

Abstract
Copy number variation (CNV) is pervasive in the human genome and can play a causal role in genetic diseases. The functional impact of CNV cannot be fully captured through linkage disequilibrium with SNPs. These observations motivate the development of statistical methods for performing direct CNV association studies. We show through simulation that current tests for CNV association are prone to false-positive associations in the presence of differential errors between cases and controls, especially if quantitative CNV measurements are noisy. We present a statistical framework for performing case-control CNV association studies that applies likelihood ratio testing of quantitative CNV measurements in cases and controls. We show that our methods are robust to differential errors and noisy data and can achieve maximal theoretical power. We illustrate the power of these methods for testing for association with binary and quantitative traits, and have made this software available as the R package CNVtools.
The advent of technologies to probe DNA copy number genome-wide has led to rapid progress in the understanding of how segments of the genome can vary in copy number between individuals1,2. In addition, there are multiple strands of evidence indicating that this copy number variation (CNV) can have an appreciable biological impact. CNVs frequently have a causal role in severe developmental syndromes and familial diseases3, CNVs can perturb gene expression within and flanking the CNV4 and CNVs can confer susceptibility to infectious and complex diseases5-7.
In light of the ever-increasing rate of discovery of CNVs throughout the human genome, and the growing appreciation for their potential role in complex disease, rapid growth in studies investigating associations between CNVs and complex diseases is likely. The development of this nascent field is critically dependent on robust statistical strategies for identifying meaningful associations. The statistical challenges inherent within CNV-association testing are substantially different from those for CNV discovery8.
A review of the sparse literature that exists on CNV-disease associations reveals that the underlying data are often substantially noisier than for SNP genotyping, largely as a result of poor discrimination of the underlying discrete copy numbers, and yet the statistical methods being applied are typically less sophisticated. Although some CNV-disease association studies simply assay presence or absence of specific copy number alleles9, most published studies rely on quantitative assessments, often crude, of the diploid copy number6,7,10. Most frequently, real time-PCR assays for known CNVs are applied to case and control groups and individuals are then binned into copy number classes using pre-defined thresholds. These classes represent diploid copy numbers (that is, the sum of the number of copies on each allele) rather than genotypes. Subsequently, nonparametric statistical tests (for example, χ2 test, trend test, Mann-Whitney test) are applied to the frequencies of the different copy number classes in the different groups. One previous study has shown that in the context of association with quantitative traits, it has been possible to identify robust associations by simply correlating the trait with the underlying quantitative CNV measurements without inferring copy number genotypes4. Although approaches based on direct testing of quantitative CNV measurements will often be appropriate for association with a quantitative trait in a single group of subjects, they are often not robust to the presence of differential errors between groups due to differences in DNA quality or handling. Thus, they will often be inappropriate in a case-control setting. Given a quantitative measurement of copy number, different diploid copy numbers are manifested as peaks, or clusters, in the distribution of measurements; the distribution of measurements will be a mixture of (often overlapping) bell-shaped curves. Direct tests of copy number measurements are sensitive to shifts in the mean and/or variance of the underlying distributions, and scoring copy number by simple binning will, in the presence of such shifts, lead to differential misclassification. Such analyses could generate many false-positive findings, especially in the context of genome-wide studies testing thousands of variants.
It is emerging that such shifts in the distribution of measurements occur widely in practice, even after careful normalization and calibration procedures have been applied to the raw observations. For example, Figure 1 shows examples of differential errors in CNV measurements from three different technologies: SNP genotyping, array comparative genome hybridization (array CGH) and a variant of quantitative PCR known as the paralog ratio test (PRT)11. In each case, shifts in the location of the clusters representing specific copy numbers between groups are readily apparent. In the first example, where both distributions are drawn from control groups from the same population, and no difference in copy number frequencies from either population structure or genetic association is expected, simple nonparametric statistical tests on the CNV measurement distributions show highly significant differences (Mann-Whitney test: P = 1.5 × 10−6; t-test: P=4.2×10−6). Similar bias has been shown to affect lower-quality SNP assays within SNP association studies12 and effective treatment is critical to avoid false-positive associations in poorerquality assays13. CNV data are typically of lower quality and present an additional challenge in that they often have more than two alleles, thus giving rise to more than three possible diploid copy numbers.
Figure 1.

Example of CNV data showing poor clustering quality and differential errors. (a) Comparison of the distribution of quantitative CNV measurements for a single CNV (W8177) in the two control groups of the WTCCC from Affymetrix 500K SNP genotyping data. (b) Comparison of the distribution of quantitative CNV measurements in array-CGH data (clone Chr15tp-11F12 on the Whole Genome TilePath array1) between the HapMap panel and the Human Genome Diversity Panel (HGDP). (c) Distribution of quantitative CNV measurements from a paralog-ratio-test assay for the β-defensin locus in Dutch and German control cohorts11.
RESULTS
We have considered six methods of increasing sophistication for testing for CNV-disease association. The first method is a nonparametric test (Mann-Whitney U test) for location shift between cases and controls of the distribution of quantitative CNV measurements (‘method 1’). The remaining five methods attempt to classify the individuals into different copy number classes, and to test for differences between the frequencies of these copy number classes between cases and controls (Fig. 2 shows these five methods schematically). The simplest of these classification methods involves simple binning of individuals into copy number classes on the basis of predefined thresholds, together with an association test on the resulting contingency table (‘method 2’).
Figure 2.

Methods for performing CNV-association testing. (a) In association studies, inference of genotypes from data and association testing of genotypic data are generally treated as separate statistical problems; however, the two underlying models can be combined into a single, integrated procedure. (b) Five different case-control association methods are represented schematically on simulated copy number intensity data in case and control groups. The first three methods classify individuals into copy number classes before performing nonparametric testing. Classification is achieved by either a priori binning or assignment on the basis of maximal a posteriori probability from mixture models fitted to the underlying intensity data. The new likelihood ratio test integrates classification and association testing into a single procedure by comparing mixture model fits under nested hypotheses.
Our next three methods closely mirror the conventional analyses for SNP genotyping; the quantitative CNV measurement distribution is modeled using a Gaussian mixture model and individuals are then assigned to copy number classes on the basis of their maximum a posteriori probability (see Methods for details). Again, an association test is then applied to the resultant contingency table. In the first of these, the Gaussian mixture model was fitted to the cases and controls combined and each subject was assigned a copy number, irrespective of the confidence of assignment (‘method 3’). This strategy was then adapted to address the problem of differential misclassification by fitting the mixture model independently in cases and controls, thus allowing for shifts in the underlying measurement distributions between groups (‘method 4’). The intuition that differential misclassification bias is removed by simply scoring cases and controls independently (‘method 4’) potentially carries a subtle flaw; in fitting the mixture model, differences in copy number frequency between groups is tacitly assumed, and this could lead to spurious inflation of any differences unless uncertainty implicit in the mixture modelling is correctly propagated into the later association test (discussed in detail in ref. 12). The next strategy was to assign copy numbers to subjects only if the posterior probability for the assignment exceeded a threshold (0.95, ‘method 5’). This last strategy was explored because it is widely used. Its use is probably suggested by the intuition that, by removing the most uncertain data, the bias caused by measurement error is minimized. However, experience of SNP genotyping brings this intuition into question, as application of stringent call quality filters can generate a different sort of bias as a result of nonrandom missingness.
To address the problems associated with fitting mixture models to cases and controls separately or independently, we developed a method to integrate CNV scoring (data model) and association testing (genetic model) into a single statistical model, and test for association using a likelihood ratio test (‘method 6’). Figure 3 illustrates the elements of this integrated model. It allows for direct influences of case or control group (phenotype) on the CNV measurement distribution, as well as the indirect association via copy number frequency. The likelihood ratio test compares maximized likelihoods for the model with or without an association between copy number and phenotype (shown as a broken arrow in the figure).
Figure 3.
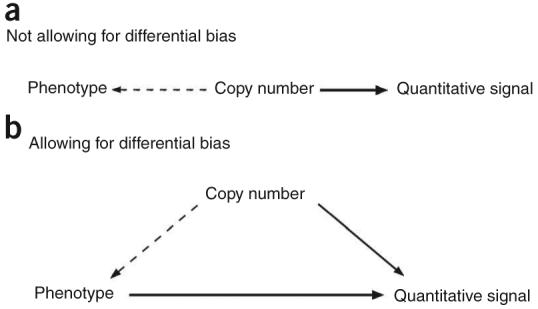
Modelling the dependency between copy number and disease. (a) Naïve model in which any dependency between disease phenotype and quantitative measurements of copy number is assumed to be due to differences in the distribution of copy number between cases and controls. (b) A more elaborate model that allows for other differences in measurement distribution between cases and controls due, for example, to differences in DNA qualities.
Method 1 is a test for a simple monotone relationship between disease risk and diploid copy number, and, for comparability, the remaining five methods have been implemented to be maximally sensitive to this type of relationship by computing trend tests (with 1 degree of freedom (d.f.)). Thus, in methods 2 through 5, we use the Cochran-Armitage test for trend in the contingency table obtained by assignment of subjects to diploid copy number classes. Similarly, method 6 yields a 1-d.f. test when the relationship between copy number and phenotype specified in the model has a simple linear-logistic form. These methods could also be adapted to test for nonlinear effects, analogous to ‘dominance’ terms in the classical biometric model. However, as when testing conventional markers, power is lost when such terms are small and the relationship is monotonic. Here we concentrate on 1-d.f. tests, although our integrated model approach can be generalized to other genetic models, and these methods are implemented in our software package.
We also note that systematic errors due to differing measurement distributions are not only related to case or control group membership. Experimental batch effects are often evident. Such effects can also lead to spurious associations, and should also be taken into account in the analysis. Our likelihood ratio testing framework can explicitly model these batch effects when the batches are large enough that the parameters of the mixture model within each batch can be robustly estimated.
False-positive rates of different CNV-association procedures
We carried out simulations to explore how differential errors and clustering quality influence the type 1 error of the six association testing methods outlined above (Supplementary Methods online).
We generated datasets that varied in signal-to-noise ratio (clustering quality, denoted by Q), as measured by the ratio of the separation of cluster means to the cluster s.d. (Supplementary Methods). We also explored the sensitivity of the type 1 error rate to small differences in cluster means and variances. Figure 4 shows to what degree the test statistics for all six methods are inflated when compared with their expected distributions. Even small location differences in the CNV measurement distribution between cases and controls can lead to massively inflated type 1 error if Mann-Whitney testing or a priori binning are used. Copy number assignment using mixture models performs better, particularly when cases and controls are scored independently (method 4), but appreciable overdispersion remains. This inflation of the test statistic in method 4 results from over-estimating the confidence of copy number assignment through constructing a contingency table, and from overestimating the differences in copy number frequencies between cases and controls through fitting the mixture model to cases and controls separately, which allows the nuisance parameters to vary between the two models. This is effectively equivalent to fitting mixture models under the alternate hypothesis that copy number frequencies do indeed differ between cases and controls. As a result, the true variance of the score test statistic is greater than the naïve estimate. By contrast, the likelihood ratio test (method 6) estimates all parameters under the null (no copy number differences) and alternate (copy number differences exist) hypotheses and thus provides the most robust test. As expected, imposing stringent call thresholds (method 5) does not remove the overdispersion, but rather exacerbates it.
Figure 4.

Sensitivity of 1-d.f. association testing methods to clustering quality and differential errors between cases and controls in simulated data. Six alternative association methods are considered: (i) Mann-Whitney testing for difference in location of CNV measurement distributions, (ii) χ2 trend tests on data binned with a priori thresholds, (iii) χ2 trend tests on mixture model assignment of case and controls together (MM-C), (iv) χ2 trend tests on mixture model assignment of case and controls separately (MM-S), (v) χ2 trend tests on high confidence mixture model assignment of case and controls separately (MM-S95) and (vi) likelihood ratio trend test. Overdispersion (λ) is estimated robustly from a linear fit to the first 90% of quantile-quantile plots from 1,000 simulated datasets. (a) Overdispersion is estimated for alternative association methods at ten different clustering qualities. Density plots for three clustering qualities are shown at the bottom. (b) Overdispersion is estimated for alternative association methods at ten different values of differential shift of means. Density plots for three values of differential shift are shown at the bottom with case and control groups in red and gray. (c) Overdispersion is shown for alternative association methods at ten different values of differential shifts in variance. Density plots for three values of differential shift are shown at the bottom with case and control groups in red and gray.
We also investigated the performance of these methods in empirical CNV data in which we expected no true associations to exist. For this purpose, we analyzed 95 known CNVs (Supplementary Table 1 online) from Affymetrix 500K SNP genotyping data collected on two UK control populations, each of ∼1,500 individuals, as part of the Wellcome Trust Case Control Consortium (WTCCC)14. In one group (the 1958 Birth Cohort sample) DNA was obtained from EBV-transformed cell lines while, from the other (UK National Blood Service controls), DNA was from fresh blood. Association tests on these 95 CNVs, which differ in numbers of alleles, clustering quality and allele frequencies, show substantially less overdispersion of χ2 statistics using the likelihood ratio trend test (λ = 1.1) as compared to Cochran-Armitage testing of separate mixture model assignment of the same CNVs with (λ = 1.74) or without (λ = 1.58) allowing for differential errors (Supplementary Fig. 1 online). This overdispersion is significantly lower for the likelihood ratio trend test (P < 0.05 for Wilcoxon signed rank test comparing test statistics produced by the likelihood ratio test and either mixture model assignment method), but is not significantly different between the two mixture model assignment methods (P > 0.05). The small degree of overdispersion observed in the likelihood ratio test statistics was not statistically significant.
Within the integrated model framework it is also possible to carry out a likelihood ratio test for difference in cluster means and variances between the two groups (Supplementary Methods). We first showed, in simulations in which there were no CNV measurement distribution differences, that this likelihood ratio test statistic had the expected χ2 distribution. In contrast, in the WTCCC data these statistics are considerably overdispersed, demonstrating that differential errors are pervasive in this empirical example (Supplementary Fig. 2 online). The same test also shows highly significant differential bias in the array-CGH and PRTexamples shown in Figure 1 (P = 1.2×10−6 and 1.1 × 10−12 for panels B and C, respectively). These observations confirm that the features of copy number data modeled in the simulations are indeed present in empirical data from different platforms, including SNP genotyping, array-CGH and PRT datasets, and that accounting for differential errors is essential for robust CNV-association testing.
Maximizing information from probes in the same CNV
All the association methods described above require a single measure to discriminate between different copy numbers. However, many CNV assay methods use multiple probes in each CNV (for example, each CNV in the Affymetrix 500K SNP genotyping data is identified by multiple SNP probe sets), so some method for summarizing these measurements is necessary. The obvious approaches are to use the mean or median. However, these are not optimal, as different probes differ in their informativeness, not least because copy number region boundaries can be uncertain. We developed two improved procedures to weight the information from each probe within each CNV region (Supplementary Methods). The first procedure is to use the first principal component from the intensity data from different probes. This, by downweighting probe intensities uncorrelated with the remainder, generally gives a better separation of different copy numbers than the mean or median of all of the probe intensities. However, we suspected that the weights would still not be optimal, and developed a one-step refinement of these scores: we fitted the Gaussian mixture model to the principal-component scores and then used the estimated CNV assignments to compute an optimal linear discriminant function of probe intensities. We demonstrated that these procedures resulted in improved clustering quality across these 95 CNVs in the Affymetrix 500K data from the WTCCC (Fig. 5a and Supplementary Fig. 3 online). We suggest that such procedures will have general utility for many applications (for example, array CGH) where multiple probes identify the same variant. As expected, the improved summary methods provide considerable protection against overestimation of CNV boundaries (Supplementary Fig. 4 online). Given that the vast majority of known CNVs do not currently have precisely mapped breakpoints, being able to overestimate the extent of CNVs without seriously downgrading measurement quality is a significant advantage.
Figure 5.
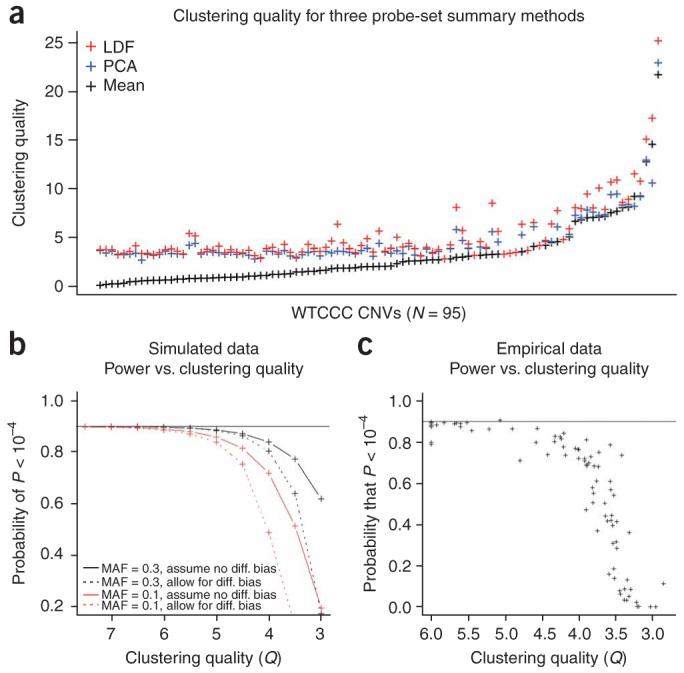
Statistical power of the likelihood ratio trend test. (a) Clustering quality resulting from alternative probe summary methods for 95 CNVs: linear discriminant function (LDF), principal components analysis (PCA) and arithmetic mean (mean). (b) Statistical power of the LR trend test in simulated data of varying clustering quality is shown for two minor allele frequencies (MAF) with odds ratios (OR) set to equalize maximal theoretical power at 90%. Power is estimated for 2,000 cases and 2,000 controls under two conditions: (i) a model that assumes no differential errors and (ii) a model allowing for differential errors. (c) Statistical power of the LR trend test in empirical data from 95 CNVs of varying clustering quality. Power is estimated for 2,000 cases and 2,000 controls, with odds ratios (OR) set to equalize maximal theoretical power at 90%. For ease of display, where the clustering quality (Q) of a CNV exceeds a value of 6, it has been set to 6.
Power estimation
We then assessed the statistical power of the likelihood ratio trend test using simulated data across a range of signal-to-noise ratios. The statistical power of the likelihood ratio trend test can be estimated by a quadratic approximation of the profile likelihood (see Methods). We observed that when copy number clusters are discrete the likelihood ratio trend test achieves the maximum theoretical power, but that power falls off rapidly with decreasing clustering quality (Fig. 5b). The loss of power is much more pronounced when the model allows for different measurement properties between cases and controls, which reflects the increasing difficulty in distinguishing between association and differences in measurement properties as clustering quality declines. This result is replicated in the empirical data on 95 CNVs described above (Fig. 5c). We noted that this marked fall-off in power is even more pronounced when copy number frequencies are low, owing to the increased difficulty of accurately modelling measurement distributions.
Although it could be argued that the more serious loss of power due to the need to model differential errors points to a need for better study design rather than additional statistical sophistication, such effects are very difficult (and often impossible) to exclude. Rarely can cases and controls be approached in strictly comparable circumstances that ensure identical DNA handling. Moreover, prospective group studies will rarely yield sufficient numbers of cases of disease to detect modest effect sizes. Family-based association studies will, perhaps, face fewer difficulties in this respect.
Quantitative traits
We generalized the likelihood ratio trend test for use in quantitative trait association by replacing the logistic regression for dependency between copy number and phenotype in our model by a simple linear regression (LR-QT test). Although studies of quantitative traits are often carried out in a manner that effectively excludes the differential errors that largely concern us here, this may not always be the case; notably, differential errors can be introduced by experimental batches, which may be confounded with the trait (for example, when extremes of the trait distribution are targeted). Although careful study design may control type 1 errors, we have also shown by simulation (Supplementary Fig. 5 online) that our approach, without allowance for measurement distribution differences, is more powerful than simple tests on the CNV measurements based on linear regression4. This advantage is maintained over a wide range of signal-to-noise ratios (clustering qualities).
Empirical examples of positive associations
We explored the performance of the likelihood ratio trend test on known CNV associations for both binary disease traits and quantitative traits in empirical data. Type 1 diabetes (T1D) is known to be strongly associated to the MHC class I region, which contains several CNVs and across which there is long-range linkage disequilibrium. Therefore, we should expect to see indirect association between T1D and these CNVs. We confirmed that the likelihood ratio test does indeed identify a highly significant association (P = 0.001) with a CNV in the class I MHC that can be detected in the WTCCC data (Fig. 6a).
Figure 6.

Examples of empirical CNV associations. (a) Association with a binary disease trait, type 1 diabetes (T1D). The red shaded area represents a density plot of copy number measurement in each group. The two WTCCC control groups come from the 1958 Birth Group (1958BC) and the National Blood Service (NBS). The colored lines reflect the posterior probability distribution for each mixture in the fitted mixture model. The P value derives from the LR trend test comparing case and control groups. (b) The first panel shows normalized expression of gene LOC288077 against copy number measurement, with a linear regression shown in blue. The second panel shows normalized gene expression against mixture model assignment, with a linear regression shown in blue. The P values in these two plots represent the nominal P values on the regression. The third panel shows a histogram of copy number measurement and the colored lines represent the posterior probability distribution for each of the five copy number classes in the fitted mixture model used in the LR trend test.
We also applied our likelihood ratio method to published case-control data used to support an association between copy number of β-defensin genes and psoriasis11. These PRT data comprise cases and controls from two populations, Dutch and German. We observed clear evidence for different measurement properties (batch effect) in the German controls relative to Dutch controls (Fig. 1c). Moreover, the German but not the Dutch data show significant P values for differential bias between cases and controls (3.6×10−6 and 0.38, respectively). Nevertheless, both Dutch and German populations show significant evidence of association (P = 0.002 and 0.02, respectively). Upon joint modelling of all four collections, in which we allow the locations and variances of copy number components to vary between all four groups and assume that the magnitude of effect is the same in both populations, we obtain a P value of 6.5 × 10−5. Thus, our analysis suggests that although differential errors between groups are clearly present in these data, the published association is not attributable to differential errors, and illustrates the potential for increasing power by applying our method to combining case-control data across datasets.
Finally, we applied the likelihood ratio test for quantitative trait association to a previously published association4 between a multiallelic CNV detected by array CGH and gene expression in the HapMap lymphoblastoid cell lines, and we demonstrated that the likelihood ratio test gives increased power over and above the nominal P values obtained from linear regression of normalized gene expression against either the intensity data or the mixture model assignment (Fig. 6b), thus corroborating our simulation results.
DISCUSSION
In summary, we have shown that, in a case-control setting, existing strategies for detecting meaningful CNV association with binary disease phenotypes are confounded by differential errors and poor clustering quality, and we have developed novel methods that are robust to both these factors. However, we have also shown that allowance for differential errors can come at a heavy cost in reduced power unless the signal-to-noise ratio of the assay is high. Our method for summarizing measurements from multiple probes in a CNV region is also highly effective and potentially of general utility in other settings where independent continuous measurements are made of an underlying discrete genetic variant. Software to perform all these analyses is freely available in the form of an R package (‘CNVtools’) and its computational efficiency is such that it would be feasible to use this software to test for CNV associations genome-wide (>10,000 loci).
Our methods accommodate both biallelic and multiallelic CNVs, have been generalized for association with quantitative traits, and are suitable for all methods of assaying CNV quantitatively, including quantitative PCR, SNP genotyping intensities and array CGH. Although in principle CNVs can be assayed quantitatively, by a broad range of experimental methods15, or qualitatively, through the presence or absence of specific-allelic structures, in practice qualitative assays tend to be difficult to design and not easily amenable to multiplexing16,17, and so we envisage that quantitative CNV assay methods will dominate as we move toward methods for assaying CNV genome-wide.
Our new methods can model large batch effects that do not follow group membership, which is an important consideration in large-scale association studies. This enables data from different sources, and indeed different platforms, to be combined, for example, if data on cases and controls have been collected using different assays, or indeed if data within a group comes from different sources. This provides added flexibility for performing CNV association studies using common control datasets.
Two minor potential limitations of our new methods are that, in common with general experience of complex mixture models, they require a reasonable signal-to-noise ratio and large sample sizes. However, we have demonstrated that the power to detect common CNV-disease associations becomes negligible before the stability of our estimation procedure breaks down, so that these problems should not be a constraint in studies with sufficient sample sizes to detect realistic effect sizes for common diseases. We would argue that, given the sensitivity to differential errors of simpler approaches, it is highly unlikely that any statistical method will provide robust CNV-association testing on intensity data in the case-control setting when appropriate mixture models cannot be robustly fitted.
As noted, the integrated LR testing framework that we have developed can be further adapted to incorporate different models for the relationship between diploid copy number and risk. This flexibility is implemented in our software. Large measurement variations between relatively small batches remain a difficulty; one could envisage extension of our method to incorporate random batch effects, but this would impose a considerable additional computation burden and would only be applicable in order to increase power in settings where the design ensured that batch effects would not confound the case-control comparison. We also foresee that integrating imputation from neighboring SNPs18 with copy number intensity data into unified mixture models should provide additional improvements in CNV-association testing in settings where tagging of CNVs is imperfect and high-quality assays are not available.
Given the nascent nature of the CNV-association field, and the great potential for reports of spurious associations, we recommend that any future CNV association study should explore the potential for bias due to different CNV measurement properties between groups and, where possible, correct this by use of procedures such as we have described here. As the technologies for assaying CNV mature, data quality should improve markedly, lessening the danger of spurious association. However, although it is likely that the effect of such advances will be to render more CNVs testable, there will still be many CNVs for which the signal-to-noise ratio is borderline for safe inference.
METHODS
Data
The Affymetrix 500K data were drawn from the Wellcome Trust Case Control Study (WTCCC) and are described in more detail elsewhere14. The two control groups were each of ∼1,500 subjects. One was drawn from the 1958 Birth Cohort (also known as the National Child Development Study) of all births in Great Britain during one week in 1958, and the other was a sample of blood donors recruited as part of the WTCCC. The 2,000 type 1 diabetes (T1D) cases were recruited from pediatric and adult diabetes clinics across Great Britain. In the case of the 1958BC and T1D samples, DNA was extracted from Epstein-Barr virus (EBV)-transformed cell lines whereas, for the blood donors, DNA was extracted from white cells filtered from blood donations. SNP genotyping was done with the commercial release of the GeneChip 500K arrays at Affymetrix Services Lab. Data normalization is described in Supplementary Methods.
The Gaussian mixture model
We shall use the notation [] to denote a probability or probability density distribution. Parameters will follow a semicolon and will be denoted by Greek letters. We will denote the composite fluorescence measurement, combined over probe sets (see below), by x; the phenotype by y; the unobserved copy number by n; and extraneous covariates by z. Our model is based on the following factorization:
Thus the model has three component parts, which we shall refer to as the ‘signal model’, the ‘phenotype model’ and the ‘copy number model’. These are further defined as follows.
Signal model, [x|n, y, z; γ, δ]
For given copy number, the signal, x, is assumed to be normally distributed with mean and variance which depend on the copy number, n, but which may also depend on phenotype, y, and extraneous covariates z in order to allow for differences in DNA source and processing and for batch effects in array processing. The ‘signal mean model’ is a generalized linear model (GLM)19 with Gaussian errors and identity link function, whereas the ‘signal variance model’ is a GLM with gamma errors and logarithmic link functions. Different link functions could be chosen but have not been investigated in the work reported here. The parameters of these two GLMs are denoted by γ and δ. For example, a simple model would have
Phenotype model, [y|n, z; β]
The distribution of phenotype conditional copy number and, possibly, extraneous covariates, is again a GLM. For case-control data, binomial errors are assumed (because the phenotype is dichotomous) and the logit link is also assumed as this model remains invariant under case-control sampling (save for a change of intercept). Use of this ‘prospective’ model (that is, treating case-control status as the response variable) is counterintuitive but has a long history in epidemiology and has been shown to lead to correct asymptotic inference20. For a dichotomous phenotype the simple ‘trend’ model is
For a quantitative phenotype, this logistic regression model is replaced by a classical regression model with Gaussian errors and identity link function.
Copy number model, [n|z; α]
Copy number can take on values 0,1,...,N. Its distribution is assumed to be multinomial. In general we can envisage this distribution as depending on extraneous covariates (such as geographical region) via a multinomial regression model, but, currently, we have only implemented dependence on a single stratification.
Model fitting and association testing
The model can be fitted by the method of maximum likelihood. As copy number is unobserved, the log likelihood is
where i( = 1,...,S) indexes subjects. This can be maximized conveniently by a variation on the EM algorithm, sometimes termed the ECM algorithm21. As usual, the E step consists of taking expectations over the ‘posterior’ distribution of the missing data, ni(i = 1,...,S), given observed data and the current parameter estimates. In the M step, however, rather than maximizing the resultant averaged log likelihood with respect to all four sets of parameters, we execute one cycle of a Gauss-Seidal iteration, maximizing with respect to each set of parameters in turn. The complete algorithm is
E step
With current parameter estimates, calculate posterior probabilities for each possible value of the unobserved copy number, n, for each subject:
M step
Maximize the posterior expectation of the ‘complete data’ log likelihood
with respect to each set of parameters in turn.
To fit the signal mean model, maximize the expected log likelihood with respect to γ, by fitting the GLM for x with Gaussian errors and variance given by the signal variance model.
To fit the signal variance model, maximize the expected log likelihood with respect to δ by fitting a GLM for with gamma errors ( represents ‘fitted values’ for x from the signal mean model).
To fit the phenotype model, maximize the expected log likelihood with respect to β by fitting the GLM for phenotype given copy number.
To fit the copy number model, maximize the expected log likelihood with respect to α. Currently α are simply multinomial proportions and are estimated by summing posterior probabilities for each value of n over subjects (possibly within strata).
These calculations were originally implemented entirely in R22. We replicate the data for each subject N + 1 times and store the posterior probabilities, Pin, as an additional column in the expanded data matrix. The first three M steps can then be carried out by simple calls to the glm() function, with the posterior probabilities incorporated into the prior weights. For greater efficiency, the computationally intensive steps were later implemented in C/C++, while retaining the R interface.
For each test for disease association, we first fit the model assuming the null hypothesis for the phenotype model (for example, in the simple case of the trend model, H0:β1 = 0). Because the EM algorithm can converge to local maxima, this step is repeated multiple times from different starting points. At this stage, the value of N to be used is chosen, applying the Bayesian information criterion23. The alternative model is then fitted, and twice the increase in the log likelihood provides the asymptotic χ2 test statistic.
Approximate power calculations
Power calculations would normally be carried out by repeated simulations of the alternative hypothesis. However, for the 1-d.f. freedom test corresponding to the trend alternative, a simple approximate method is available. In large samples, the profile log likelihood for the trend parameter, β, can be assumed to be approximately quadratic over the range of interest, so that its second derivative is approximately constant. With a change of sign this second derivative is termed the observed information and will be denoted by I. Then, the log likelihood ratio χ2 test statistic, T, is approximately equal to , where is the maximum likelihood estimate (MLE) of β. Consequently, the information may be estimated from an observed value of the log likelihood ratio test and the corresponding MLE:
Standard asymptotic theory shows that, under the null hypothesis H0:β = 0, T is distributed as χ2 with 1-d.f. The same theory shows that, under the alternative hypothesis H1:β = βT where βT is not too large, T is approximately distributed as a noncentral χ2 variate with noncentrality parameter . In our real examples, I was estimated from the observed test values and MLEs as described above while the theoretical maximum value of the information, achieved when the copy number, n, is known, is given by
where S denotes sample size, and y is the dichotomous phenotype.
Supplementary Material
Note: Supplementary information is available on the Nature Genetics website.
ACKNOWLEDGMENTS
C.B., T.F., R.R. and M.E.H. are funded by the Wellcome Trust (WT), J.M. is funded by the WT and the National Institute of General Medical Sciences, V.P. is supported by a Juvenile Diabetes Research Foundation (JDRF) fellowship, and D.C. is supported by a JDRF/WT fellowship. The authors would like to thank the Wellcome Trust Case Control Consortium, D. Conrad, A. Moses, N. Carter, M. Dermitzakis, B. Stranger, J. Armour and E. Hollox for data access and helpful discussions.
Footnotes
URLs. Fitting code available as an R package, http://cnv-tools.sourceforge.net/.
Published online at http://www.nature.com/naturegenetics/
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 3.Lupski JR, Stankiewicz P, editors. Genomic Disorders: The Genomic Basis of Disease. Humana Press; Totowa, New Jersey: 2006. [DOI] [PubMed] [Google Scholar]
- 4.Stranger BE, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flint J, et al. High frequencies of alpha-thalassaemia are the result of natural selection by malaria. Nature. 1986;321:744–750. doi: 10.1038/321744a0. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez E, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 7.Aitman TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulone-phritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 8.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat. Genet. 2007;39:S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 2007;80:1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fellermann K, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollox EJ, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clayton DG, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat. Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 13.Plagnol V, Cooper JD, Todd JA, Clayton DG. A method to address differential bias in genotyping in large-scale association studies. PLoS Genet. 2007;3:e74. doi: 10.1371/journal.pgen.0030074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armour JA, Barton DE, Cockburn DJ, Taylor GR. The detection of large deletions or duplications in genomic DNA. Hum. Mutat. 2002;20:325–337. doi: 10.1002/humu.10133. [DOI] [PubMed] [Google Scholar]
- 16.Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 2000;95:360–362. [PubMed] [Google Scholar]
- 17.Newman TL, et al. High-throughput genotyping of intermediate-size structural variation. Hum. Mol. Genet. 2006;15:1159–1167. doi: 10.1093/hmg/ddl031. [DOI] [PubMed] [Google Scholar]
- 18.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 19.McCullagh P, Nelder JA. Generalized Linear Models. Chapman and Hall; London: 1989. [Google Scholar]
- 20.Prentice RL, Pyke R. Logistic disease incidence models and case-control studies. Biometrika. 1979;66:403–411. [Google Scholar]
- 21.Meng X-L, Rubin DB. Maximum likelihood estimation via the ECM algorithm: A general framework. Biometrika. 1993;80:267–278. [Google Scholar]
- 22.R Development Core Team R: A Language and Environment for Statistical Computing. 2007. < http://cran.r-project.org/doc/manuals/refman.pdf> .
- 23.Schwarz G. Estimating the dimension of a model. Annals of Statistics. 1978;6:461–464. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Genetics website.