAP-induced changes occur in the endothelium, they would be expected to improve local regulation of vascular resistance by increasing the bioavailability of nitric oxide and facilitating prostacyclin release (44). We speculate that enhanced endothelial function and reduced sympathetic outflow both contribute to CPAP-related improvements in forearm vascular regulation, whereas enhanced endothelial function is likely to be more important in the brain, where the influence of the sympathetic nervous system is relatively weak. Our finding that 6 weeks of CPAP treatment caused a small, but statistically significant, decrease in the MAP response to hypoxia suggests that reduction in sympathetic tone and/or augmentation of local hypoxic vasodilation in resistance arteries has a potentially important effect on systemic hemodynamics.
Clinical Implications
We found that impaired vascular responses to alterations in arterial O2 and CO2 were present in most, but not all, patients with OSA. These impairments, which seem to be dependent on OSA severity, may be important pathogenetic links between OSA and vascular disease. Moreover, impaired blood flow regulation could limit the delivery of oxygen and energy substrates during acute episodes of OSA and also under physiologic conditions such as exercise. Diminished cerebrovascular responsiveness to CO2 may exacerbate breathing instability during sleep. Our study demonstrates that OSA-related alterations in vascular responsiveness to hypoxia, which seem to be dependent on OSA severity, can be reversed relatively rapidly when nightly exposure to intermittent hypoxia is eliminated with CPAP. These beneficial effects can be observed even when CPAP is used for as little as 4 hours per night.
Supplementary Material
Acknowledgments
The authors thank Glen Leverson, Ph.D., for assistance with statistical analysis, Ruth Benca, M.D., Ph.D., and the sleep technicians at Wisconsin Sleep for help with subject recruitment; Beth Dunlap, R.N., for help with study coordination, and Mr. David Pegelow and Mr. Dominic Puleo for technical assistance.
Supported by NHLBI grant R01 HL074072. K.J.R. was supported by NHLBI training grant T32 HL07654 and J.M.D. was supported by UW-Madison Institutional Clinical and Translational Science award K12 RR025012. This research was also supported by the Office of Research and Development, Clinical Science R&D Service, Department of Veterans Affairs.
This article has an online supplement, which is accessible from the issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.200903-0393OC on September 10, 2009
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Golbin JM, Somers VK, Caples SM. Obstructive sleep apnea, cardiovascular disease, and pulmonary hypertension. Proc Am Thorac Soc 2008;5:200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khayat R, Patt B, Hayes D Jr. Obstructive sleep apnea: the new cardiovascular disease. I. Obstructive sleep apnea and the pathogenesis of vascular disease. Heart Fail Rev 2008;14:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest 1993;103:1763–1768. [DOI] [PubMed] [Google Scholar]
- 4.Waradekar NV, Sinoway LI, Zwillich CW, Leuenberger UA. Influence of treatment on muscle sympathetic nerve activity in sleep apnea. Am J Respir Crit Care Med 1996;153:1333–1338. [DOI] [PubMed] [Google Scholar]
- 5.Imadojemu VA, Gleeson K, Quraishi SA, Kunselman AR, Sinoway LI, Leuenberger UA. Impaired vasodilator responses in obstructive sleep apnea are improved with continuous positive airway pressure therapy. Am J Respir Crit Care Med 2002;165:950–953. [DOI] [PubMed] [Google Scholar]
- 6.Ip MS, Tse HF, Lam B, Tsang KW, Lam WK. Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med 2004;169:348–353. [DOI] [PubMed] [Google Scholar]
- 7.Nieto FJ, Herrington DM, Redline S, Benjamin EJ, Robbins JA. Sleep apnea and markers of vascular endothelial function in a large community sample of older adults. Am J Respir Crit Care Med 2004;169:354–360. [DOI] [PubMed] [Google Scholar]
- 8.Carlson JT, Rangemark C, Hedner JA. Attenuated endothelium-dependent vascular relaxation in patients with sleep apnoea. J Hypertens 1996;14:577–584. [DOI] [PubMed] [Google Scholar]
- 9.Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 2000;102:2607–2610. [DOI] [PubMed] [Google Scholar]
- 10.Lattimore JL, Wilcox I, Skilton M, Langenfeld M, Celermajer DS. Treatment of obstructive sleep apnoea leads to improved microvascular endothelial function in the systemic circulation. Thorax 2006;61:491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remsburg S, Launois SH, Weiss JW. Patients with obstructive sleep apnea have an abnormal peripheral vascular response to hypoxia. J Appl Physiol 1999;87:1148–1153. [DOI] [PubMed] [Google Scholar]
- 12.Foster GE, Hanly PJ, Ostrowski M, Poulin MJ. Effects of CPAP on cerebral vascular response to hypoxia in obstructive sleep apnea patients. Am J Respir Crit Care Med 2007;175:720–725. [DOI] [PubMed] [Google Scholar]
- 13.Loeppky JA, Miranda FG, Eldridge MW. Abnormal cerebrovascular responses to CO2 in sleep apnea patients. Sleep 1984;7:97–109. [DOI] [PubMed] [Google Scholar]
- 14.Urbano F, Roux F, Schindler J, Mohsenin V. Impaired cerebral autoregulatoin in obstructive sleep apnea. J Appl Physiol 2008;105:1852–1857. [DOI] [PubMed] [Google Scholar]
- 15.Foster GE, Hanly PJ, Ostrowski M, Poulin MJ. Ventilatory and cerebrovascular responses to hypercapnia in patients with obstructive sleep apnoea: effect of CPAP therapy. Respir Physiol Neurobiol 2009;165:73–81. [DOI] [PubMed] [Google Scholar]
- 16.Dopp J, Reichmuth K, Puleo D, Hayes D, Skatrud J, Morgan B. Vascular responses to hypercapnia in patients with obstructive sleep apnea [abstract]. Am J Respir Crit Care Med 2006;173:A517. [Google Scholar]
- 17.Przybylowski T, Bangash MF, Reichmuth K, Morgan BJ, Skatrud JB, Dempsey JA. Mechanisms of the cerebrovascular response to apnoea in humans. J Physiol 2003;548:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenfield AD, Whitney RJ, Mowbray JF. Methods for the investigation of peripheral blood flow. Br Med Bull 1963;19:101–109. [DOI] [PubMed] [Google Scholar]
- 19.Snedecor GW, Cochran WG. Statistical methods, 8th ed. Ames, IA: Iowa State University Press; 1989.
- 20.Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery 1993;32:737–741. [PubMed] [Google Scholar]
- 21.Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke 2000;31:1672–1678. [DOI] [PubMed] [Google Scholar]
- 22.Kirkham FJ, Padayachee TS, Parsons S, Seargeant LS, House FR, Gosling RG. Transcranial measurement of blood velocities in the basal cerebral arteries using pulsed Doppler ultrasound: velocity as an index of flow. Ultrasound Med Biol 1986;12:15–21. [DOI] [PubMed] [Google Scholar]
- 23.Narkiewicz K, Kato M, Pesek CA, Somers VK. Human obesity is characterized by a selective potentiation of central chemoreflex sensitivity. Hypertension 1999;33:1153–1158. [DOI] [PubMed] [Google Scholar]
- 24.Heistad DD, Wheeler RC, Aoki VS. Reflex cardiovascular responses after 36 hr of hypoxia. Am J Physiol 1971;220:1673–1676. [DOI] [PubMed] [Google Scholar]
- 25.Heistad DD, Abboud FM, Mark AL, Schmid PG. Impaired reflex vasoconstriction in chronically hypoxemic patients. J Clin Invest 1972;51:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaak EE, van Baak MA, Kemerink GL, Pakbiers MTW, Heidendal GAK, Saris WHM. Total forearm blood flow as an indicator of skeletal muscle blood flow: effect of subcutaneous adipose tissue blood flow. Clin Sci 1994;87:559–566. [DOI] [PubMed] [Google Scholar]
- 27.Diomedi M, Placidi F, Cupini LM, Bernardi G, Silvestrini M. Cerebral hemodynamic changes in sleep apnea syndrome and effect of continuous positive airway pressure treatment. Neurology 1998;51:1051–1056. [DOI] [PubMed] [Google Scholar]
- 28.Slutsky AS, Rebuck AS. Heart rate response to isocapnic hypoxia in conscious man. Am J Physiol 1978;234:H129–H132. [DOI] [PubMed] [Google Scholar]
- 29.Gandevia SC, McCloskey DI, Potter EK. Inhibition of baroreceptor and chemoreceptor reflexes on heart rate by afferents from the lungs. J Physiol 1978;276:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narkiewicz K, van de Borne PJH, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation 1999;99:1183–1189. [DOI] [PubMed] [Google Scholar]
- 31.Spicuzza L, Bernardi L, Balsamo R, Ciancio N, Polosa R, Di Maria G. Effect of treatment with nasal continuous positive airway pressure on ventilatory response to hypoxia and hypercapnia in patients with sleep apnea syndrome. Chest 2006;130:774–779. [DOI] [PubMed] [Google Scholar]
- 32.Wang D, Grunstein RR, Teichtahl H. Association between ventilatory response to hypercapnia and obstructive sleep apnea–hypopnea index in asymptomatic subjects. Sleep Breath 2007;11:103–108. [DOI] [PubMed] [Google Scholar]
- 33.Fredricks KT, Liu Y, Rusch NJ, Lombard JH. Role of endothelium and arterial K+ channels in mediating hypoxic dilation of middle cerebral arteries. Am J Physiol 1994;267:H580–H586. [DOI] [PubMed] [Google Scholar]
- 34.Frisbee JC, Maier KG, Falck JR, Roman RJ, Lombard JH. Integration of hypoxic dilation signaling pathways for skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol 2002;283:R309–R319. [DOI] [PubMed] [Google Scholar]
- 35.Bevan RD, Dodge J, Nichols P, Penar PL, Walters CL, Wellman T, Bevan JA. Weakness of sympathetic nerual control of human pial compared with superficial temporal arteries reflects low innervation density and poor sympathetic responsiveness. Stroke 1998;29:212–221. [DOI] [PubMed] [Google Scholar]
- 36.Iadecola C, Zhang F. Nitric oxide–dependent and –independent components of cerebrovasodilation elicited by hypercapnia. Am J Physiol 1994;266:R546–R552. [DOI] [PubMed] [Google Scholar]
- 37.Pelligrino DA, Santizo RA, Wang Q. Miconazole represses CO2-induced pial arteriolar dilation only under selected circumstances. Am J Physiol 1999;277:H1484–H1490. [DOI] [PubMed] [Google Scholar]
- 38.Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res 2001;88:600–608. [DOI] [PubMed] [Google Scholar]
- 39.Ainslie PN, Ashmead JC, Ide K, Morgan BJ, Poulin MJ. Differential responses to CO2 and sympathetic stimulation in the cerebral and femoral circulations in humans. J Physiol 2005;566:613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan BJ, Crabtree DC, Palta M, Skatrud JB. Combined hypoxia and hypercapnia evokes long-lasting sympathetic activation in humans. J Appl Physiol 1995;79:205–213. [DOI] [PubMed] [Google Scholar]
- 41.Møller DS, Lind P, Strunge B, Pedersen EB. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am J Hypertens 2003;16:274–280. [DOI] [PubMed] [Google Scholar]
- 42.Alonso-Fernández A, García-Río F, Arias MA, Hernanz A, de la Peña M, Piérola J, Barceló A, López-Collazo E, Agustí A. Effects of CPAP upon oxidative stress and nitrate deficiency in sleep apnoea: a randomized trial. Thorax 2008;64:581–586. [DOI] [PubMed] [Google Scholar]
- 43.Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski J, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea: impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 2000;162:566–570. [DOI] [PubMed] [Google Scholar]
- 44.Wollin MS. Interactions of oxidants with vascular signaling systems. Arterioscler Thromb Vasc Biol 2000;20:1430–1442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.