

Abstract
The shorter “M23” isoform of the glial cell water channel aquaporin-4 (AQP4) assembles into orthogonal arrays of particles (OAPs) in cell plasma membranes, whereas the full-length “M1” isoform does not. N-terminal residues are responsible for OAP formation by AQP4-M23 and for blocking of OAP formation in AQP4-M1. In investigating differences in OAP formation by certain N-terminus mutants of AQP4, as measured by freeze-fracture electron microscopy versus live-cell imaging, we discovered reversible, temperature-dependent OAP assembly of certain weakly associating AQP4 mutants. Single-particle tracking of quantum-dot-labeled AQP4 in live cells and total internal reflection fluorescence microscopy showed >80% of M23 in OAPs at 10–50°C compared to <10% of M1. However, OAP formation by N-terminus cysteine-substitution mutants of M1, which probe palmitoylation-regulated OAP assembly, was strongly temperature-dependent, increasing from <10% at 37°C to >70% at 10°C for the double mutant M1-C13A/C17A. OAP assembly by this mutant, but not by native M23, could also be modulated by reducing its membrane density. Exposure of native M1 and single cysteine mutants to 2-bromopalmitate confirmed the presence of regulated OAP assembly by S-palmitoylation. Kinetic studies showed rapid and reversible OAP formation during cooling and OAP disassembly during heating. Our results provide what to our knowledge is the first information on the energetics of AQP4 OAP assembly in plasma membranes.
Introduction
Aquaporin-4 (AQP4) is a water-selective channel expressed in glial cells in brain and various epithelial cells in kidney, airways, exocrine glands, and other tissues (1–3). In the central nervous system, AQP4 plays an important role in brain water balance, neuroexcitation, and glial cell migration (4). Two predominant isoforms of AQP4 are expressed, a full-length (“M1”) isoform, and a shorter (“M23”) isoform with translation initiation at methionine 23 (1) (Fig. 1 A). AQP4 molecules form tetramers in membranes (5,6). AQP4-M23 tetramers can further assemble in supramolecular complexes called orthogonal arrays of particles (OAPs), which are regular, square arrays of intramembrane particles seen by freeze-fracture electron microscopy (FFEM) in brain, kidney and other tissues (7–11). AQP4-M1 tetramers do not form OAPs. Our lab discovered the involvement of AQP4 in OAPs by showing the presence of OAPs in AQP4-M23-transfected cells (12), and the absence of OAPs in AQP4 knock-out mice (13). The biological importance of AQP4 assembly in OAPs is not known. There is speculation that OAPs might enhance AQP4 water permeability (14,15) or play a role in AQP4 polarization to astrocyte foot processes (16). It has also been suggested that OAPs are sites of intercellular contact (5), although a more recent study could not confirm involvement of AQP4 in cell-cell adhesion (17). Correlations have been reported between OAP abundance and disease processes such as toxic encephalopathies (18), muscular dystrophy (19) and neuromyelitis optica (20).
Figure 1.
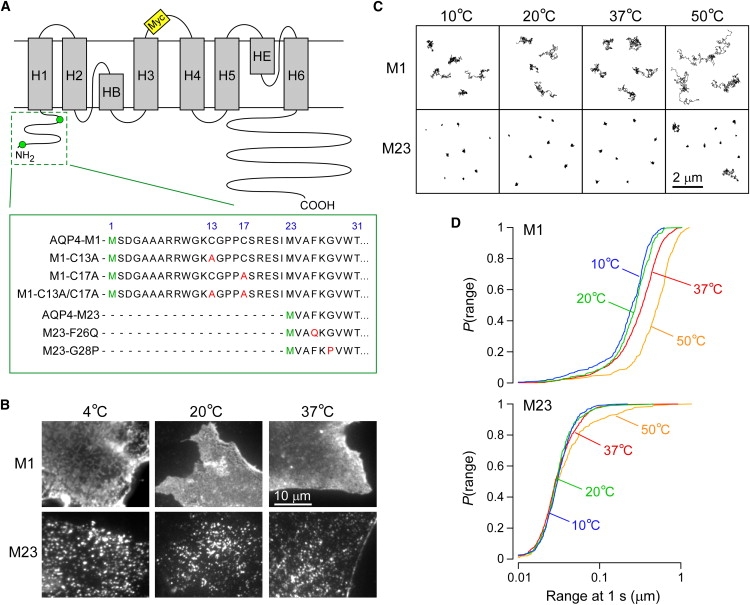
Temperature-independent assembly of native AQP4 isoforms M1 and M23. (A) AQP4 schematic showing transmembrane helices (gray), the positions of the inserted Myc sequence (yellow) in the second extracellular loop, and Met1 and Met23 (green) in the cytoplasmic N-terminal domain. N-terminus sequences of the AQP4 mutants used in this study are shown in the expanded green box. (B) TIRF micrographs of Alexa-labeled COS-7 cells transfected with M1 (upper) or M23 (lower) and fixed at the indicated temperature. Scale bar, 10 μm. (C) Representative trajectories from AQP4 isoforms M1 (upper) and M23 (lower) diffusing in the membrane of live COS-7 cells at the indicated temperatures. Scale bar, 2 μm. (D) Cumulative probability distribution of ranges at 1 s (P(range)) for AQP4 isoforms M1 (upper) and M23 (lower) recorded at 10°C (blue), 20°C (green), 37°C (red), and 50°C (orange).
We recently established the molecular determinants of OAP formation by quantum-dot single-particle tracking (SPT) of the M1 and M23 isoforms of AQP4 in live cells at physiological temperature, 37°C. We found that AQP4-M23 diffusion in membranes is highly confined because of its assembly in OAPs, whereas AQP4-M1 is freely diffusible (21). Diffusion measurements and total internal reflection fluorescence imaging of various AQP4 mutants and chimeras showed that OAP formation by AQP4-M23 involves hydrophobic intermolecular interactions of N-terminal AQP4 residues just downstream of Met23, and that AQP4-M1 is prevented from forming OAPs by blocking of these interactions by N-terminal residues just upstream of Met23 (22). It was assumed in these studies that strong AQP4-M23 tetramer-tetramer interactions produce stable OAPs.
Here, we report the discovery that OAP assembly can be strongly modulated by temperature and membrane density, with certain AQP4 mutants undergoing rapid, reversible, and near-complete interconversion between OAPs and nonassociated tetramers with temperature change. Our original motivation for studying temperature-dependent OAP assembly was to investigate apparently contradictory data on N-terminus AQP4 mutants that abolished putative palmitoylation, where freeze-fracture electron microscopy showed OAPs (23) but live-cell imaging at 37°C did not (22). We discovered that these and various other AQP4 mutants assembled weakly and reversibly into OAPs, such that OAP assembly/disassembly could be driven by changes in temperature or membrane density, allowing biophysical investigation of the energetics of OAP formation in live cells.
Methods
Cell culture and transfections
DNA constructs used in this study encoded rat AQP4 (M1 and M23 isoforms, and mutants thereof (Fig. 1 A)), into which was inserted a 10-residue Myc epitope (NH2-EQKLISEEDL-COOH) in the second extracellular loop, as previously described (22). COS-7 (American Type Culture Collection code CRL-1651) cell cultures were maintained at 37°C in 5% CO2/95% air in DME-H21 medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Six hours before transfection, cells were transferred to 12-well plates containing glass coverslips 18 mm in diameter using antibiotic-free medium. Cells were transfected 12–24 h before experiment using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. In some cases, cells were incubated overnight with 100 μM 2-bromopalmitate (Sigma, St. Louis, MO) in normal medium, beginning 6–12 h after transfection.
Labeling for fluorescence microscopy
Labeling of AQP4 with quantum dots (Qdots) was done at room temperature. Cells were washed with 3 mL of phosphate-buffered saline (PBS) containing 6 mM glucose and 1 mM pyruvate (GP buffer) and incubated for 5 min in blocking buffer (GP buffer containing 1% bovine serum albumin), and for another 5 min with 70 ng/mL mouse anti-myc antibody (Covance, Emeryville, CA) in blocking buffer. Cells were then rinsed with GP buffer, incubated for 5–8 min with 0.1 nM goat F(ab′)2 anti-mouse IgG-conjugated Qdot 655 (Invitrogen) in blocking buffer, then rinsed again with GP buffer. Coverslips were transferred to a custom-built perfusion chamber and maintained in GP buffer throughout the experiment.
For heavy fluorescence labeling for total internal reflection fluorescence microscopy (TIRFM), cells were incubated for 30 min at 37°C or 4°C, rinsed with GP buffer, and then fixed for 30 min with 4% paraformaldehyde at 37°C or 4°C. After fixation, all labeling was conducted at room temperature. Fixed cells were rinsed in PBS, then incubated first for 30 min in blocking buffer, then for 1 h with 1.4 μg/mL anti-myc antibody in blocking buffer. Cells were again rinsed with PBS and incubated for 1 h with 10 μg/mL goat F(ab′)2 anti-mouse IgG-conjugated Alexa Fluor 488 (Invitrogen) in blocking buffer. Cells were then rinsed extensively in PBS, and coverglasses were mounted with VectaMount hard-set medium for microscopy (Vector Laboratories, Burlingame, CA).
Quantitative surface staining of AQP4 was performed at room temperature. Live cells were incubated for 10 min in blocking buffer followed by 20 min with 0.7 μg/mL anti-myc antibody in blocking buffer. Cells were then rinsed with GP buffer and fixed with 4% paraformaldehyde for 30 min. Fixed cells were then rinsed in PBS and incubated again for 10 min in blocking buffer followed by 20 min with 2 μg/mL secondary-linked Alexa Fluor 488 in blocking buffer. Cells were rinsed with PBS and coverglasses were mounted with hard-set medium for microscopy.
Instrumentation and measurements
SPT was performed on a Nikon Eclipse TE2000S inverted epifluorescence microscope (Nikon, Melville, NY) equipped with a Nikon 100× TIRF oil immersion objective (numerical aperture 1.45) and a deep-cooled charge-coupled device (CCD) camera (Hamamatsu EM-CCD, Bridgewater, NJ). Qdot fluorescence was excited using an E460SPUV excitation filter and 475DCXRU dichroic mirror, and detected through a D655/40m emission filter (Chroma, Rockingham, VT). For experiments at constant temperature, cells were incubated for at least 10 min in a temperature-controlled chamber with both heating and cooling capacity (Harvard Apparatus, Holliston, MA), and data were acquired continuously at 11 ms/frame (91 Hz) for 6 s. For kinetic analysis, cells were placed in a custom-built perfusion chamber under constant flow of warm or cold perfusate. Temperature was recorded by a solid-state temperature probe (Warner Instruments, Hamden, CT) positioned at the fluid exit port of the chamber. The spatial resolution of the system, determined from the standard deviation of x,y coordinates of immobilized Qdots on a coverglass (24), was 18 nm at 91 Hz. Intermittent blinking to background levels confirmed that only single Qdots were tracked.
Wide-field fluorescence for quantitative analysis of AQP4 membrane expression was performed with the microscope, objective, and camera described above. Alexa Fluor 488 was excited and observed through Chroma filter set 41001, with a neutral density filter (optical density 0.6) inserted for the highest AQP4 concentrations to prevent saturation of the CCD.
TIRFM was done using a Nikon Eclipse TE2000E microscope with a through-objective TIRF attachment and a 100× TIRF oil immersion objective (numerical aperture 1.49) mounted on a perfect focus module (Nikon). An argon-ion laser (Spectra Physics, Mountain View, CA) on a custom-built launch was coupled through a fiberoptic to the TIRF module. Alexa Fluor 488-labeled AQP4 was excited using a Z488/10× excitation filter and Z488RDC dichroic mirror, and detected through an ET525/50m emission filter (Chroma). Images were acquired using a QuantEM 512SC deep-cooled CCD camera (Photometrics, Tucson, AZ).
SPT analysis
Image sequences were analyzed and trajectories constructed using IDL software (Research Systems, Boulder, CO), with algorithms available as shareware at http://www.physics.emory.edu/faculty/weeks/idl/. Blinking of individual Qdots was accounted for during reconstruction of trajectories. A trajectory was considered to be continuous if a blinking Qdot was rediscovered within a 4-pixel radius and 20-frame window. Trajectories of at least 200 steps in length were then selected and analyzed. The mean-squared displacement (MSD) as a function of time 〈r2(t)〉 was constructed for each trajectory, and the diffusion coefficient, D, and offset (due to noise) were determined by a linear fit of the first three time steps of the MSD:
| (1) |
The offset was subtracted from each point, and the first 25% of the curve (25) was fitted, using a weighted Levenberg-Marquardt nonlinear least-squares fitting algorithm, to a combined quadratic, polynomial, and exponential function (26) with fitting parameters a1, a2, a3 ≥ 0:
| (2) |
The fit was weighted by the variance in the MSD at each time step, and the range of an individual particle at specified time t was computed as
| (3) |
Results from this study are primarily reported as cumulative distributions of ranges at 1 s (Fig. 1 D), where P(range) is defined as the probability that a particle's range is less than or equal to a given distance at t = 1 s. Based on the finding that >95% of AQP4 is present in OAPs in M23-expressing cells (21,23,27), we estimated the fraction of AQP4 isoforms and mutants assembled in OAPs at various temperatures as P(76 nm), the range at which P(range) = 0.95 for native AQP4-M23 at 20°C. The fraction of free tetramers, f, in equilibrium with OAPs is then given by
| (4) |
The thermodynamic model used to compute the standard Gibbs free energy of association of AQP4 mutants is given in the Appendix. SPT data sets were composed of at least 200 total trajectories gathered from at least 20 cells. Plotted error bars in all figures represent the mean ± SE, where n is the number of cells. Statistical significance of differences in estimated OAP fractions was determined using the Student's t-test.
Results
Temperature-independent assembly of native M1 and M23 AQP4 isoforms
As previously reported (21), AQP4 assembly in OAPs is readily seen by TIRFM in fixed cells and by SPT in live cells. Fig. 1 shows differences in behavior observed for the naturally occurring AQP4 isoforms M1 and M23. TIRF micrographs of M1-transfected COS-7 cells at 37°C show a smooth, near-uniform distribution of AQP4 over the cell membrane, quite different from the distinct fluorescent clusters, corresponding to OAPs, in M23-transfected cells (Fig. 1 B). Reducing the temperature to 4°C had little effect on the appearance of the M1 and M23 isoforms, except that in some cells, M1 was seen in channel-like structures, which may be related to bulk membrane phase separation and AQP4 partitioning into ordered or disordered lipid phases. In live cells, the diffusion of M1 was rapid and free compared to that in M23 at all temperatures studied, with representative single-particle trajectories shown in Fig. 1 C. The average diffusion coefficient of M1 varied from ∼3 × 10−10 cm2/s at 10°C to ∼1 × 10−9 cm2/s at 50°C, whereas the average diffusion coefficient of M23 was <7 × 10−11 cm2/s at all temperatures.
Fig. 1 D shows plots of the cumulative probability distribution of ranges (at 1 s) for all measured trajectories. These plots indicate the remarkable difference in the diffusion of individual AQP4 tetramers (M1) versus those confined within OAPs (M23). The median range at 1 s for M1 varied from 252 nm at 10°C to 524 nm at 50°C, whereas the median range for M23 was only 30 nm at 50°C. At temperatures up to 37°C, nearly all M23 trajectories showed extreme confinement. At the nonphysiological temperature 50°C, a small fraction (∼10%) of more rapidly diffusing M23 was seen (Fig. 1 C), suggesting partial thermal disruption of M23 OAPs. The distribution of ranges of M23 did not change between 10°C and 37°C; however, the small mobile population at 50°C produced a visible change in the distribution (Fig. 1 D).
Temperature-dependent assembly of “weakly associating” M1 and M23 AQP4 mutants
Reversible palmitoylation at N-terminal cysteines is a common posttranslational modification in membrane proteins. AQP4-M1 contains potential palmitoylation sites at Cys13 and Cys17 (Fig. 1 A). It was reported that a palmitoylation-null mutant of M1, in which both cysteines were mutated to alanine (M1-C13A/C17A), could produce OAPs as seen by FFEM, whereas little or no OAP formation was seen for single cysteine mutants (23). However, SPT analysis of these mutants in live cells showed no difference in their diffusion when compared to native M1 (22). More recently, a study using blue native gel electrophoresis showed bands corresponding to possible OAP formation for the same double mutant (28). To investigate technical factors that might be responsible for the different findings, we tested the temperature dependence of OAP formation, speculating that differences might be found in live cells at 37°C versus fixed samples or cell lysates, which are often prepared at reduced temperatures.
Fig. 2 shows that formation of OAPs in live cells by the double cysteine mutant of M1 is highly temperature-dependent. As temperature was reduced from 37°C to 10°C, a remarkable shift was seen in the distribution of ranges for M1-C13A/C17A (Fig. 2 A). In contrast, the diffusion of single cysteine mutants was similar to that of native M1, with only a small shift in the distribution of ranges of M1-C13A at 10°C, and no difference for M1-C17A at any temperature. FFEM and immunolabeling previously showed that >95% of AQP4-M23 is present in OAPs (23,27), and diffusion analysis indicated that 90–96% of M23 could be classified as restricted or immobile (21). The range at which P(range) = 0.95 for M23 (76 nm at 20°C) was therefore taken as an estimate of the maximum range of an AQP4 tetramer that is confined within an OAP; P(76 nm) was thus used to calculate the approximate fraction of AQP4 tetramers in OAPs, as done previously (22). Accordingly, the fraction of M1-C13A/C17A forming OAPs increased from <10% at 37°C to 72 ± 5% at 10°C (Fig. 2 B). Mutation of Cys13 to alanine produced an OAP fraction of 17 ± 3% at 10°C, whereas mutation of Cys17 did not increase OAP formation when compared to native M1. M23 diffusion did not change between 10°C and 37°C, corresponding to an OAP fraction between 99% and 95%, but decreased to 87 ± 3% at 50°C (Fig. 2 B).
Figure 2.
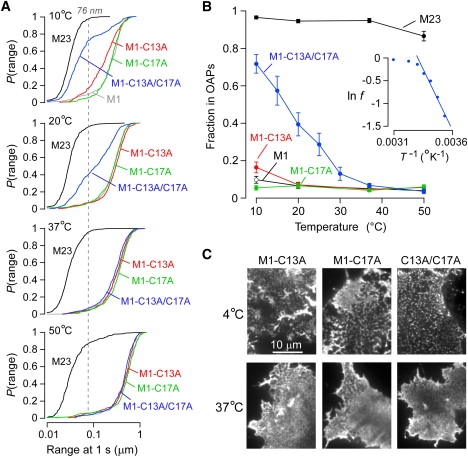
Temperature-dependent OAP assembly of a double cysteine mutant of AQP4-M1. (A) P(range) for single cysteine mutants M1-C13A (red) and M1-C17A (green), and double cysteine mutant M1-C13A/C17A (blue) at indicated temperatures. P(range) for M23 (black) and M1 (gray) are shown for reference. Dashed line indicates the 95th percentile of the range of M23 at 20°C. (B) Estimated fraction of M23 (black), M1 (white), M1-C13A (red), M1-C17A (green), and M1-C13A/C17A (blue) forming OAPs versus temperature, based on the 95th percentile of the range for M23 at 20°C. Error bars represent the mean ± SE. (Inset) Natural log of the fraction of free M1-C13A/C17A tetramers plotted against the inverse of absolute temperature. (C) TIRF micrographs of Alexa-labeled COS-7 cells transfected with M1-C13A (left), M1-C17A (middle), or M1-C13A/C17A (right) and fixed at 4°C (upper) or 37°C (lower). Scale bar, 10 μm.
The ability to estimate the fraction of AQP4 tetramers that are free versus the fraction bound in OAPs allowed computation of the standard Gibbs free energy, ΔG°, for addition of one tetramer to a preexisting OAP. Fig. 2 B (inset) shows a plot of the log of the fraction of free AQP4 tetramers, ln f, versus 1/T (see Appendix). For the double cysteine mutant M1-C13A/C17A, the relationship yielded a linear fit below 25°C, where large OAPs coexist with free tetramers, giving a ΔG° of −11 ± 1 kcal/mol over this temperature range. At higher temperatures, where OAP content is low or existing OAPs are considerably smaller, the model is not valid.
To confirm that the reduced M1-C13A/C17A diffusion at reduced temperature was due to OAP formation, TIRF microscopy was done on fixed, heavily labeled cells. Fixation at 4°C versus 37°C gave grossly different patterns. As in the case of native M1, cells transfected with single cysteine mutants showed the appearance of channel-like structures at 4°C, but without the distinct clusters seen in M23-transfected cells. The double cysteine mutant, however, showed well demarcated, diffraction-limited puncta, corresponding to submicron-sized OAPs (Fig. 2 C, upper). In cells fixed at 37°C, all cysteine mutants showed a similar appearance (Fig. 2 C, lower), similar to that seen in Fig. 1 B (upper) for native M1.
Having established the ability of a mutant of M1 to form OAPs at low temperature, we investigated whether previously discovered OAP-null mutants of M23 (22) could also be induced to form OAPs at low temperature. We postulated that certain mutations in M23 might weaken their intermolecular associations and hence prevent OAP formation at 37°C but allow OAP formation at reduced temperatures. Two M23 mutants were studied: M23-F26Q and M23-G28P (Fig. 1 A). At 37°C, <20% of M23-F26Q and <10% of M23-G28P assembled in OAPs, but at 10°C the fractions increased to 86 ± 5% and 56 ± 6%, respectively (Fig. 3, A and B). The computed average ΔG° was −13 ± 2 kcal/mol for assembly of M23-F26Q at temperatures below 30°C (Fig. 3 B, inset). TIRF microscopy confirmed the formation of OAP clusters at reduced temperature for these two “weakly associating” M23 mutants (Fig. 3 C).
Figure 3.
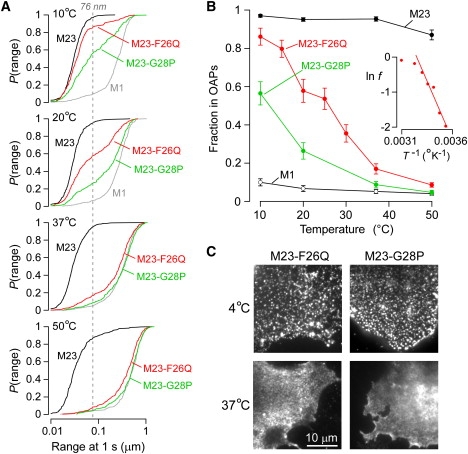
Temperature-dependent OAP assembly of weakly associating mutants of AQP4-M23. (A) P(range) for point mutants M23-F26Q (red) and M23-G28P (green) at the indicated temperature. P(range) for M23 (black) and M1 (gray) are shown for comparison. Dashed line indicates 95th percentile of the range of M23 at 20°C. (B) Estimated fraction of M23 (black), M1 (white), M23-F26Q (red), and M23-G28P (green) forming OAPs as a function of temperature. Error bars represent the mean ± SE. (Inset) Natural log of the fraction of free M23-F26Q tetramers plotted against the inverse of absolute temperature. (C) TIRF micrographs of Alexa-labeled COS-7 cells transfected with M23-F26Q (left) or M23-G28P (right) and fixed at 4°C (upper) or 37°C (lower). Scale bar, 10 μm.
Role of palmitoylation in native AQP4-M1
The appearance of OAPs at reduced temperature for the AQP4 mutant M1-C13A/C17A suggests the involvement of S-palmitoylation in OAP disruption by native M1. To further investigate the significance of palmitoylation, we measured OAP formation by M1 after exposure to 2-bromopalmitate (BrPA), which reduces palmitoylation of many membrane-associated proteins (29). After overnight exposure to BrPA, the fraction of native M1 associating into OAPs increased from <10% to 22 ± 3% at 10°C (Fig. 4 A). These data suggest that BrPA produced only partial depalmitoylation of M1. We reasoned that a larger effect might be seen after BrPA treatment of single cysteine mutants of M1. It was interesting to find that the effects of BrPA depended on the position of the remaining cysteine residue. With M1-C13A, incubation with BrPA modestly increased the OAP fraction from 17 ± 3% to 29 ± 5% at 10°C (Fig. 4 B). In contrast, BrPA exposure greatly increased OAP content of M1-C17A at 10°C from <10% to 47 ± 7% (Fig. 4 C). BrPA had no effect on native M1 or the single cysteine mutants at 37°C (Fig. 4). These data support the conclusion that S-palmitoylation is responsible for the differences observed between native M1 and cysteine mutants at low temperature. The differences observed between the individual cysteine mutants after exposure to BrPA provide insight into the mechanisms of AQP4-M1 palmitoylation (see Discussion).
Figure 4.
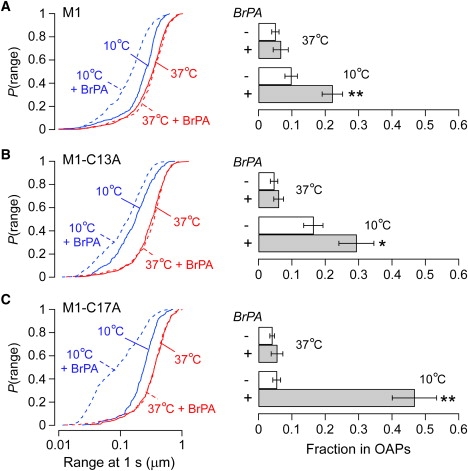
S-palmitoylation affects temperature-dependent assembly of AQP4-M1. (A) (Left) P(range) of native M1 at 37°C (red) and 10°C (blue) under control conditions (solid line) and after overnight incubation with 2-bromopalmitate (dashed line). (Right) Estimated fractions of AQP4 tetramers in OAPs with or without 2-bromopalmitate. ∗P < 0.05; ∗∗P < 0.01. (B) The same experiment as in A, but with single cysteine mutant M1-C13A. (C) The same experiment as in A, but with single cysteine mutant M1-C17A. Error bars represent the mean ± SE.
Membrane-density-dependent assembly of AQP4 in OAPs
The ability to modulate the assembly of certain AQP4 mutants with temperature suggests that OAP formation follows the thermodynamic principles of mass action. We postulated that OAP assembly of these AQP4 mutants should thus also be sensitive to their concentration in the plasma membrane. To modulate membrane density, cells were transfected with AQP4-encoding DNAs diluted with different amounts of empty plasmid, producing cells with differing levels of AQP4 in the plasma membrane. For M23, DNA dilutions by up to 100-fold did not change diffusion, even at 50°C (Fig. 5 A, left). In contrast, dilution of M1-C13A/C17A produced a remarkable increase in the diffusion at 10°C (Fig. 5 A, right), corresponding to significantly reduced OAP content (Fig. 5 B, lower). Dilution of native M23 or mutant M1 had no effect at 37°C (Fig. 5 B). Surface staining of (nonpermeabilized) transfected cells confirmed that dilution of the AQP4-encoding DNA produced an approximately equal reduction in AQP4 membrane expression (Fig. 5 C). Thus, the OAP assembly of a weakly associating AQP4 mutant could be modified by both temperature and membrane density.
Figure 5.
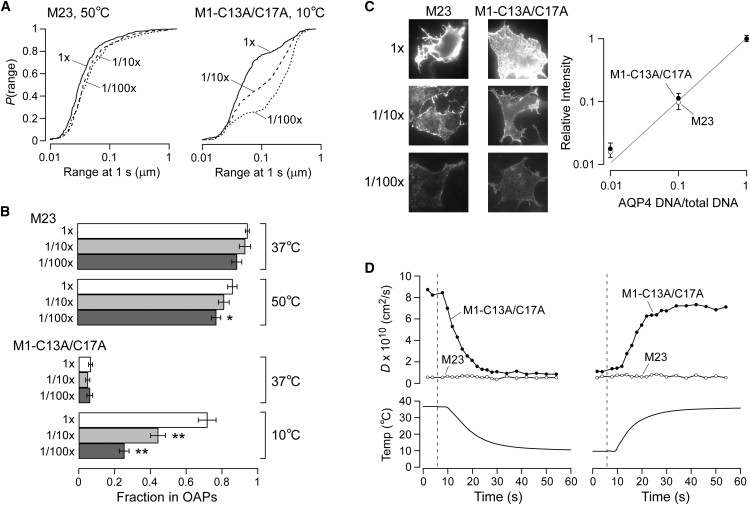
Membrane density-dependence and kinetics of assembly of AQP4 in OAPs. (A) P(range) of native M23 at 50°C (left) and M1-C13A/C17A at 10°C (right) without (solid line) and with (dashed line) dilution of the AQP4-encoding plasmid DNA by 10× (dashed line) or 100× (dotted line) with empty plasmid. (B) Estimated fraction of M23 (upper) and M1-C13A/C17A (lower) assembled in OAPs at indicated temperature and relative AQP4 concentration. ∗P < 0.05; ∗∗P < 0.01. Error bars represent the mean ± SE. (C) (Left) Fluorescence micrographs after surface labeling of COS-7 cells with Alexa Fluor 488 (intensity enhanced to visualize cells at 100× dilution). (Right) Average relative intensities versus AQP4 dilution ratios for M23 (open circles) and M1-C13A/C17A (solid circles). Error bars represent the mean ± SE. (D) Average diffusion coefficient (upper) of M23 (open circles) and M1-C13A/C17A (solid circles), and solution temperature (lower), versus time during rapid temperature drop or rise. Dashed lines indicate the time of switching between warm and cold perfusates.
Rapid kinetics of OAP assembly and disassembly
Last, we investigated the kinetics of OAP assembly and disassembly. A perfusion chamber, previously described (30), was modified to allow for rapid changes in perfusate temperature, with simultaneous monitoring of AQP4 diffusion in real time. Perfusates were rapidly switched between solutions maintained at high and low temperatures. A thermister probe was positioned in line with the solution exiting the chamber to monitor temperature. Fig. 5 D shows the kinetics of M1-C13A/C17A diffusion in response to temperature changes. It is interesting that the assembly and disassembly of OAPs was very rapid, occurring on a timescale of seconds or less, within the resolution time of our ability to change temperature. These results indicate rapid reversibility of OAP assembly in live cells.
Discussion
We found here that certain variants of AQP4, including AQP4 mutants and posttranslationally modified native AQP4, can undergo rapid and reversible OAP assembly at the plasma membrane in live cells. The identification of weakly associating AQP4 tetramers challenges the generally accepted view that OAPs are fixed and stable. OAP assembly of the weakly associating AQP4 tetramers could be modulated by temperature and membrane density, providing the first information, to our knowledge, about the energetics of OAP assembly in live cells.
We previously reported that cysteine replacement mutants of AQP4-M1 diffuse identically to native M1 in live cells at 37°C (22), providing evidence against OAP formation by the palmitoylation-null mutant M1-C13A/C17A. We found here that M1-C13A/C17A can form OAPs at reduced temperatures in live cells (Fig. 2). Studies with an inhibitor of S-palmitoylation, BrPA, confirmed that OAP formation by the cysteine mutants at low temperature is due to reduced palmitoylation (Fig. 4). The mechanism by which palmitoylation prevents OAP formation by M1 at low temperature is not known. We previously speculated that residues upstream from Met23 nonspecifically block tetramer-tetramer contacts near Met23, and that these interactions occur close to the cytoplasmic face of the plasma membrane (22). Lipid additions in the N-terminus of M1 might anchor the disrupting domain to the face of the membrane, which could efficiently interfere with the OAP-forming interactions near Met23.
A single palmitoylatable cysteine can prevent OAP formation by M1 at low temperature; however, the efficiency of Cys13 in disrupting OAPs differed from that of Cys17 (Fig. 4). At 10°C, M1-C13A showed more restricted diffusion than M1-C17A and native M1, suggesting that under normal conditions Cys17 is only partially palmitoylated, whereas Cys13 is largely or fully palmitoylated. Furthermore, incubation with BrPA resulted in only an ∼2-fold increase in OAP content with M1-C13A and native M1, as compared to an eightfold increase with M1-C17A. Together, these data suggest that Cys17 is less prone to (de)palmitoylation than Cys13. The mechanism of S-(de)palmitoylation is not well understood: some data suggest that the reaction occurs spontaneously in the absence of enzymes, whereas conflicting data indicate the necessity of an acyltransferase or thioesterase (31). It is established that the target cysteine must be deprotonated before transfer of the acyl group from palmitoyl-coenzyme A. Lys12 may provide a local base for more efficient deprotonation at Cys13 compared to Cys17. Another possibility is that the position of Cys17 makes it less accessible to an acyltransferase or thioesterase.
These findings may explain the apparent difference of our initial observation that M1-C13A/C17A does not form OAPs in live cells from observations of groups using FFEM in fixed cells (23) or blue native gel electrophoresis of cell lysates (28), who reported OAP formation by this mutant. Cell fixation or lysis at reduced temperature would explain the observed OAPs in those studies. We also show here that OAP formation by M1-C13A/C17A can be modulated by membrane concentration (Fig. 5). Cells expressing very high levels of mutant AQP4 might form some OAPs, even with samples prepared at 37°C. Our data show that under physiological conditions of AQP4 expression and temperature, prevention of OAP formation by N-terminal residues in M1 does not require palmitoylation. Further studies are needed to explore the possibility that lipid modification of M1 modulates OAP formation by M23 at physiological temperature.
Weakly interacting point mutants of AQP4-M23 can also be induced to form OAPs at reduced temperature (Fig. 3). These mutants presumably disrupt efficient N-terminus tetramer-tetramer interactions responsible for the assembly of native M23 into stable, strongly associating OAPs. Because the M23-F26Q mutation significantly reduces the overall hydropathy of the M23 N-terminus, we previously speculated that the interaction responsible for OAP formation is primarily hydrophobic (22). Our data here support that notion, as hydrophobic interactions can be highly temperature-dependent (32). Proline substitution at Lys27 or Gly28 fully disrupts OAP formation at 37°C (22), suggesting that formation of secondary structure, such as a short amphipathic helix, may be involved in OAP formation. We show here that at 10°C M23-G28P can assemble in OAPs (Fig. 3), providing evidence against the involvement of a helix in the OAP-forming tetramer-tetramer interaction, since proline mutation would prevent helix formation at all temperatures. More likely, proline forms a rigid kink in the unstructured N-terminus of M23, preventing proper positioning of hydrophobic residues for interaction with adjacent tetramers. At low temperatures, the steric impairment may be overcome partially by the increased favorability of hydrophobic contact, resulting in weakly assembled OAPs.
An interesting finding is that assembly and disassembly of M1-C13A/C17A into OAPs is very rapid (Fig. 5 D), occurring over a few seconds or less, faster than the kinetics of temperature change in our system. OAP assembly requires the diffusion of individual AQP4 tetramers to coalesce, nucleate, and grow into large stable arrays. In a previous study using FFEM of M1 and M23 in COS-7 cells, we showed a density of ∼73 AQP4 tetramers/μm2 (21), corresponding to an average separation of ∼141 nm between the centers of randomly distributed tetramers. AQP4 tetramers assembled in OAPs have a separation of ∼8 nm between centers. Quantitative surface staining of M1-C13A/C17A indicated AQP4 expression levels similar to native M23. Using a measured average diffusion coefficient for unassembled AQP4 of 4 × 10−10 cm2/s, we compute that AQP4 tetramers diffuse an average range of 1400 nm in 8 s, which is the half-time for the temperature drop in Fig. 5 D. AQP4 tetramer diffusion is thus sufficiently rapid to account for the rapid OAP assembly and disassembly. Nucleation and growth of OAPs must also occur over a few seconds or less.
The ability of mutant and posttranslationally modified AQP4 molecules to assemble into OAPs at reduced temperature provides insight into the energetics of AQP4 supramolecular assembly in live cells. The intermolecular interactions between native M23 tetramers responsible for OAP formation must be considerably stronger than those of the mutants studied here (∼12 kcal/mol), as ∼90% of M23 tetramers assembled in OAPs at 37°C even at a very low membrane density of <1 AQP4 tetramer/μm2 (Fig. 5 B). Also, native M23 OAPs are largely resistant to modulation by temperature (Fig. 1). FFEM has provided useful data on AQP4 OAPs in primary and transfected cell cultures for >30 years, and blue native gel electrophoresis was recently introduced as a technically simple alternative to deduce AQP4 OAP assembly (33). Our results suggest that these techniques, which often involve fixation or cell lysis at reduced temperature, are suitable for the study of OAPs formed by native AQP4 isoforms, but may be unsuitable for the study of certain mutant and translationally modified AQP4 water channels. Atomic force microscopy has been applied to generate high-resolution images of AQP0 arrays in intact lens membranes (34) and so might provide another approach to study AQP4 OAPs in a temperature-controlled, nonfixed system.
Appendix
Consider the stepwise formation of an orthogonal array An, where n is the number of AQP4 tetramers in the assembly:
| (5) |
The association constant Keq for adding one tetramer to a preexisting array is then
| (6) |
where Ci is the concentration of arrays of size i. If the membrane is populated with many large arrays, the addition of a single tetramer to an existing array will minimally alter the overall array concentration, such that Cn+1 ≈ Cn. Equation 6 then becomes
| (7) |
where f is the fraction of AQP4 tetramers that are free, CT is the total concentration of AQP4 tetramers in the membrane, ΔG° is the standard Gibbs free energy, R is the universal gas constant, and T is the absolute temperature. Rearrangement of Eq. 7 gives
| (8) |
A linear fit to a plot of ln f versus 1/T has slope ΔG°/R and intercept −ln CT, provided that the fit is restricted to the temperature range in which large OAPs coexist with individual tetramers, where Eq. 7 is valid. This linear range is below 25°C for M1-C13A/C17A and below 30°C for M23-F26Q (Figs. 2 B and 3 B, insets).
Acknowledgments
This work was supported by National Institutes of Health grants EB00415, HL73856, DK35124, EY13574, DK86125, and DK72517, a grant from the Guthy-Jackson Charitable Foundation, and Research Development Program and Drug Discovery grants from the Cystic Fibrosis Foundation. Dr. Crane was supported in part by National Institutes of Health, National Research Service award GM808512.
References
- 1.Hasegawa H., Ma T., Skach W., Matthay M.A., Verkman A.S. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J. Biol. Chem. 1994;269:5497–5500. [PubMed] [Google Scholar]
- 2.Jung J.S., Bhat R.V., Preston G.M., Guggino W.B., Baraban J.M. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA. 1994;91:13052–13056. doi: 10.1073/pnas.91.26.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frigeri A., Gropper M.A., Turck C.W., Verkman A.S. Immunolocalization of the mercurial-insensitive water channel and glycerol intrinsic protein in epithelial cell plasma membranes. Proc. Natl. Acad. Sci. USA. 1995;92:4328–4331. doi: 10.1073/pnas.92.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkman A.S., Binder D.K., Bloch O., Auguste K., Papadopoulos M.C. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim. Biophys. Acta. 2006;1758:1085–1093. doi: 10.1016/j.bbamem.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 5.Hiroaki Y., Tani K., Kamegawa A., Gyobu N., Nishikawa K. Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 6.Ho J.D., Yeh R., Sandstrom A., Chorny I., Harries W.E. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc. Natl. Acad. Sci. USA. 2009;106:7437–7442. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landis D.M., Reese T.S. Arrays of particles in freeze-fractured astrocytic membranes. J. Cell Biol. 1974;60:316–320. doi: 10.1083/jcb.60.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellisman M.H., Rash J.E., Staehelin L.A., Porter K.R. Studies of excitable membranes. II. A comparison of specializations at neuromuscular junctions and nonjunctional sarcolemmas of mammalian fast and slow twitch muscle fibers. J. Cell Biol. 1976;68:752–774. doi: 10.1083/jcb.68.3.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orci L., Humbert F., Brown D., Perrelet A. Membrane ultrastructure in urinary tubules. Int. Rev. Cytol. 1981;73:183–242. doi: 10.1016/s0074-7696(08)61289-9. [DOI] [PubMed] [Google Scholar]
- 10.Bordi C., Amherdt M., Perrelet A. Orthogonal arrays of particles in the gastric parietal cell of the rat: differences between superficial and basal cells in the gland and after pentagastrin or metiamide treatment. Anat. Rec. 1986;215:28–34. doi: 10.1002/ar.1092150105. [DOI] [PubMed] [Google Scholar]
- 11.Wolburg H. Orthogonal arrays of intramembranous particles: a review with special reference to astrocytes. J. Hirnforsch. 1995;36:239–258. [PubMed] [Google Scholar]
- 12.Yang B., Brown D., Verkman A.S. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J. Biol. Chem. 1996;271:4577–4580. [PubMed] [Google Scholar]
- 13.Verbavatz J.M., Ma T., Gobin R., Verkman A.S. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J. Cell Sci. 1997;110:2855–2860. doi: 10.1242/jcs.110.22.2855. [DOI] [PubMed] [Google Scholar]
- 14.Yang B., van Hoek A.N., Verkman A.S. Very high single channel water permeability of aquaporin-4 in baculovirus-infected insect cells and liposomes reconstituted with purified aquaporin-4. Biochemistry. 1997;36:7625–7632. doi: 10.1021/bi970231r. [DOI] [PubMed] [Google Scholar]
- 15.Silberstein C., Bouley R., Huang Y., Fang P., Pastor-Soler N. Membrane organization and function of M1 and M23 isoforms of aquaporin-4 in epithelial cells. Am. J. Physiol. Renal Physiol. 2004;287:F501–F511. doi: 10.1152/ajprenal.00439.2003. [DOI] [PubMed] [Google Scholar]
- 16.Amiry-Moghaddam M., Otsuka T., Hurn P.D., Traystman R.J., Haug F.M. An α-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. USA. 2003;100:2106–2111. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H., Verkman A.S. Evidence against involvement of aquaporin-4 in cell-cell adhesion. J. Mol. Biol. 2008;382:1136–1143. doi: 10.1016/j.jmb.2008.07.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatton J.D., Ellisman M.H. Orthogonal arrays are redistributed in the membranes of astroglia from alumina-induced epileptic foci. Epilepsia. 1984;25:145–151. doi: 10.1111/j.1528-1157.1984.tb04170.x. [DOI] [PubMed] [Google Scholar]
- 19.Schotland D.L., Bonilla E., Wakayama Y. Freeze fracture studies of muscle plasma membrane in human muscular dystrophy. Acta Neuropathol. 1981;54:189–197. doi: 10.1007/BF00687741. [DOI] [PubMed] [Google Scholar]
- 20.Nicchia G.P., Mastrototaro M., Rossi A., Pisani F., Tortorella C. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. 2009;57:1363–1373. doi: 10.1002/glia.20855. [DOI] [PubMed] [Google Scholar]
- 21.Crane J.M., Van Hoek A.N., Skach W.R., Verkman A.S. Aquaporin-4 dynamics in orthogonal arrays in live cells visualized by quantum dot single particle tracking. Mol. Biol. Cell. 2008;19:3369–3378. doi: 10.1091/mbc.E08-03-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crane J.M., Verkman A.S. Determinants of aquaporin-4 assembly in orthogonal arrays revealed by live-cell single-molecule fluorescence imaging. J. Cell Sci. 2009;122:813–821. doi: 10.1242/jcs.042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki H., Nishikawa K., Hiroaki Y., Fujiyoshi Y. Formation of aquaporin-4 arrays is inhibited by palmitoylation of N-terminal cysteine residues. Biochim. Biophys. Acta. 2008;1778:1181–1189. doi: 10.1016/j.bbamem.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Fujiwara T., Ritchie K., Murakoshi H., Jacobson K., Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 2002;157:1071–1081. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxton M. Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 1997;72:1744–1753. doi: 10.1016/S0006-3495(97)78820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haggie P., Kim J., Lukacs G., Verkman A. Tracking of quantum dot-labeled CFTR shows near immobilization by C-terminal PDZ interactions. Mol. Biol. Cell. 2006;17:4937–4945. doi: 10.1091/mbc.E06-08-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furman C.S., Gorelick-Feldman D.A., Davidson K.G., Yasumura T., Neely J.D. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc. Natl. Acad. Sci. USA. 2003;100:13609–13614. doi: 10.1073/pnas.2235843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strand L., Moe S.E., Solbu T.T., Vaadal M., Holen T. Roles of aquaporin-4 isoforms and amino acids in square array assembly. Biochemistry. 2009;48:5785–5793. doi: 10.1021/bi802231q. [DOI] [PubMed] [Google Scholar]
- 29.Webb Y., Hermida-Matsumoto L., Resh M.D. Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem. 2000;275:261–270. doi: 10.1074/jbc.275.1.261. [DOI] [PubMed] [Google Scholar]
- 30.Solenov E., Watanabe H., Manley G.T., Verkman A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Cell Physiol. 2004;286:C426–C432. doi: 10.1152/ajpcell.00298.2003. [DOI] [PubMed] [Google Scholar]
- 31.Dietrich L.E., Ungermann C. On the mechanism of protein palmitoylation. EMBO Rep. 2004;5:1053–1057. doi: 10.1038/sj.embor.7400277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dill K.A., Bromberg S. Garland Science; New York: 2003. Molecular Driving Forces: Statistical Thermodynamics in Chemistry and Biology. [Google Scholar]
- 33.Sorbo J.G., Moe S.E., Ottersen O.P., Holen T. The molecular composition of square arrays. Biochemistry. 2008;47:2631–2637. doi: 10.1021/bi702146k. [DOI] [PubMed] [Google Scholar]
- 34.Buzhynskyy N., Hite R.K., Walz T., Scheuring S. The supramolecular architecture of junctional microdomains in native lens membranes. EMBO Rep. 2007;8:51–55. doi: 10.1038/sj.embor.7400858. [DOI] [PMC free article] [PubMed] [Google Scholar]