

Abstract
In Alzheimer's disease, calcium permeability through cellular membranes appears to underlie neuronal cell death. It is increasingly accepted that calcium permeability involves toxic ion channels. We modeled Alzheimer's disease ion channels of different sizes (12-mer to 36-mer) in the lipid bilayer using molecular dynamics simulations. Our Aβ channels consist of the solid-state NMR-based U-shaped β-strand-turn-β-strand motif. In the simulations we obtain ion-permeable channels whose subunit morphologies and shapes are consistent with electron microscopy/atomic force microscopy. In agreement with imaged channels, the simulations indicate that β-sheet channels break into loosely associated mobile β-sheet subunits. The preferred channel sizes (16- to 24-mer) are compatible with electron microscopy/atomic force microscopy-derived dimensions. Mobile subunits were also observed for β-sheet channels formed by cytolytic PG-1 β-hairpins. The emerging picture from our large-scale simulations is that toxic ion channels formed by β-sheets spontaneously break into loosely interacting dynamic units that associate and dissociate leading to toxic ionic flux. This sharply contrasts intact conventional gated ion channels that consist of tightly interacting α-helices that robustly prevent ion leakage, rather than hydrogen-bonded β-strands. The simulations suggest why conventional gated channels evolved to consist of interacting α-helices rather than hydrogen-bonded β-strands that tend to break in fluidic bilayers. Nature designs folded channels but not misfolded toxic channels.
Introduction
Alzheimer's disease (AD), like many other protein conformational diseases, including Huntington's disease, Parkinson's disease, type II diabetes, and cystic fibrosis result from protein misfolding that alters their three-dimensional conformations from native (generally soluble) to nonnative (insoluble) folded structures (1). In AD, β-amyloid (Aβ) peptides form β-sheet-rich ordered aggregates (2) with Aβ oligomers being the toxic species (3,4). Although it is generally accepted that toxicity relates to ion transport through the cellular membrane, the mechanism has been a debated issue (5). On the one hand, using a variety of methods, numerous studies have consistently observed that Aβ peptides assemble to form ion channels in the cell membrane inducing selective ion permeation (1,6–20). On the other hand, other studies did not observe any channels. These studies (21,22) observed nonselective ion leakage that they argued occurs through membrane thinning and lowered dielectric barrier induced by the interaction of the Aβ oligomers with the membrane. Recent investigations of the Aβ-induced ion flux by current recordings across the membranes seem to have resolved the controversy (23). These studies reexamined the previous postulate that Aβ-induced membrane thinning caused gradual increase in cellular ion flux (21,22). They showed that even in the absence of Aβ, trace amounts of the hexafluoro-isopropanol remaining in the solvent used to prepare Aβ oligomers led to membrane thinning causing a gradual increase in cellular current. They further showed that elimination of the hexafluoro-isopropanol solvent leads to the characteristic Aβ-induced stepwise ion flux across the planar lipid bilayer (1,6,13,18), suggesting that Aβ forms an ion channel.
Increasing evidence lends support to ion channel formation (24–26). Electron microscopy (EM) shows doughnut-like Aβ pores (27,28), and atomic force microscopy (AFM) presents heterogeneous ion channels that directly mediate ion conductance (1,16). The AFM-imaged Aβ channels in the 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) membrane have a central pore of ∼2.0 nm diameter and 8–12 nm in outer diameter (1). The channels are subunit assemblies, with shapes varying from rectangular with four subunits to hexagonal with six, and show cation-selective conductance. As AFM probes the topmost portion of the channel, the observed structures may refer either to conducting pores or pores collapsed at the bottom, thus not providing information about the functionality of the observed structure. Our previous molecular dynamics simulations (19,20) of nonamyloidogenic Aβ peptides (29) in the bilayer predicted ion-permeable channels formed by loosely attached mobile subunits with shapes, morphologies, and dimensions similar to the AFM-imaged channels (1,16). The simulations focused on the Aβ17–42 small protofibril (pentamer) coordinate set based on two-dimensional NMR in combination with solid state NMR (ssNMR), mutational data and EM (30). In addition, Aβ9–42 was also simulated, using the ssNMR-based coordinates (31). In the modeled channels the Aβ peptides retained the U-shaped β-strand-turn-β-strand motif observed in both coordinate sets. This motif was first predicted by modeling the Aβ16–35 (32). To date, similar U-shaped motifs were observed in the ssNMR structure of a β2-microglobulin fragment (33) and in the CA150 WW domain (34), suggesting that U-shaped motifs are a general feature of amyloid organization (35–37). These oligomers may also form channels in the membrane (38–40). This motif associates into stacked β-sheets, with the intermolecular β-strands H-bonded to each other.
Antimicrobial peptide protegrin-1 (PG-1), a β-hairpin peptide, also forms channels in the membrane. The channel is cytotoxic, leaking chloride ions. This unregulated leakage leads to target cells death. The PG-1 β-hairpins associate to form a β-sheet channel, also with loosely attached subunits (41), thus sharing a common morphological motif with other β-sheet channels (1,19,20).
This study aims to characterize toxic AD ion channels and to obtain the preferred channel sizes and apparent molecular mass. In particular, through this characterization and comparison with experiment, it further aims to obtain some of the hallmarks differentiating toxic amyloid channels from “normally” folded gated (e.g., Na+, K+, and Ca2+) ion channels that are optimized by evolution. Both misfolded toxic amyloid channels and the normally folded channels are gated; however, in the first the gating is unregulated and is the outcome of the membrane dynamics, sharply contrasting the second. Toxic amyloid channels possess features distinct from the physiological gated ion channels that robustly regulate the ion flux (42,43). Functional gated channels i), contain mostly α-helices; ii), fold into their native state; iii), are stable and long-lived membrane proteins with membrane supporting channel conformations; iv), have a fixed number of subunits that are specifically associated; and v), selectively transport ions that are essential to cell metabolism via orchestrated conformational changes. In contrast, nonphysiological toxic amyloid channels i), can contain mostly β-sheets; ii), are not present in a unique size; iii), can have a different number of subunits that are nonspecifically associated; iv), induce rapid ion leakage; v), are associated with many diseases and lead to cell death; and vi), consist of misfolded states with the channels varying in size, shape, and morphology in membranes (1,16). Our modeling captures the distinct features of amyloid channels. These features are further observed in the cytotoxic β-sheets channel of antimicrobial peptide PG-1.
Using our previous protocols (19,20) we comprehensively test a range of Aβ channel sizes to characterize these on the atomic scale. Without structural information from experiments, modeling the three-dimensional structures of amyloid ion channels is highly challenging. Our channel modeling starts with the experimentally available Aβ9–42 monomer conformation in the oligomeric state (34 residues; ∼3.6 kDa/monomer; adding Ile41 and Ala42 (31)). Channels with increasing number of Aβ9–42 monomers (12-, 16-, 20-, 24-, and 36-mer) were constructed and explicitly simulated in the DOPC bilayer (see Fig. S1 in the Supporting Material). Further, the 24-mer channel was also simulated in the anionic bilayer containing palmitoyl-oleyl-phosphatidylcholine (POPC) and palmitoyl-oleyl-phosphatidylglycerol (POPG) (POPC/POPG = 4:1, molar ratio) to see the lipid effects on the channel formation. Because previous experimental data relate to Aβ1–42 and Aβ1–40 (1,16), comprehensive experiments were conducted to test the Aβ9–42 model. Additional ongoing work targets the Aβ17–42, including AFM, calcium uptake by APP deficient cells, single channel conductance, and inhibition by zinc validating the candidate models (H. Jang, F. Teran Arce, R. Capone, S. Ramachandran, R. Lal, and R. Nussinov, unpublished). We note that as in any modeling study, our work in this study can only suggest candidates that would eventually need direct experimental tests.
Materials and Methods
A monomer conformation of the Aβ fragment Aβ9–42 based on solid state NMR of small Aβ9–40 protofibril coordinates (31) was used to construct channels with annular shapes adding Ile41 and Ala42. Annular shapes were obtained through rotations of the monomer 12, 16, 20, 24, and 36 times with respect to the pore axis (Fig. S1). Depending on the direction of the rotation, two channel topologies were constructed (Fig. S2): i), CNpNC (where C and N represent C- and N- terminal β-strands respectively, and p denotes a central pore); and ii), NCpCN Aβ channels. The former has a central pore enclosed by the N-terminal β-strands, whereas the latter by the C-terminal β-strands. In this study, we present only results for the pore-preserving CNpNC topology because the NCpCN with the charged N-terminal facing the bilayer and the hydrophobic C-terminal facing the solvated pore collapse (19).
Each annular channel is minimized with a rigid body motion of the peptides to enhance the formation of backbone hydrogen bonds (H-bonds) within a β-sheet and then embedded in the DOPC bilayer. In particular, the 24-mer channel is simulated in the anionic bilayer containing POPC and POPG with a molar ratio of 4:1. A unit cell containing two layers of lipids with 90,000 to 200,000 atoms, depending on the channel size, is constructed (Table S1). The bilayers containing 200–500 lipids constitute the unit cell with TIP3P waters added at both sides. The system contains MgCl, KCl, CaCl, and ZnCl at the same concentration of 25 mM to satisfy a total cations concentration near 100 mM. The CHARMM program (44) was used to construct the set of starting points and to relax the systems to a production-ready stage. Simulations of the initial construction and the preequilibration were carried out on the constant number of atoms, pressure, surface area, and temperature ensemble. Production runs were 30 ns for the 12- to 24-mer channels and 50 ns for the 36-mer channels. The NAMD code (45) on a Biowulf cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov) was used for the starting point. In the production simulations, the dynamics were carried out on both the constant number of atoms, pressure, surface area, and temperature and the constant number of atoms, pressure, surface tension, and temperature ensembles with γ = 0. However, no significant differences in the results were found for the different ensembles. Averages were taken after 10 ns discarding initial transient conformations.
Results
Optimal channel sizes
We use the experiment-based Aβ oligomer coordinate set to construct channels of different sizes. Our conceptual design for the initial channel is based on a perfect annular shape. Depending on the direction of the monomer rotation, two types of annular channel topologies are obtained (Fig. S2). We focus only on the pore-preserving topology with the hydrophobic C-terminal β-strands interfacing the bilayer and the N-terminal β-strands containing polar and charged residues facing into, and forming, the solvated pore (19,20). Size exclusion chromatography indicated that medium-to-large globulomers of (at least) 90–110 kDa lead to membrane permeability (21). No channels were observed for these particles. In our simulation, the molecular mass of 36-mer Aβ9–42 (130 kDa) channel is >90 kDa, the upper limit of channel formation (Fig. S1). We classify the channels into three groups: small (12-mer), intermediate (16-, 20-, 24-mer), and large (36-mer).
The Aβ channels increase the outer and the inner pore diameters during the simulations (Fig. 1). The figure shows the averaged channel structures embedding the averaged pore structures calculated by the HOLE program (46). For the 16-, 20-, and 24-mer Aβ9–42 channels, the outer diameter of simulated channels ranges between ∼7.4–8.3 nm, and the pore diameter between ∼1.6–2.5 nm in accord with experimental channels (1,16). However, the 12-mer pore diameter ∼0.6 nm is smaller, whereas the 36-mer pore diameter ∼3.7 nm is larger than the experimental channels.
Figure 1.
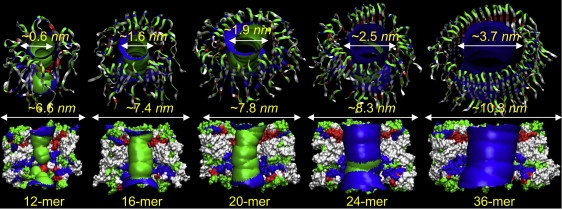
Averaged pore structures calculated by the HOLE program (46) embedded in the averaged channel conformations during the simulations for the 12-, 16-, 20-, 24-, and 36-mer Aβ9–42 channels. The 12-, 16-, 20-, and 36-mer structures were obtained from the simulations in the zwitterionic DOPC bilayer. The 24-mer structure was obtained from the anionic bilayer containing POPC and POPG with a molar ratio of 4:1. In the angle views of the pore structure (upper row), whole channel structures are shown with the ribbon representation. In the lateral views of the pore structure (lower row), cross-sectioned channels are given in the surface representation. For the pore structures in the surface representation, the degree of the pore diameter is indicated by the color codes in the order of red < green < blue, but the scale of these colors is relative to each channel. In the channel structures, hydrophobic residues are shown in white, polar and Gly residues are shown in green, positively charged residues are shown in blue, and negatively charged residues are shown in red.
Channel shape and subunit organization
As a starting point, the annular channel was simulated in the fully relaxed and hydrated lipid bilayer environment. The initial annular structure is highly frustrated, because in the CNpNC topology, the pore lining N-terminal strands form a β-sheet, whereas lipid-contacting outer C-terminal strands do not form a β-sheet due to larger curvature at the channel periphery. However, the initial frustration in the annular conformation is removed gradually via relaxation of the lipid bilayer (19). As the simulations progress, localized β-sheets begin to form in regions of the outer rim leading to the formation of “subunits” within the oligomeric channel, with the subunit appearance occurring after the initial 5 ns of the simulation. For the small and intermediate Aβ9–42 channels, the outer β-sheet, absent in the initial structure due to the larger curvature at the channel periphery, is recovered at certain regions dividing into several small subunits with or without disordered monomers in-between. Disordered monomers dismiss the U-shaped motif with large RMSD values, whereas the peptides participating in the subunits retain the U-shaped motif with low RMSD values (Fig. S3). Although the inner β-sheet backbone H-bonds hold tightly within the subunits, these H-bonds are straddled between these subunits, barely preventing subunit dissociation. In the 36-mer Aβ9–42 channel with smaller curvature, an outer β-sheet was present; however, during the simulations it still broke into several small more optimized β-sheets. The discontinuous β-sheet network can determine the boundary between the ordered subunits in the channels. In the Aβ9–42 channels, the ordered subunits can be defined by several criteria (Fig. 2): straightness of the strands by the β-strand order parameters using , where θα is the angle between the positional vectors connecting two Cα atoms, and N is the total number of vector pairs (Fig. 2, upper left); % of β-sheet content based on the intermolecular backbone H-bonds between β-stands (Fig. 2, lower left); the averaged β-strand B-factor or temperature-factor calculated from the RMS fluctuations relative to the starting point during the simulations with a simple correlation of (47) (Fig. 2, upper right); and descriptions of secondary structure by STRIDE (48) (Fig. 2, lower right). Well-ordered β-strands have large value of Sβ with high percentage forming the β-sheet, whereas they have small value of β-strand B-factor.
Figure 2.
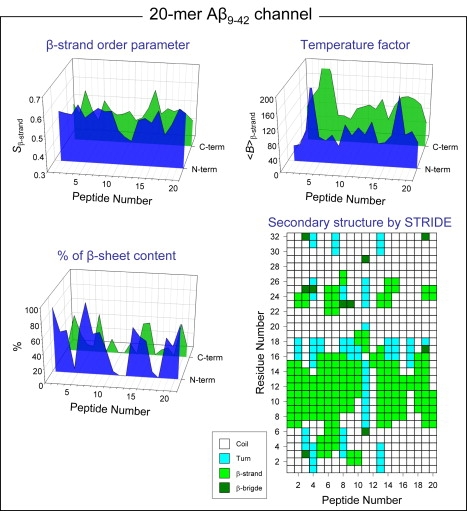
Parameters to define the ordered subunits for the 20-mer Aβ9–42 channel: the β-strand order parameter Sβ-strand (upper left), % of β-sheet content (lower left), the averaged β-strand B-factor (upper right), and the description of secondary structure by STRIDE (48) (lower right). The β-strand order parameter, the β-sheet content based on the intermolecular backbone H-bonds, and the averaged β-strand B-factor from the β-strand RMSF are calculated for the N-terminal (blue area) and C-terminal (green area) β-strands separately as a function of peptide number. Peaks in the Sβ-strand and % of β-sheet content curves indicate well ordered β-strand, whereas troughs in the β-strands B-factor curves denote the ordered β-strand. The secondary structure by STRIDE was calculated for the averaged structure.
The 16-mer Aβ9–42 channel has pentagonal shape with five subunits. The 20-mer channel has triangular shape with three subunits, and 24-mer channels have rectangular shapes with four ordered subunits (Fig. 3 a, upper row). The average positions of the β-strands calculated from the contour map of the x,y coordinates of the β-strands projected onto the two-dimensional bilayer surface verify the shapes and number of subunits (Fig. 3 a, lower row). The 24-mer channel obtained the subunits in the anionic bilayer (POPC/POPG = 4:1). Our previous 24-mer Aβ9–42 channel simulation obtained five subunits in the DOPC bilayer (20), suggesting that even for the same channel, subunit formations strongly depend on the fluidic bilayer dynamics. The 12-mer Aβ9–42 channel has two marginally defined subunits with collapsed pore (Fig. 1), suggesting that at least three subunits are required to form a water pore. The 36-mer Aβ9–42 channel has an ellipsoidal shape with two broken parts (Fig. S4). A hemifission may take place at the disordered narrowing neck. The disconnected end of the detached subunits may migrate and fuse with nearby channels, with the fission decaying spontaneously (49). The fission of the 36-mer channel suggests that the lipid bilayer does not support such large channel formation. The subunit organization observed for the Aβ9–42 channels is shared by other β-sheet channels (Fig. 3 b). Our recent simulations for the PG-1 channels (41) have shown that the octameric PG-1 channels with both antiparallel and parallel β-sheet arrangements divided into 4–5 subunits, indicating that although sequence composition, lengths, and detailed monomer conformations are different, the β-sheet channel formation via subunits association-dissociation is determined by the bilayer dynamics.
Figure 3.
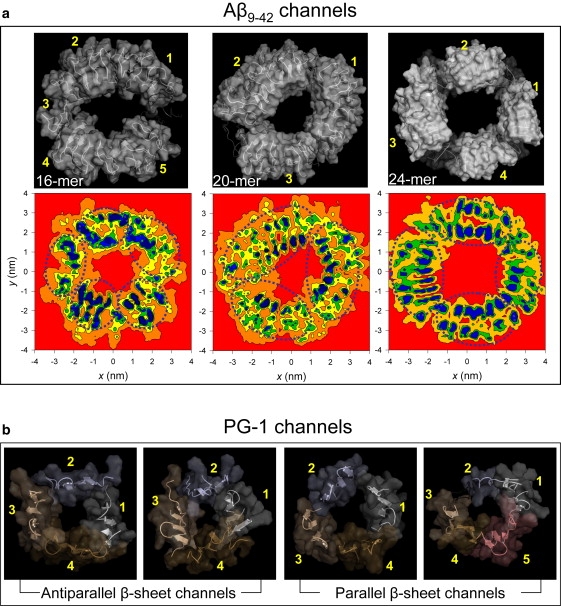
Simulated channel structures with highlighted subunits (upper row) and mapping of x,y coordinates of the β-strands of each peptide onto the x-y plane (lower row) for the (a) Aβ9-42 channels. The 12-, 16-, 20-, and 36-mer structures were obtained from the simulations in the zwitterionic DOPC bilayer. The 24-mer structure was obtained from the anionic bilayer containing POPC and POPG with a molar ratio of 4:1. The averaged channels in the surface representation are shown in the view along the membrane normal. In the β-strands mapping, the contour lines enclose the high frequency regions in the order of red < orange < yellow < green < blue, but the scale of these colors is relative to each channel. (b) The simulated channel structures with highlighted subunits for the antiparallel and parallel β-sheet channels of PG-1 (41).
Comparative peptide interaction
The interaction of the individual peptide is inhomogeneous, leading to heterogeneous Aβ channel shapes (Fig. S5). Of particular interest is the peptide interaction energy depending on the channel size. We calculated the interaction energy for each peptide with other peptides, lipids, and waters. The peptide interaction energy was then averaged over the time and the number of peptides in the channel (Fig. 4). For intermediate Aβ9–42 channels, well-balanced peptide interactions with the environment support the channel conformations; especially the channels have similar energy values for the peptide-water interaction. The peptide interaction energies suggest that in the channels the Aβ peptides favor interaction with the surroundings in the order of waters > other peptides > lipids. Strong peptide-water interaction indicates that the peptide has various ways of exposure to waters. In addition to bulk water, these include the central water pore and the hydrated cavity beneath each U-turn. For the peptide-peptide interaction, the interaction energy mainly derives from interaction between neighboring peptides (Fig. S6); the peptides have strong intermolecular backbone H-bonds between the β-strands in the ordered subunits. The peptide-lipid interaction is relatively weaker than the peptide-peptide interaction, suggesting that the subunits as rigid particles are mobile in the fluidic lipid bilayer.
Figure 4.
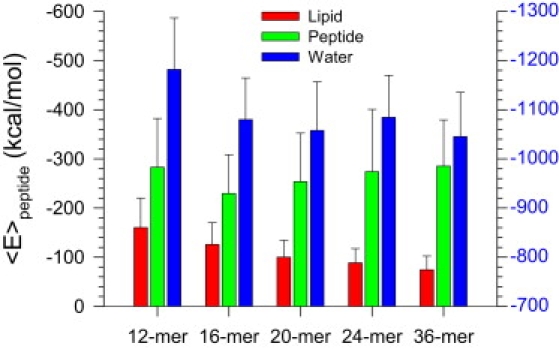
Averaged peptide interaction energy for the Aβ9-42 channels. All peptide-lipid interactions were calculated for the peptides interacting with DOPC lipids including 24-mer channels. The peptide-lipid (red bars) and peptide-peptide (green bars) interaction use the scale of interaction energy on the left (black labels). The peptide-water (blue) interaction uses the scale of interaction energy on the right (blue labels).
The small 12-mer Aβ9–42 channel has relatively strong peptide-water and peptide-lipid interactions. The peptides in the 12-mer channels are mainly disordered; lacking the intermolecular backbone H-bonds, they present increased interactions with lipids and waters. The 12-mer channel should have relatively weak peptide-peptide interaction compared to intermediate channels, because the trend of interaction energy decreases as the channels size decreases. However, the relatively strong peptide-peptide interaction in the 12-mer channel is due to the collapsed pore, inducing additional peptide-peptide interaction for non-neighboring pairs (Fig. 1). In contrast to the 12-mer channel, the 36-mer channel has relatively weak peptide-water and peptide-lipid interactions, and relatively strong peptide-peptide interaction. In the subunits of 36-mer channels, β-strand interactions are fully saturated with the H-bonding producing strong interactions between neighboring peptides. The ordered subunits become fibril-like rigid oligomers. The relatively weak interaction between peptides and lipids makes these subunits mobile in the lipid bilayer, inducing fissile channels.
Ion permeable channels
The channels preserve a large pore, wide enough for conducting water and ions. The Aβ9–42 channels confirm that cations are easily trapped by the negatively charged Glu22 side chains at the top bilayer leaflet, creating a cationic ring (19). In the pore of the Aβ9–42 channel, anions are attracted to the positively charged His14 and Lys16 side chains at the bottom bilayer leaflet. To observe the ions' behavior in the pore, the potential of mean force (PMF) representing the relative free energy profile for each ion across the bilayer is calculated (Fig. 5). The ion-density-based PMF calculation is useful to estimate rough relative free energy changes for ions, providing a general outline for ion permeation through the pore (41,50,51). The details of the PMF calculation are described in the Supporting Material.
Figure 5.
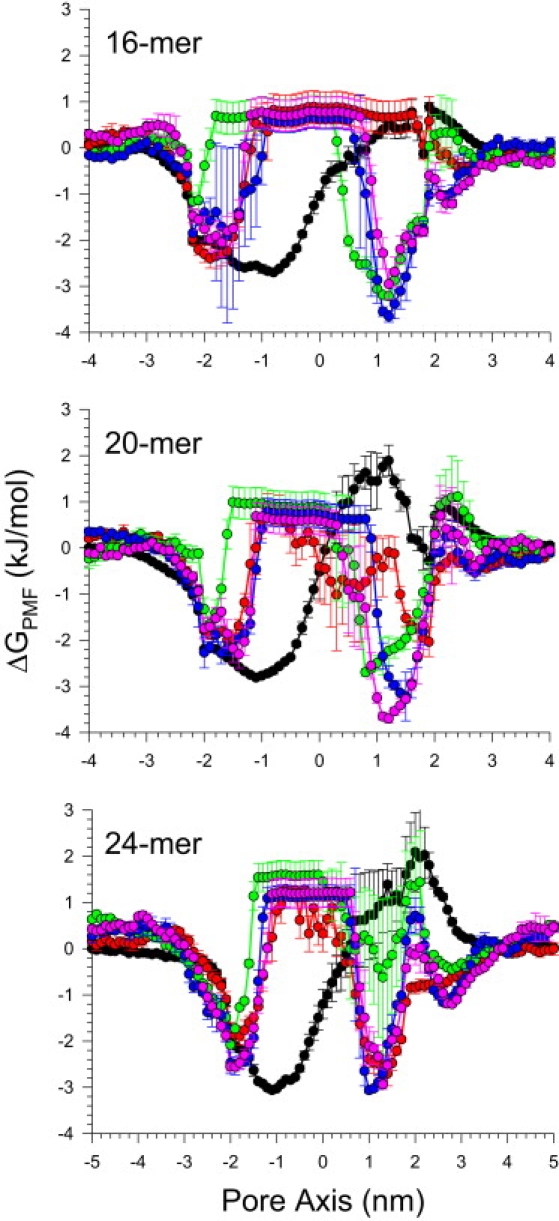
PMF, ΔGPMF, calculated using the equation ΔGPMF = −kBT ln(ρz/ρbulk), where kB is the Boltzmann constant, T is the simulation temperature, ρz is the ion density at the position z along the pore axis, and ρbulk is the ion density in the bulk region, representing the relative free energy profile for Mg2+ (green symbols and lines), K+ (red), Ca2+ (blue), Zn2+ (pink), and Cl− (black) as a function of the distance along the pore center axis for Aβ9–42 channels. The PMF results for the 12-, 16-, 20-, and 36-mer were obtained from the simulations in the zwitterionic DOPC bilayer. The PMF result for the 24-mer was obtained from the anionic bilayer containing POPC and POPG with a molar ratio of 4:1.
In the simulations, the channel systems contain four cations, Mg2+, Ca2+, K+, and Zn2+ at the same concentration (25 mM), and an anion, Cl−. On the top bilayer surface, the cations interact with the phosphate headgroups at the lipid/water interface. Just below the interface, the negatively charged Glu22 side chains in the pore form cation binding sites. At the lower bilayer leaflet, the cations interact with the phosphate headgroups and the Aβ C termini. In the pore, both Ca2+ and Zn2+ exhibit a low mobility at the binding site, indicating that they are easily trapped by the side chains, whereas Mg2+ and K+ are very mobile. Without Zn2+, Ca2+ are dominantly trapped by the side chains (19), but there are less trapped Ca2+ in the presence of Zn2+. Zn2+ inhibits Aβ toxicity, serving as channel blocker by reducing calcium transport (1,6–17,19,20). The 16-, 20-, and 24-mer channels binding sites provide a relatively low free energy profile for Ca2+ or Zn2+ compared to other cations. However, in the 12-mer channel calcium binding is not observed at the Glu22 side chains (Fig. S7), because the collapsed pore blocks Ca2+ entering to the pore with a high energy barrier. The 36-mer channel binding sites provide a relatively low free energy profile for all cations, suggesting nonselective ion leakiness due to the large pore (Fig. S7). In the lower pore portion of Aβ9–42 channel, the positively charged His14 and Lys16 side chains provide low free energy profile for Cl−, creating anionic binding sites.
Discussion and Conclusions
Explicit solvent molecular dynamics simulations of ssNMR-based Aβ oligomer coordinates (31) in the bilayer modeled as perfectly annular structures obtain ion-permeable channels. AFM images provide a consistent picture of ion channels for a series of disease-related amyloid species (1). The channels present a range of sizes and morphologies. Remarkably, the 16-, 20-, and 24-mer simulations obtain channels with similar subunit organization and dimensions as imaged by AFM (1,16). The 24-mer channel was simulated in both the zwitterionic DOPC bilayer and the anionic bilayer containing POPC and POPG with a molar ratio of 4:1. It is expected that the channel interacts more strongly with the anionic bilayer than the zwitterionic bilayer due to the electrostatic interaction. However, no significant differences in the results of the subunit formation in the channel conformation were found. The smaller (12-mer) channel collapses and the larger (36-mer) channel is not supported by the bilayer, allowing us to derive the apparent molecular mass of the AFM-imaged channels that we confirm to be less than what is predicted for large Aβ oligomers (21). The range of channel sizes observed for the Aβ, 16-, 20-, and 24-mer, contrasts the functional regulated native state gated channels that have a single size, a precise number of subunits, and one specific way of subunit organization.
Normally folded functional gated ion channels selectively permeate ions across membranes with electrical currents in two steady-state levels corresponding to the open and closed states (42,43). In contrast, toxic Aβ channels catalyze the diffusion of ions across membranes presenting stepwise fluctuations of electrical currents with typical ion channel behavior; however, the fluctuating large amplitude spikes in the current indicate distinct channels (23). This experimentally observed behavior derives from Aβ channel formation in the dynamic membrane: after insertion into the cell membranes, small Aβ oligomer subunits assemble to form dynamic, leaky pore-like structures (Fig. 6 a). In the 16-, 20-, and 24-mer channel simulations we do not observe a movement toward dissociation; what we observe are dynamic structures. Such a dynamic leaky behavior is in contrast to functional gated channels that fold into specific native structures optimized by evolution (Fig. 6 b). In normally folded controlled gated channels good contacts are maintained between the α-helical subunits, robustly preventing ion leakage. Aβ channel formation is cytotoxic, similar to the channels formed by β-cytolytic peptides such as PG-1 antimicrobial peptides (41), suggesting that amyloid channel formation may induce membrane defects such as those induced by PG-1 membrane pore formation (52). We note that toxic α-antimicrobial peptides do not form channels. Rather, the α-helices interactions are lipid-mediated (Fig. 6 c). Our results suggest why controlled gated channels consist of α-helices rather than the consistently observed breakage-prone ion-leaking β-sheet channels.
Figure 6.
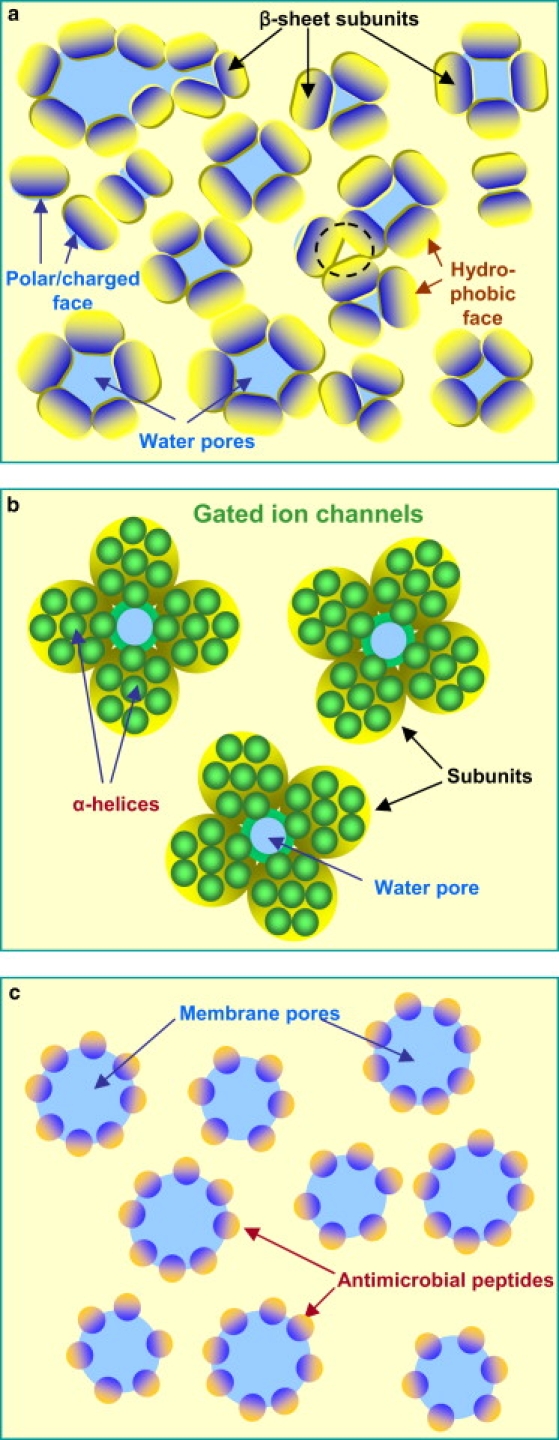
(a) Cartoon representing the dynamics of Alzheimer ion channel formation in the fluidic lipid bilayer. The mobile subunit has the hydrophobic face (yellow) and polar/charged face (blue). A channel with water filled pore can be formed when at least three subunits assemble their polar/charged faces together. The pore-preserving channels provide ion conductance with selectivity across the membrane. Large pores with nonselective ion conductance are not supported by the bilayer membranes and tend to reduce the size (top left). Misassembled subunits do not conduct ions due to the collapsed pore as denoted by the dotted circle. Cartoons representing (b) functional gated ion channels and (c) membrane pore formations by toxic α-antimicrobial peptides. The gated ion channel diagram was inspired by the K+ channel with four identical subunits, but the number of α-helices in the subunit diagram is arbitrary.
Our simulations suggest that the Aβ9–42 channels induce a barrel-stave membrane pore where the channel is parallel to the lipid tails (Fig. S8). However, this is not necessarily the case for all amyloid channels. Amyloid channels can also induce other membrane pore types such as toroidal depending on the outward-facing amino acid sequence interacting with the bilayer (53,54). A charged/polar surface preferentially interacting with lipid headgroups may induce such a curved membrane surface (52). Under such circumstances, ion leakage can be not only through the pore but via the solvated interface between the channel and the bilayer. In both toxic barrel-stave channels as observed here and toroidal channels with subunit organizations, solvated intersubunits areas can also leak ions.
To conclude, our simulations provide atomic resolution models for Aβ channels based on the NMR β-strand-turn-β-strand motif. We have simulated the channels in an explicit membrane environment to obtain a range of potential sizes to characterize, the apparent molecular mass and the preferred channel size and organization and compared with experiment. We have further simulated channels in different membrane environments, in DOPC, and in an anionic bilayer. We note that these models are consistent with experimental data; however, as in all modeling studies (55), other models consistent with available data are always possible. Moreover, currently it is increasingly realized that although still presenting the β-strand-turn-β-strand motif, amyloid seeds and fibrils are polymorphic (56–58), suggesting that small oligomers penetrating the bilayer can also present a polymorphic variability affecting channel conformations. Nonetheless, we believe that the dynamic toxic channel picture painted by our models should hold for a polymorphic range. Our studies provide what we believe are conformational details useful for drug development (24–26). The Aβ channels have β-sheet structure that is reasonable for both amyloid formation (36,37,59) and channel formation (40). It is interesting to note that antimicrobial cytolytic β-sheet channels similarly break into dynamic subunits (41), suggesting that the fluidic membrane does not support intact cylindrical β-sheets channels, regardless of the sequence, length, and conformational details. The H-bonds break in some regions leading to optimization of the H-bonds in the subunits. All these channels can leak ions through the solvated channel pores. We propose that this could be a main evolutionary reason why gated functional channels tend to be made of tightly interacting α-helices rather than of β-sheet organization.
Supporting Material
Potential mean force calculations, references, a table, and eight figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01460-X.
Supporting Material
Acknowledgments
We thank Dr. Robert Tycko for providing us with the coordinates of the Aβ9–40 oligomer. This study used the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). Initial coordinates of the channels are available at http://protein3d.ncifcrf.gov/hbj/coor/.
This work was supported by the National Cancer Institute, National Institutes of Health (N01-CO-12400), and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
References
- 1.Quist A., Doudevski I., Lin H., Azimova R., Ng D. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe D.J. Alzheimer's disease. In the beginning. Nature. 1991;354:432–433. doi: 10.1038/354432a0. [DOI] [PubMed] [Google Scholar]
- 3.Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 4.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 5.Kagan B.L. Amyloidosis and protein folding. Science. 2005;307 doi: 10.1126/science.307.5706.42b. 42–43, author reply 42–43. [DOI] [PubMed] [Google Scholar]
- 6.Arispe N., Pollard H.B., Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arispe N., Pollard H.B., Rojas E. β-Amyloid Ca(2+)-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell. Biochem. 1994;140:119–125. doi: 10.1007/BF00926750. [DOI] [PubMed] [Google Scholar]
- 8.Arispe N., Pollard H.B., Rojas E. Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc. Natl. Acad. Sci. USA. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawahara M., Arispe N., Kuroda Y., Rojas E. Alzheimer's disease amyloid β-protein forms Zn(2+)-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys. J. 1997;73:67–75. doi: 10.1016/S0006-3495(97)78048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawahara M., Kuroda Y. Molecular mechanism of neurodegeneration induced by Alzheimer's β-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res. Bull. 2000;53:389–397. doi: 10.1016/s0361-9230(00)00370-1. [DOI] [PubMed] [Google Scholar]
- 11.Kawahara M., Kuroda Y., Arispe N., Rojas E. Alzheimer's β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- 12.Rhee S.K., Quist A.P., Lal R. Amyloid β protein-(1–42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem. 1998;273:13379–13382. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- 13.Hirakura Y., Lin M.C., Kagan B.L. Alzheimer amyloid Aβ1–42 channels: effects of solvent, pH, and Congo Red. J. Neurosci. Res. 1999;57:458–466. [PubMed] [Google Scholar]
- 14.Hirakura Y., Yiu W.W., Yamamoto A., Kagan B.L. Amyloid peptide channels: blockade by zinc and inhibition by Congo red (amyloid channel block) Amyloid. 2000;7:194–199. doi: 10.3109/13506120009146834. [DOI] [PubMed] [Google Scholar]
- 15.Lin H., Zhu Y.J., Lal R. Amyloid β protein (1–40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry. 1999;38:11189–11196. doi: 10.1021/bi982997c. [DOI] [PubMed] [Google Scholar]
- 16.Lin H., Bhatia R., Lal R. Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Y.J., Lin H., Lal R. Fresh and nonfibrillar amyloid β protein(1–40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J. 2000;14:1244–1254. doi: 10.1096/fasebj.14.9.1244. [DOI] [PubMed] [Google Scholar]
- 18.Kourie J.I., Henry C.L., Farrelly P. Diversity of amyloid β protein fragment [1–40]-formed channels. Cell. Mol. Neurobiol. 2001;21:255–284. doi: 10.1023/a:1010995121153. [DOI] [PubMed] [Google Scholar]
- 19.Jang H., Zheng J., Nussinov R. Models of β-amyloid ion-channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007;93:1938–1949. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang H., Zheng J., Lal R., Nussinov R. New structures help the modeling of toxic amyloid β ion channels. Trends Biochem. Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Kayed R., Sokolov Y., Edmonds B., McIntire T.M., Milton S.C. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 22.Sokolov Y., Kozak J.A., Kayed R., Chanturiya A., Glabe C. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J. Gen. Physiol. 2006;128:637–647. doi: 10.1085/jgp.200609533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capone R., Quiroz F.G., Prangkio P., Saluja I., Sauer A.M. Amyloid-β induced ion flux in artificial lipid bilayers and neuronal cells: resolving a controversy. Neurotox. Res. 2009;16:1–13. doi: 10.1007/s12640-009-9033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz J.C., Simakova O., Jacobson K.A., Arispe N., Pollard H.B. Small molecule blockers of the Alzheimer Aβ calcium channel potently protect neurons from Aβ cytotoxicity. Proc. Natl. Acad. Sci. USA. 2009;106:3348–3353. doi: 10.1073/pnas.0813355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arispe N., Diaz J.C., Flora M. Efficiency of histidine-associating compounds for blocking the Alzheimer's Aβ channel activity and cytotoxicity. Biophys. J. 2008;95:4879–4889. doi: 10.1529/biophysj.108.135517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simakova O., Arispe N.J. Early and late cytotoxic effects of external application of the Alzheimer's Aβ result from the initial formation and function of Aβ ion channels. Biochemistry. 2006;45:5907–5915. doi: 10.1021/bi060148g. [DOI] [PubMed] [Google Scholar]
- 27.Lashuel H.A., Hartley D., Petre B.M., Walz T., Lansbury P.T. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 28.Lashuel H.A., Hartley D.M., Petre B.M., Wall J.S., Simon M.N. Mixtures of wild-type and a pathogenic (E22G) form of Aβ40 in vitro accumulate protofibrils, including amyloid pores. J. Mol. Biol. 2003;332:795–808. doi: 10.1016/s0022-2836(03)00927-6. [DOI] [PubMed] [Google Scholar]
- 29.Thinakaran G., Koo E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luhrs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petkova A.T., Yau W.M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma B., Nussinov R. Stabilities and conformations of Alzheimer's β-amyloid peptide oligomers (Aβ16–22, Aβ16–35, and Aβ10–35): sequence effects. Proc. Natl. Acad. Sci. USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwata K., Fujiwara T., Matsuki Y., Akutsu H., Takahashi S. 3D structure of amyloid protofilaments of β2-microglobulin fragment probed by solid-state NMR. Proc. Natl. Acad. Sci. USA. 2006;103:18119–18124. doi: 10.1073/pnas.0607180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferguson N., Becker J., Tidow H., Tremmel S., Sharpe T.D. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA. 2006;103:16248–16253. doi: 10.1073/pnas.0607815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J., Ma B., Nussinov R. Consensus features in amyloid fibrils: sheet-sheet recognition via a (polar or nonpolar) zipper structure. Phys. Biol. 2006;3:1–4. doi: 10.1088/1478-3975/3/3/P01. [DOI] [PubMed] [Google Scholar]
- 36.Zheng J., Jang H., Ma B., Tsai C.J., Nussinov R. Modeling the Alzheimer Aβ17–42 fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys J. 2007;93:3046–3057. doi: 10.1529/biophysj.107.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng J., Jang H., Ma B., Nussinov R. Annular structures as intermediates in fibril formation of Alzheimer Aβ17–42. J. Phys. Chem. B. 2008;112:6856–6865. doi: 10.1021/jp711335b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin M.C., Mirzabekov T., Kagan B.L. Channel formation by a neurotoxic prion protein fragment. J. Biol. Chem. 1997;272:44–47. doi: 10.1074/jbc.272.1.44. [DOI] [PubMed] [Google Scholar]
- 39.Hirakura Y., Azimov R., Azimova R., Kagan B.L. Polyglutamine-induced ion channels: a possible mechanism for the neurotoxicity of Huntington and other CAG repeat diseases. J. Neurosci. Res. 2000;60:490–494. doi: 10.1002/(sici)1097-4547(20000515)60:4<490::aid-jnr7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Hirakura Y., Kagan B.L. Pore formation by β-2-microglobulin: a mechanism for the pathogenesis of dialysis associated amyloidosis. Amyloid. 2001;8:94–100. doi: 10.3109/13506120109007350. [DOI] [PubMed] [Google Scholar]
- 41.Jang H., Ma B., Lal R., Nussinov R. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic Alzheimer β-amyloid ion channels. Biophys. J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roux B. Ion conduction and selectivity in K(+) channels. Annu. Rev. Biophys. Biomol. Struct. 2005;34:153–171. doi: 10.1146/annurev.biophys.34.040204.144655. [DOI] [PubMed] [Google Scholar]
- 43.Tombola F., Pathak M.M., Isacoff E.Y. How does voltage open an ion channel? Annu. Rev. Cell Dev. Biol. 2006;22:23–52. doi: 10.1146/annurev.cellbio.21.020404.145837. [DOI] [PubMed] [Google Scholar]
- 44.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM—a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 45.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smart O.S., Goodfellow J.M., Wallace B.A. The pore dimensions of gramicidin A. Biophys. J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wriggers W., Mehler E., Pitici F., Weinstein H., Schulten K. Structure and dynamics of calmodulin in solution. Biophys. J. 1998;74:1622–1639. doi: 10.1016/S0006-3495(98)77876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frishman D., Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 49.Kozlovsky Y., Kozlov M.M. Membrane fission: model for intermediate structures. Biophys. J. 2003;85:85–96. doi: 10.1016/S0006-3495(03)74457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Groot B.L., Grubmuller H. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science. 2001;294:2353–2357. doi: 10.1126/science.1066115. [DOI] [PubMed] [Google Scholar]
- 51.Leontiadou H., Mark A.E., Marrink S.J. Ion transport across transmembrane pores. Biophys. J. 2007;92:4209–4215. doi: 10.1529/biophysj.106.101295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 53.Smith P.E., Brender J.R., Ramamoorthy A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: the case of islet amyloid polypeptide. J. Am. Chem. Soc. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong P.T., Schauerte J.A., Wisser K.C., Ding H., Lee E.L. Amyloid-β membrane binding and permeabilization are distinct processes influenced separately by membrane charge and fluidity. J. Mol. Biol. 2009;386:81–96. doi: 10.1016/j.jmb.2008.11.060. [DOI] [PubMed] [Google Scholar]
- 55.Durell S.R., Guy H.R., Arispe N., Rojas E., Pollard H.B. Theoretical models of the ion channel structure of amyloid β-protein. Biophys. J. 1994;67:2137–2145. doi: 10.1016/S0006-3495(94)80717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meinhardt J., Sachse C., Hortschansky P., Grigorieff N., Fandrich M. Aβ(1–40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J. Mol. Biol. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paravastu A.K., Leapman R.D., Yau W.M., Tycko R. Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang R., Hu X., Khant H., Ludtke S.J., Chiu W. Interprotofilament interactions between Alzheimer's Aβ1–42 peptides in amyloid fibrils revealed by cryoEM. Proc. Natl. Acad. Sci. USA. 2009;106:4653–4658. doi: 10.1073/pnas.0901085106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buchete N.V., Tycko R., Hummer G. Molecular dynamics simulations of Alzheimer's β-amyloid protofilaments. J. Mol. Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.