

Abstract
Contact-dependent communication between immune cells generates protection, but also facilitates viral spread. We found that macrophages formed long-range actin-propelled conduits in response to negative factor (Nef), a human immunodeficiency virus type-1 (HIV-1) protein with immunosuppressive functions. Conduits attenuated immunoglobulin G2 (IgG2) and IgA class switching in systemic and intestinal lymphoid follicles by shuttling Nef from infected macrophages to B cells through a guanine exchange factor-dependent pathway involving the amino-terminal anchor, central core and carboxy-terminal flexible loop of Nef. By showing stronger virus-specific IgG2 and IgA responses in patients harboring Nef-deficient virions, our data suggest that HIV-1 exploits intercellular highways as a “Trojan horse” to deliver Nef to B cells and evade humoral immunity systemically and at mucosal sites of entry.
Cell-to-cell communication is essential for the immune system to generate protective responses. Contact-independent communication entails transmission of information via diffusion of soluble factors, whereas contact-dependent communication involves physical connections through specialized areas of contact. Short-range connections comprise gap junctions and synapses, whereas long-range connections encompass nanotubules and filopodial bridges1. Similar to cytonemes originating from the Drosophila imaginal disc, long-distance intercellular conduits allow complex messages to travel between distant cells without crossing the plasma membrane and without decreasing in strength in relationship to the distance traveled2. While nanotubules can traffic both surface and cytoplasmic proteins, filopodial bridges transfer surface proteins only3. By establishing intercellular connectivity networks, these cell protrusions may enable immune cells to amplify protective responses without relying on the slower process of diffusion4.
A downside of contact-dependent communication systems is that pathogens may hijack them to undergo cell-to-cell spreading. This process is exemplified by human immunodeficiency virus type-1 (HIV-1), the etiologic agent of acquired immunodeficiency syndrome (AIDS). HIV-1 utilizes a specialized area of immune contact known as virological synapse to spread from dendritic cells or macrophages to CD4+ T cells5. In addition, HIV-1 uses intercellular conduits for efficient cell-to-cell transmission as other retroviruses do6,7. These conduits might also spread viral subunits with immunosuppressive function, which could allow HIV-1 to cause extensive damage to immune cells without needing to infect them8,9. One viral immunosuppressive protein possibly implicated in intercellular transportation is negative factor (Nef), a membrane-targeting viral protein involved in AIDS development and immune dysfunction10,11.
Nef targets cell membranes and elicits cytoskeletal rearrangement, organelle formation, and synapse destabilization12-15, all events involved in protein trafficking via intercellular conduits3. In addition, Nef accumulates in macrophages, a conduit-forming cell type often proximal to Nef-containing B cells in infected follicles8. Since B cells are generally not infected by HIV-1, they might acquire Nef from neighboring infected macrophages via nanotubules or filopodial bridges. This transfer could attenuate production of immunoglobulin G (IgG) and IgA8, two antibody isotypes with antiviral activity16.
HIV-1 causes multiple B cell abnormalities, including hyperproduction of nonspecific IgG and IgA antibodies17,18. In addition, HIV-1 elicits robust virus-specific IgG responses. While IgG to viral envelope (Env) glycoproteins remains sustained throughout the disease, IgG to group antigen (Gag) and other viral proteins progressively declines19,20. This decline involves especially the IgG2 subclass, which is involved in antiviral defense, whereas IgG1 and IgG3 subclasses are less affected21. Additional B cell abnormalities include poor IgA responses to both Env and Gag proteins, defective IgG and IgA responses to opportunistic pathogens and vaccines, and loss of memory B cells, a subset of class-switched and high-affinity B cells originating from the germinal center (GC) of secondary lymphoid follicles22-24.
In general, HIV-1-induced humoral defects are thought to originate from activation-induced B cell exhaustion and loss of CD4+ T cells, a subset of T cells that provides help to follicular B cells via CD40 ligand (CD40L)-CD40 interaction25-28. Yet, normalization of CD4+ T cells by antiretroviral therapy does not fully restore antigen-specific IgG and IgA responses and memory B cells29,30, suggesting the additional involvement of B cell-intrinsic defects. Consistent with this possibility, B cells from infected patients show decreased responsiveness to CD4+ T cell help via CD40L31.
Here we show that HIV-1-infected macrophages formed long-range B cell-targeting conduits in response to Nef. These conduits inhibited systemic and intestinal IgG2 and IgA class switching by translocating membrane-bound Nef as well as Nef-containing endosomes from macrophages to follicular B cells through an actin-propelled [http://www.signaling-gateway.org/molecule/query?afcsid=A000191], Vav-mediated [http://www.signaling-gateway.org/molecule/query?afcsid=A002361] and GTPase-dependent mechanism involving the myristoylated anchor, central core and flexible loop domains of Nef. These findings together with additional in vivo data obtained from patients infected by Nef-sufficient or Nef-deficient virions suggest that HIV-1 utilizes long-distance intercellular highways as a “Trojan horse” to deliver Nef to B cells and evade protective virus-specific IgG2 and IgA responses systemically and at mucosal sites of entry.
Results
HIV-1-infected macrophages transfer Nef to B cells
Nef enhances HIV-1-induced antibody abnormalities by attenuating CD40L expression on CD4+ T cells, hindering CD4+ T cell interaction with antigen-presenting cells, and augmenting nonspecific B cell activation via macrophages15,27,32. These effects promote nonspecific hypergammaglobulinemia while hampering specific T cell-dependent (TD) IgG and IgA responses. Nef also invades follicular B cells, thereby interfering with the initiation of class switch-inducing signals from CD40L8. Since macrophages are critical to translocate viral antigens to follicular B cells33, we hypothesized that B cells acquire Nef through a contact-dependent mechanism involving macrophages.
Immunohistochemistry showed abundant CD68+ macrophages containing p24 and Nef in follicular and extrafollicular areas of infected systemic and intestinal lymphoid tissues (Fig. 1a). Together with HIV-1-trapping follicular dendritic cells (FDCs), these infected macrophages correlated with less abundant IgG2 and IgA in GCs. Of note, infected macrophages were often proximal to pre-switched IgD+ B cells containing Nef, but not p24 (Fig. 1b,c and Supplementary Fig. 1 online). Flow cytometry confirmed the presence of Nef but not p24 in purified B cells from infected lymphoid follicles (Fig. 1d).
Figure 1. Infected primary macrophages transfer Nef to B cells and inhibit TD class switching.
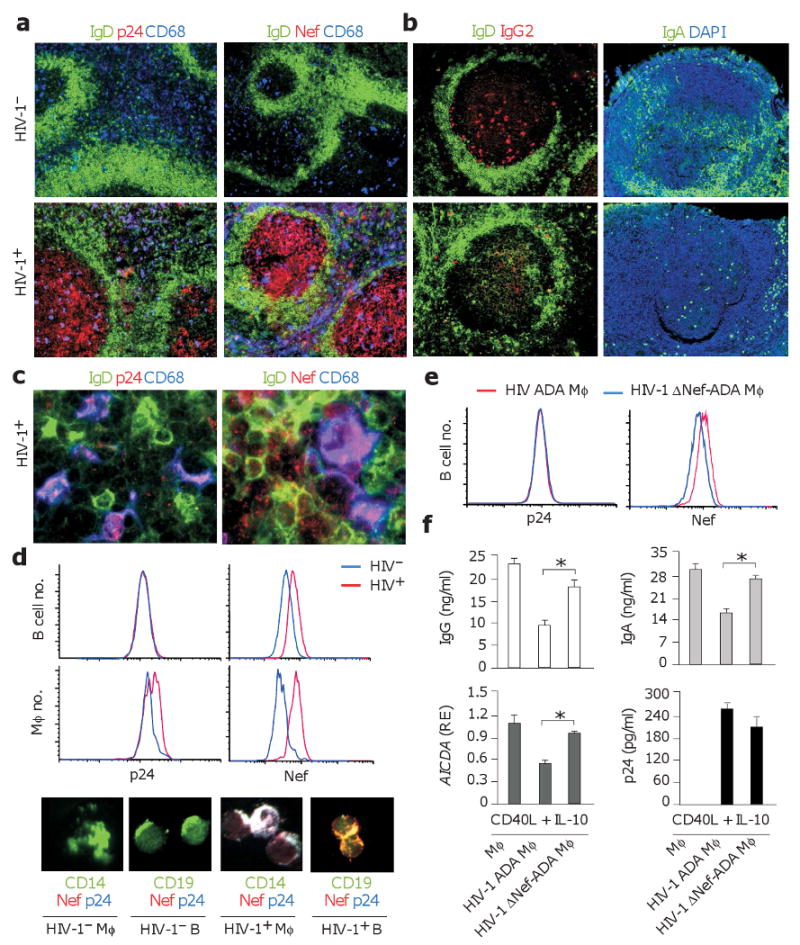
(a, b) Immunohistology of HIV-1− and HIV-1+ follicles from systemic lymph nodes or intestinal Peyer's patches stained for IgD or IgA (green), p24, Nef or IgG2 (red), and CD68 (blue). Original magnification, ×10. (c) Primary macrophages and B cells from HIV-1+ lymph nodes stained for IgD (green), p24 or Nef (red), and CD68 (blue). Original magnification, ×63. (d) Flow cytometry of p24 and Nef in primary CD19+ B cells and CD14+ macrophages (Mφ) sorted from HIV-1− (blue histograms) or HIV-1+ (red histograms) spleens. Similar cells were analyzed by immunofluorescence analysis upon staining for CD19 or CD14 (green), Nef (red), and p24 (blue). Original magnification, ×40. (e) p24 and Nef in CD19+ B cells sorted from a 24-h co-culture with primary macrophages infected with either wt ADA HIV-1 (red histograms) or ΔNef-ADA HIV-1 (blue histograms). (f) QRT-PCR of AICDA transcripts and ELISAs of IgG and IgA from IgD+ B cells co-cultured with primary uninfected, wt ADA-infected or ΔNef-ADA-infected macrophages in the presence of CD40L and IL-10 for 7 d. AICDA mRNA was quantified in B cells sorted after 4 d and normalized to ACTB mRNA. Relative expression (RE) compared to B cells stimulated with CD40L and IL-10 in the absence of macrophages. ELISA of p24 was performed 7 d after mock or ADA infection. Panels a-e show 1 of 5 experiments yielding similar results, whereas panel f summarizes 3 experiments (bars indicate mean s.d.; asterisk is p < 0.05).
Flow cytometry and immunofluorescence analysis demonstrated that IgD+ B cells acquired Nef but not p24 upon 24-h exposure to macrophages harboring either ADA, an R5 strain of HIV-1 that utilizes the chemokine receptor CCR5 to enter macrophages and CD4+ T cells, or LAI, an X4 strain of HIV-1 that utilizes CXCR4 to infect CD4+ T cells as well as macrophages (Fig. 1e). Similar results were obtained by co-culturing U1 macrophage-like cells harboring a tumor necrosis (TNF)-inducible virus with IgD+ B cells pre-labeled with CellTracker (Supplementary Fig. 2 online). This compound passes freely through cell membranes, but after internalization is transformed into a cell-impermeant adduct that remains inside live cells for several generations. This strategy allowed us to monitor and quantify Nef and p24 transfer to B cells through confocal microscopy. Unlike unstimulated HIV-1− U1 cells or (not shown) parental HIV-1− U937 cells, TNF-α-stimulated HIV-1+ U1 cells contained Nef and p24, released p24, and transferred Nef but not p24 to B cells in a time-dependent manner. Taken together, these data indicate that B cells acquire Nef from infected macrophages.
HIV-1-infected macrophages inhibit class switching
Since accumulation of Nef-containing macrophages correlates with attenuated IgG2 and IgA production in HIV-1-containing GCs, we wished to verify whether infected macrophages inhibit class switching via Nef. IgD+ B cells were first co-cultured for 24 hours with uninfected or HIV-1-infected macrophages and then sorted and exposed to TD signals such as CD40L and interleukin-10 (IL-10). Class switching was evaluated through enzyme-linked immunosorbent assay (ELISA) and quantitative-reverse transcription polymerase chain reaction (QRT-PCR). Compared to B cells exposed to non-infected macrophages (Fig. 1f), B cells exposed to infected macrophages secreted less IgG and IgA in response to CD40L and IL-10, and expressed fewer transcripts for activation-induced cytidine deaminase (AICDA), a hallmark of ongoing class switch DNA recombination (CSR)34.
These inhibitory effects were reversed when the B cells were incubated with macrophages harboring Nef-deficient (ΔNef) HIV-1. Restoration of class switching was not due to differences in viral replication, because macrophages infected with Nef-sufficient and ΔNef virions released comparable amounts of p24. Similar results were obtained by incubating B cells with HIV-1+ U1 macrophage-like cells or with 293 epithelial cells expressing wyld type or ΔNef HIV-1 plasmids (Supplementary Fig. 3 online). Supernatants from HIV-1+ U1 cells or (not shown) primary macrophages did not affect class switching, indicating that infected macrophages render B cells less sensitive to TD class switch-inducing signals through a contact-dependent mechanism involving Nef.
Macrophages inhibit class switching via Nef
To verify whether Nef is sufficient for macrophages to acquire class switch-inhibiting functions, macrophage-like THP-1 cells expressing Nef-enhanced green fluorescent protein (Nef-eGFP) were incubated with IgD+ B cells marked with CellTracker. Transfer of control eGFP or Nef-eGFP to B cells was tracked and quantified through fluorescence microscopy. We found that B cells cultured with eGFP-THP-1 cells did not accumulate eGFP, whereas B cells incubated with Nef-eGFP-THP-1 cells did in a time-dependent manner (Fig. 2a,b). Next, we sorted B cells from co-cultures to establish whether acquisition of Nef was associated with attenuation of CD40-dependent class switching. Compared to B cells incubated with eGFP-THP-1 cells, B cells incubated with Nef-eGFP-THP-1 cells induced less byproducts of IgA CSR8,26, such as germline Iα-Cα, AICDA and switch circle Iα-Cμ transcripts, in response to CD40L and IL-10 (Fig. 2c-e). These B cells also induced less byproducts of IgG CSR (not shown) and secreted less IgA and IgG but comparable IgM proteins (Fig. 2f). Supernatants from eGFP- or Nef-eGFP-THP-1 cells did not affect CD40-mediated class switching (Fig. 2g), confirming that B cells become less sensitive to TD class switch-inducing signals upon acquiring Nef in a contact-dependent manner.
Figure 2. Nef is sufficient for macrophage-like cells to acquire class switch-inhibiting functions.

(a) Imaging of CellTracker-loaded IgD+ B cells (blue) co-cultured with eGFP-THP-1 or Nef-eGFP-THP-1 macrophage-like cells (Mφ, green) for 24 h. Original magnification, ×5. (b) Time course analysis of eGFP-THP-1 or Nef-eGFP-THP-1 in IgD+ B cells cultured as in a for 48 h. (c-e) QRT-PCR of Iα1-Cα1, Iα2-Cα2, AICDA and Iα-Cμ transcripts from IgD+ B cells cultured as in b, sorted and then incubated with or without CD40L and IL-10 for additional 48 h. Iα1-Cα1, Iα2-Cα2 and AICDA mRNAs were normalized to ACTB mRNA and Iα-Cμ mRNA to Iμ-Cμ mRNA. RE, relative expression. (f) ELISAs of IgG, IgA and IgM proteins from IgD+ B cells cultured as in c-e, except that the activation step was carried out for 7 days. (g) IgG, IgA and IgM proteins from IgD+ B cells incubated with supernatants from eGFP-THP-1- or Nef-eGFP-THP-1 cells in the presence or absence of CD40L and IL-10 for 7 days. (h, i) Immunohistology of HIV-1− and HIV-1+ follicles from systemic lymph nodes and intestinal Peyer's patches stained for IgD (green), AID (red), and Nef (blue or dark gray). DAPI (blue) counterstains nuclei. Original magnification, ×10. Panels a, h and i show 1 of 5 experiments yielding similar results, whereas panels b-g summarize 3 experiments (bars indicate mean s.d.; asterisk is p < 0.05). Scale bar equals 10 μm.
Nef accumulation correlates with GC alterations
Given the reduced IgG2 and IgA expression in infected GCs and the AICDA-inhibiting activity of infected macrophages, we verified the expression of activation-induced cytidine deaminase (AID) in vivo by immunohistology. Compared to HIV-1− GCs, HIV-1+ GCs accumulated Nef and contained less AID (Fig. 2h,i and Supplementary Fig. 4 and Table 1 online). Attenuation of AID expression correlated with reduced expression of B cell lymphoma protein 6 (Bcl-6) and interferon responsive factor 4 (IRF4), two factors required for GC B cell differentiation and class switching35, and with increased expression of B lymphocyte-induced maturation protein (Blimp)-1 and CD138 (Supplementary Figs. 5,6 online), a Bcl-6-suppressing plasma cell-inducing protein and a plasma cell hallmark, respectively36.
Consistent with prior data indicating that Nef inhibits class switching by attenuating CD40L-mediated IKK (inhibitor of NF-κB kinase)-dependent activation of the transcription factor nuclear factor-κB (NF-κB) as well as IL-4-mediated Jak3 (Janus kinase 3)-dependent activation of STAT6 (signal transducer and activator of transcription 6) (ref. 8), HIV-1+ GCs also showed reduced IKK, Jak3 and STAT6 phosphorylation and increased expression of IκB (inhibitor of NF-κB) and SOCS (suppressor of cytokine signaling) proteins, two Nef-inducible inhibitors of NF-κB and STAT6, respectively8. These alterations were associated with loss of GC B cells, because HIV-1+ GCs expressed normal amounts of Ki-67 and the nuclear factor Pax5, two hallmarks of GC B cells36. Finally, HIV-1+ GCs had conserved or increased CD21+CD35+ follicular dendritic cells, CD4+ T cells and CD40L expression, at least in systemic lymph nodes. Thus, Nef may inhibit class switching in the context of an overall perturbation of the GC reaction.
Nef induces B cell-targeting conduits via multiple motifs
Nef reprograms endocytosis, cytoskeleton rearrangement, organelle trafficking and cell signaling to down-regulate CD4 and HLA-I on infected cells and induce immune suppression11. All these events are also involved in intercellular trafficking1,3. To elucidate the molecular requirements underlying the induction of B cell-targeting conduits in macrophages, we generated macrophage-like THP-1 cells expressing wild type Nef or Nef (G2A)-eGFP, Nef (P72-5A)-eGFP, Nef (L160-1A)-eGFP and Nef (E62-5A)-eGFP mutants with inactive MGxxxS(1), PxxPxR(72), ExxxLL(160) and EEEE(62) motifs, respectively.
MGxxxS(1) is a myristoylated motif from the amino-terminal anchor of Nef that binds Nef to the plasma membrane, an event essential for most Nef functions13. PxxPxR(72) is a proline-rich motif from the central core of Nef that induces actin cytoskeleton remodeling, endosome formation and signaling by interacting with multiple kinases, including Vav12,37. EEEE(62) is an acidic cluster from the central core of Nef that promotes recruitment of Nef-containing endosomes to the trans-Golgi network (TGN) by interacting with phosphofurin acidic cluster sorting protein-1 (PACS-1), a component of the clathrin-independent endocytic pathway14. Finally, ExxxLL(160) is a di-leucin motif from the carboxy-terminal flexible loop of Nef that elicits formation of Nef- and CD4-containing endosomes by interacting with adaptor protein-2 (AP-2), a key component of the clathrin-dependent endocytic pathway38-40.
Confocal microscopy showed that, unlike control eGFP, Nef-eGFP targeted the plasma membrane and a perinuclear compartment of THP-1 cells and induced thin protrusions bridging THP-1 cells with each other and with B cells (Fig. 3a-c and Supplementary Movies 1–3 online). Inactivation of MGxxxS(1) altered the subcellular distribution of Nef, abrogated cell protrusions, and impaired Nef transfer to B cells. Inactivation of PxxPxR(72) and ExxxLL(160) did not disrupt the subcellular localization of Nef, but attenuated cell protrusions and Nef transfer to B cells. Inactivation of EEEE(62) neither altered Nef localization nor affected cell protrusions, but attenuated Nef transfer to B cells. Thus, Nef anchoring to the plasma membrane is essential for Nef to stimulate conduit formation in macrophages and invade B cells. These effects may also require reprogramming of macrophage signaling and endocytosis.
Figure 3. Nef utilizes multiple motifs to stimulate conduit formation in macrophage-like cells.

(a) Live-cell confocal imaging of THP-1 macrophage-like cells (Mφ) 24 h after nucleofection with eGFP (control), wt Nef-eGFP, Nef (G2A)-eGFP, Nef (P72-5A)-eGFP, Nef (L160-1A)-eGFP or Nef (E62-5A)-eGFP plasmid. (b) Live-cell confocal imaging of Nef-eGFP-THP-1 cells (green) co-cultured with CellTracker-loaded IgD+ B cells (red) for 6 h. Bottom panels show Nef-eGFP-THP-1 cells after three-dimensional reconstruction. (c) Average number of protrusions on individual THP-1 cells nucleofected as in A (upper panel), and percentage of IgD+ B cells acquiring eGFP fluorescence after co-culture with THP-1 cells nucleofected as in A for 24 h. (d) Live-cell DIC (left and mid panels) and confocal (right panel) imaging of Nef-eGFP-THP-1 cells (green) co-cultured with CellTracker-loaded IgD+ B cells (red). Dashed box in left panel includes cells magnified in mid panel. (e) Live-cell DIC imaging of THP-1 cells pre-loaded with LysoTracker and co-cultured with IgD+ B cells. In upper panel, arrowheads show a long-range tubular conduit containing CellTracker, whereas dashed rectangle pinpoints short-range transfer of CellTracker from a THP-1 cell to a B cell. In bottom panels, short-range transfer is shown over time, each panel being separated by approximately 15 sec. Panels a, b, d and e show 1 of 4 experiments yielding similar results, whereas panel c summarizes 3 experiments (bars indicate mean s.d.; asterisk is p < 0.05 compared to wt Nef-eGFP value). Scale bar equals 10 μm.
Nef traffics to B cells via long-range intercellular conduits
We next wondered whether Nef-induced macrophage protrusions develop into long-range conduits. Confocal microscopy showed that Nef-containing THP-1 macrophage-like cells formed an intricate network of conduits targeting distant THP-1 cells or CellTracker-loaded IgD+ B cells (Fig. 3d and Supplementary Figs. 7,8 online). Some conduits contained Nef along their entire length, whereas others encompassed organelle-like bulges loaded with Nef. Time-lapse confocal microscopy of co-cultures comprising THP-1 cells pre-loaded with LysoTracker, a probe that marks late endosomes and lysosomes, confirmed that B cell-targeting conduits traffic organelles in addition to Nef via both short-range and long-range mechanisms (Fig. 3e and Supplementary Movie 4 online). After establishing an initial contact, some conduits remained anchored to B cells from a few seconds to a few minutes (Supplementary Fig. 9 online). During this time, B cells accumulated Nef within membrane and sub-membrane regions proximal to the conduit. Further experiments showed directional movement of Nef-eGFP from THP-1 macrophage-like cells to B cells along conduits (Supplementary Fig. 10 online).
To rule out the possibility that intercellular conduits result from the artificial activation of macrophage-like THP-1 cells by overexpressed Nef-eGFP, we studied the formation of these conduits in primary macrophages infected by HIV-1. Stainings with the membrane-targeting lectin wheat germ agglutinin (WGA) showed that macrophages formed an increased number of long-range conduits upon infection with ADA HIV-1 (Fig. 4a). This increase was less pronounced in macrophages infected with ΔNef ADA. Of note, long-range conduits generated by ADA-infected macrophages contained both Nef and p24 (Fig. 4b,c). Together with short-range conduits, these long-range conduits trafficked Nef (Fig. 4d) but not p24 (not shown) to Pax5+ B cells. Thus, infected macrophages translocate both membrane-bound and organelle-associated Nef proteins to B cells via long-range highways.
Figure 4. Infected primary macrophages transfer Nef to B cells via long-range actin-propelled conduits.

(a) Confocal imaging of uninfected, wt ADA-infected or ΔNef-ADA-infected macrophages (Mφ) stained for Nef (red) or p24 (blue) in the presence of WGA (green). Leftmost panel shows number of long (> 100 μm) tethers/50 uninfected or infected macrophages. Original magnification, ×40. (b-d) ADA-infected macrophages cultured with or without IgD+ B cells and stained for Nef (red) and p24 or Pax5 (blue) in the presence of WGA (green). Original magnification, ×5 (b and c, upper panels), ×63 (b and c, lower panels), ×10 (d). (e, f) Percentage of THP-1-Nef-eGFP macrophage-like cells (Mφ) forming tethers and percentage of IgD+ B cells acquiring Nef-eGFP from Nef-eGFP-THP-1 cells in the presence or absence of chemical inhibitors (azide, latrunculin B, colchicin, nocodazole, wortmannin, AD101 or ADMD3100) or DN inhibitors (to β-arrestin-2, dynamin Ia, Arf6, Vav2, Cdc42, Rac1 or Rho). Control value corresponds to no inhibitor or empty construct and was set at 100%. Panels a-d show 1 of 4 experiments yielding similar results, whereas panels e and f summarize 3 experiments (bars indicate mean s.d.; asterisk is p < 0.05 compared to control). Scale bar equals 10 μm.
Nef invades B cells via macrophage actin remodeling
Considering that cells require adenosine triphosphate (ATP)-dependent remodeling of their actin cytoskeleton to form conduits1,3, we wondered about the role of ATP and actin in Nef transfer to B cells. Both the ATP inhibitor azide and the actin polymerization inhibitor latrunculin-B attenuated conduit formation in THP-1 macrophage-like cells and Nef transfer to B cells, whereas the tubulin polymerization inhibitors colchicin and nocodazole did not (Fig. 4e). Since Nef utilizes phosphoinositol-3 kinase (PI3K) and adenosine diphosphate ribosylation factor 6 (ARF6) to promote clathrin-independent recruitment of HLA-I-containing Nef-negative endosomes to the trans-Golgi network (TGN)14, we also explored the role of PI3K. We found that conduit formation and Nef transfer were not affected by the PI3K inhibitor wortmannin. Since CCR5 and CXCR4 mediate intercellular communication and HIV-1 transmission5, we determined the role of these chemokine receptors as well. Consistent with the expression of CXCR4 but not CCR5 by B cells26, a CXCR4 inhibitor attenuated Nef transfer to B cells, whereas a CCR5 inhibitor did not. Neither inhibitor affected conduit formation.
Knowing that Nef induces actin rearrangement by interacting with the guanine nucleotide exchange factor Vav via PxxPxR(72) (ref. 12), we also addressed the role of Vav and downstream small guanine triphosphate (GTP)ases in Nef-induced conduit formation and B cell invasion. We found that dominant-negative constructs to Vav or Cdc42, Rac-1 and Rho small GTPases inhibited conduit formation by THP-1 cells as well as Nef-eGFP transfer from THP-1 cells to B cells (Fig. 4f). Thus, macrophages translocate Nef to B cells through a Vav-mediated small GTPase-dependent mechanism involving actin-propelled intercellular conduits. Some of these conduits may navigate toward B cells through a CXCR4-dependent mechanism.
Nef invades B cells via membrane and vesicular routes
Considering that Nef uses endosome-regulating ExxxLL(160) and EEEE(62) motifs to invade B cells and knowing that organelles can navigate along conduits1-3, we wondered whether Nef translocates from macrophages to B cells within vesicular cargoes. Dominant-negative constructs to β-arrestin-2 [http://www.signaling-gateway.org/molecule/query?afcsid=A000027] and dynamin Ia [http://www.signaling-gateway.org/molecule/query?afcsid=A000794], which regulate clathrin-dependent formation of CD4- and Nef-containing endosomes14,39,41, attenuated Nef-eGFP transfer from THP-1 macrophage-like cells to B cells, whereas dominant-negative constructs to ARF6, which regulates clathrin-independent formation of HLA-I-containing Nef-negative endosomes14, did not.
Confocal imaging documented Nef-containing organelle-like bulges positive for a membrane-specific dye along long-range conduits connecting Nef-dsRed-expressing THP-1 macrophage-like cells or HIV-1-infected primary macrophages with B cells (Fig. 5a). Some of these bulges stained positive for β-adaptin or TGN75 (Fig. 5b,c), suggesting their origin from clathrin-dependent and clathrin-independent (TGN-derived) pathways14,39, respectively. Electron microscopy confirmed that conduits comprised Nef-containing vesicular and tubular structures in addition to membrane-bound Nef and additionally showed Nef-positive structures budding from the surface of THP-1 macrophage-like cells, navigating in the extracellular space between macrophage-like cells and B cells, and docking on the surface of B cells adjacent to macrophage-like cells (Fig. 5d). Confocal microscopy showed that some of these exosome-like structures involved clathrin as they were positive for β-adaptin (Fig. 5d,e).
Figure 5. Macrophage-like cells inhibit class switching by trafficking membrane and vesicular Nef via Vav and small GTPases.
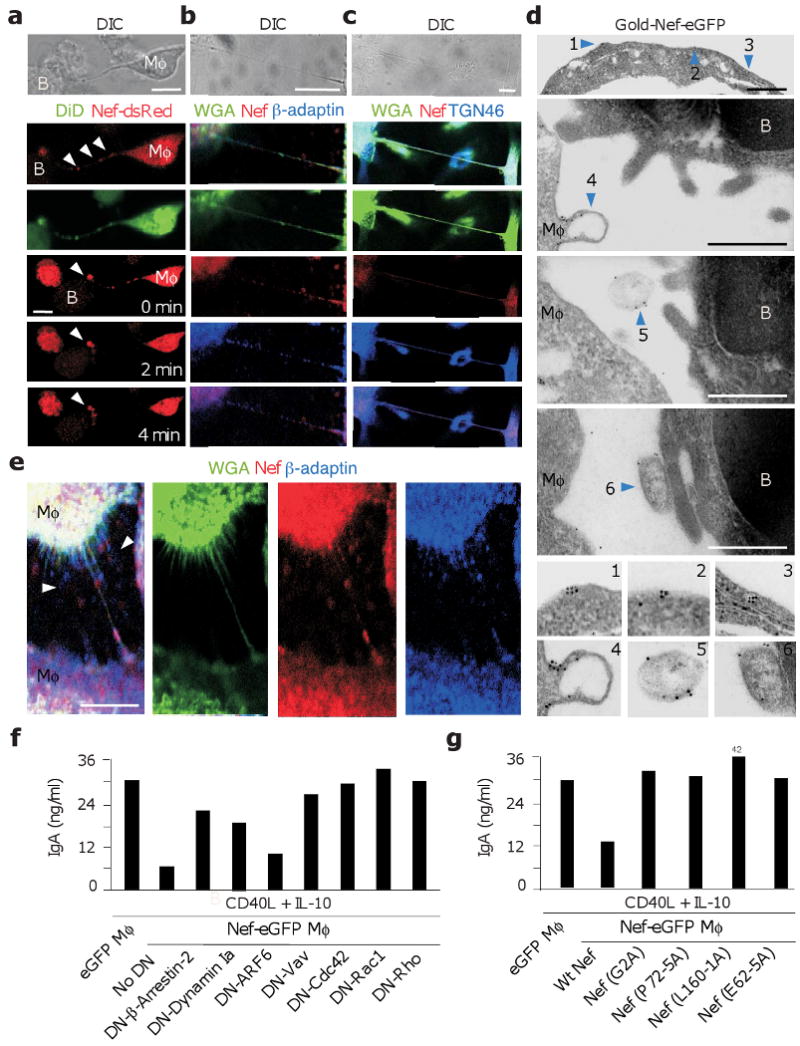
(a) Live DIC (uppermost panels) or confocal imaging of Nef-dsRed-THP-1 macrophage-like cells (Mφ, red) co-stained with the membrane-specific probe DiD (green) and incubated with unstained IgD+ B cells. Arrowheads point to organelle-like beads within an intercellular conduit. The bottom three panels from the top show intercellular transfer of Nef-dsRed over time. Arrowheads point to site of Nef-dsRed accumulation in a B cell. Original magnification, ×40. (b, c) Confocal imaging of ADA-infected primary macrophages (Mφ) stained for Nef (red) and β-adaptin or TGN46 (blue) in the presence of WGA (green). (d) Transmission electron microscopy of Nef-eGFP-THP-1 macrophage-like cells (Mφ) co-cultured with IgD+ B cells for 6 hours and stained with a gold-labeled monoclonal antibody to GFP. Arrowheads 1 and 2, membrane-bound Nef on a Mφ-derived conduit; arrowhead 3, Nef within vesicular and tubular structures from a Mφ-derived conduit; arrowheads 4-6, Nef associated with Mφ budding as well as free or B cell-docked exosome-like bodies, respectively. Original magnification, ×36000. (e) Confocal imaging of ADA-infected primary macrophages stained for Nef (red) or β-adaptin (blue) in the presence of WGA (green). Arrowheads point to extracellular bodies. (f) ELISAs of IgA from IgD+ B cells incubated with THP-1-Nef-eGFP cells nucleofected with either an empty plasmid (control) or a DN plasmid to β-arrestin-2, dynamin Ia, ARF6, Vav, Cdc42, Rac1 or Rho. After 48 h, B cells were sorted and cultured for additional 7 days with or without CD40L and IL-10. (g) ELISAs of IgA from IgD+ B cells incubated with THP-1 cells expressing eGFP, wt Nef-eGFP, Nef (G2A)-eGFP, Nef (P72-5A)-eGFP, Nef (L160-1A)-eGFP or Nef (E62-5A)-eGFP. B cells were cultured as in f. Panels a-g show 1 of 4 experiments yielding similar results. Scale bars equal 10 and 1 μm in confocal and transmission electron microscopy images, respectively.
We next wondered whether Nef-trafficking via intercellular conduits affected class switching. We found that Nef-eGFP-THP-1 cells reversed their ability to inhibit IgA and IgG2 class switching in CD40-activated IgD+ B cells in the presence of mutations inactivating membrane-anchoring (MGxxxS(1)), cytoskeleton-remodeling (PxxPxR(72)) and endosome-reprogramming (EEEE(62) or ExxxLL(160)) motifs of Nef or in the presence of dominant-negative constructs to Vav, β-arrestin-2, dynamin Ia, Cdc42, Rac-1 or Rho, but not ARF6 (Fig. 5f,g and Supplementary Fig. 11 online). Thus, inhibition of class switching by infected macrophages likely involves Nef transfer to B cells through the plasma membrane and both clathrin-dependent and clathrin-independent (but ARF6-negative) vesicular structures.
Nef inhibits virus-specific IgG2 and IgA responses in vivo
Having shown reduced IgG2 and IgA class switching in Nef-containing follicles, we wished to determine whether these lymphoid structures included Nef-trafficking B cell-targeting conduits. Systemic and intestinal follicles from HIV-1− and HIV-1+ subjects comprised B cells connected with each other or macrophages through long-range conduits bearing membrane-bound CD19 (Fig. 6a,b and Supplementary Fig. 12 online). In HIV-1+ subjects, some conduits contained Nef and targeted Nef-containing B cells (Fig. 6c). Unlike HIV-1+ follicular areas, HIV-1+ extrafollicular areas contained little or no Nef and showed florid class switching, including AID, IgA (Fig. 6d) and (not shown) IgG expression. Extrafollicular class switching correlated with CD68+ macrophages expressing ferritin and B cell-activating factor of the TNF family (BAFF) (Fig. 6e), two HIV-1-inducible molecules with innate B cell-activating function26,27,34,42-44. In spite of inducing more ferritin than non-infected macrophages (Fig. 6f), infected macrophages did not utilize ferritin to inhibit TD class switching. Rather, ferritin cooperated with BAFF to increase TD IgG and IgA, but not IgM production (Fig. 6g).
Figure 6. Nef inhibits class switching in GCs but not extrafollicular areas.

(a, b) Immunohistology of HIV-1− and HIV-1+ follicles from systemic lymph nodes stained for IgD (green), Nef (red), and CD11c (blue). DAPI (blue) counterstains nuclei. Arrowheads point to intercellular conduits. Mφ, macrophages. Original magnification, ×63. (c-e) HIV-1− and HIV-1+ follicles from systemic lymph nodes or intestinal Peyer's patches stained for IgD or IgA (green), AID, ferritin or BAFF (red), and Nef or CD68 (blue). DAPI (blue) counterstains nuclei. Original magnification, ×10. (f) ELISA of ferritin from uninfected or ADA-infected primary macrophages. (g) ELISAs of IgM, IgG and IgA from IgD+ B cells co-cultured with CD40L or BAFF and IL-10 in the presence or absence of ferritin for 7 d. Panels a-e show 1 of 4 experiments yielding similar results, whereas panels f and g summarize 3 experiments. Scale bar equals 10 μm.
Circulating ferritin was increased in long-term non-progressors (LTNPs) infected with ΔNef HIV-1 compared to LTNPs infected with wt HIV-1, further discounting the role of ferritin in the inhibition of class switching by Nef (Supplementary Fig. 13 online). In spite of having comparable viral and immunological parameters45, including low viral load, normal CD4+ T cell count and normal total serum IgM, IgG and IgA antibodies (Supplementary Fig. 13 and Table 2 online), ΔNef-HIV-1+ LTNPs had more IgG2, IgA1 and IgA2 to viral p24, Tat (transactivator of transcription) and Env gp120 than wt HIV-1+ LTNPs did (Fig. 7a). When followed over time, these HIV-1-specific antibodies as well as ferritin showed no positive correlation with the viral load (Fig. 7b and Supplementary Fig. 14 online), confirming that ΔNef-HIV-1 enhances antiviral immunity independently of viral replication. Thus, HIV-1 attenuates virus-specific IgG2 and IgA responses in Nef-rich GC areas by trafficking Nef from infected macrophages to follicular B cells via long-range conduits. Furthermore, HIV-1 boosts nonspecific IgG and IgA responses by up-regulating the production of polyclonal B cell-activating molecules such as ferritin and BAFF in macrophages from extra-GC areas (Supplementary Fig. 15 online).
Figure 7. ΔNef HIV-1 elicits more virus-specific IgG2, IgA1 and IgA2 than wt HIV-1 irrespective of viral load.

(a) Serum IgG2, IgG3, IgA1 and IgA2 to viral p24 Gag, Tat and gp120 Env in ΔNef HIV-1+ LTNPs and wt HIV-1+ LTNPs. Each value from ΔNef HIV-1+ LTNPs represents the average of 3 to 5 independent determinations performed over a time period ranging from 6 to 12 years as indicated in Supplementary Table 2 online. Values are expressed as optical density (OD) at 450 nm. (b) Linear regression analysis of viral load and p24-specific IgA1 in D36, C54, C64 and C98 ΔNef HIV-1+ LTNPs examined at multiple time points as indicated in Supplementary Table S2.
Discussion
We found that HIV-1 transferred the immunosuppressive protein Nef from macrophages to systemic and intestinal B cells through long-distance actin-propelled conduits. This transfer required the MGxxxS(1), PxxPxR(72), EEEE(62) or ExxxLL(160) motifs of Nef and involved a Vav-mediated GTPase-dependent pathway that rendered B cells less responsive to IgG2 and IgA class-inducing signals from CD4+ T cells.
HIV-1 has evolved multiple strategies to evade antibody recognition, including poor antigenicity, limited immunogenicity, great variability and conformational camouflage of Env glycoproteins, which are essential for viral entry46. Additional strategies impair antibody production by inducing CD4+ T cell depletion, activation-induced B cell anergy, polyclonal B cell expansion, and loss of memory B cells18,24,25,27,28. Although more prominent with active viral replication, these humoral defects persist in aviremic patients receiving antiretroviral therapy24,29,30, suggesting the existence of replication-independent mechanisms for antibody evasion. One of these mechanisms involves targeting of systemic and intestinal B cells by Nef-trafficking conduits emerging from infected macrophages.
IgG and IgA play an important role in the control of HIV-1 infection both systemically and at mucosal sites of entry46. By showing that ΔNef-HIV-1+ LTNPs had more virus-reactive IgG2 and IgA than wt HIV-1+ LTNPs, our studies provide in vivo evidence that HIV-1 perturbs the antiviral response of systemic and mucosal B cells through a mechanism involving Nef. This mechanism is replication-independent, because both groups of LTNPs had comparable viral load and CD4+ T cell count and showed neither B cell hyperactivation nor hypergammaglobulinemia. Furthermore, when examined over time, virus-specific antibodies did not correlate with the viral load in ΔNef-HIV-1+ LTNPs. Like patients exposed to antiretroviral therapy47, wt HIV-1+ LTNPs may archive Nef in GCs. By trafficking along intercellular conduits, Nef eventually invades antigen-specific follicular B cells, thereby dampening their responsiveness to class switch-inducing signals from CD4+ T cells. The preferential impairment of IgG2 and IgA responses could result from a higher sensitivity to Nef of discrete B cell subsets with restricted class switching potential. These subsets might include B cells with neutralizing potential.
As suggested by recent studies9,48, HIV-1 likely promotes intercellular trafficking of multiple viral subunits to hamper the function of both immune and non-immune cells. Besides Nef, HIV-1 expresses Vif (virus infectivity factor) and Vpr (viral protein R). These accessory proteins accumulate in infected GCs (Santini & Cerutti, unpublished data) and inhibit the function of AID and UNG (uracil DNA glycosylase), respectively, two enzymes essential for class switching and somatic hypermutation49-51. Should Vif and Vpr traffic to B cells as Nef does, HIV-1 would have a powerful arsenal for the inhibition of both affinity maturation and class switching in GCs. This inhibition may contribute not only to the loss of class-switched memory B cells observed in both viremic and aviremic patients24,29, but also to the paucity of rapid and sustained neutralizing IgG and IgA responses to invading virions46.
When exposed to Nef-containing macrophages, B cells acquired Nef and became less responsive to TD class switch-inducing signals in that they expressed less AID and underwent less IgG2 and IgA CSR and production in response to CD40L. These in vitro data were supported by in vivo observations showing massive Nef accumulation as well as attenuated AID expression and decreased IgG2 and IgA production in infected GCs. Of note, the inhibition of AID expression was somewhat more evident in infected lymphoid tissues than in co-cultures of B cells with infected macrophages, possibly reflecting the fact that the class switch-inhibiting mechanisms acting in vivo are more complex than those present in vitro. In this regard, Nef accumulation in GCs was associated with reduced expression of the GC-inducing factor Bcl-6 and with increased expression of the GC-inhibiting factor Blimp-136. This latter may impair class switching by silencing AID expression and triggering premature plasma cell differentiation36. Tissue data also provided in vivo correlates to prior in vitro findings showing that Nef impairs TD class switching by impeding CD40 and cytokine receptor signaling via NF-κB and STAT transcription factors through a mechanism involving up-regulation of negative feedback proteins such as IκBα and SOCS8.
GC abnormalities were dissociated from an obvious loss of follicular dendritic cells, CD4+ T cells and CD40L, further suggesting the involvement of B cell-intrinsic defects such as those brought about by Nef. This viral protein had a direct negative effect on B cells, because supernatants from HIV-1-infected macrophages did not show any inhibitory effect on class switching. In agreement with this observation, our previous work shows that B cells capturing exogenous soluble Nef or expressing transfected endogenous Nef become less responsive to class switch-inducing signals8.
Unlike GCs, extra-GC areas exhibited florid IgG and IgA class switching, which correlated with macrophage accumulation of HIV-1-inducible B cell-activating factors with innate class switch-inducing activity such as ferritin and BAFF26,27,34,42-44. Our data suggest that ferritin and BAFF cooperatively induce CD40-independent polyclonal activation of extrafollicular B cells, thereby contributing to the emergence of nonspecific hypergammaglobulinemia. This latter is an important co-factor in the pathogenesis of HIV-1-induced humoral deficiency17,26, and together with non-protective IgG1 responses to Env antigens, persists in AIDS patients with very few CD4+ T cells, further suggesting its origin from a CD40-independent pathway17,19. Such a pathway may be insensitive to Nef due to the fact that this protein preferentially targets CD40L, predominantly accumulates in GCs, and overrides its intrinsic B cell-inhibitory activity in extrafollicular areas by triggering ferritin production. Nonspecific CD40-independent responses may also involve Env, because Env augments BAFF production by engaging canonical CD4, CCR5 and CXCR4 HIV-1 receptors on macrophages26. Env further activates extrafollicular B cells by engaging BAFF-inducible mannose C-type lectin receptors with class switch-inducing activity26. These alternative HIV-1 receptors were not expressed by the resting B cells utilized in the present studies, suggesting that Env played a marginal role in the class switching events triggered by wt or ΔNef virions.
Infected GCs were also characterized by the presence of long-range intercellular conduits that appeared to shuttle Nef from HIV-1-harboring macrophages to non-infected B cells. Consistent with prior studies1 and with our finding that LPS stimulates intercellular conduit formation in uninfected macrophages, we found intercellular conduits also in non-infected follicles, suggesting that HIV-1 hijacks a physiological intercellular communication network to disseminate viral immunosuppressive proteins to non-infected B cells. Infected macrophages enhanced the generation of long-range conduits through Nef. This accessory protein utilized its membrane-anchoring MGxxxS(1) motif to progress along conduits, invade B cells and inhibit class switching. In addition to Nef, conduits emerging from infected macrophages contained p24. In agreement with recent studies7, p24 was possibly associated with “surfing” viral particles, but unlike Nef, was not transferred to B cells. This observation can be explained by the fact that conduit-associated p24-containing virions may only invade B cells expressing Env-binding mannose C-type lectin receptors26, which were rare in our cultures. It must be also considered that infected macrophages contained less p24 than Nef. Finally, p24 lacks membrane-targeting molecular moieties such as MGxxxS(1), which was essential for the intercellular navigation of Nef.
Nef invasion of B cells would be favored by the presence of membrane continuity or receptor-dependent synaptic contact between conduits and targeted B cells3. Synaptic contact could implicate CXCR4, because an inhibitor of this chemokine receptor attenuated Nef trafficking from macrophages to B cells. Nef also required Vav-mediated small GTPase-dependent cytoskeleton remodeling via PxxPxR(72) to elicit conduit formation and B cell invasion. By interacting with Vav and possibly other kinases, such as PAKs (p21-activated kinases)11,12, PxxPxR(72) could also inhibit CD40-mediated class switch-inducing signals in B cells. Consistent with this possibility, PAK-2 has been recently found to interfere with CD40 signaling in Nef-containing dendritic cells52.
Nanotubular conduits establish cytoplasmic continuity with targeted cells, allowing intercellular trafficking of cytoplasmic proteins and organelles2,3. Given its ability to interact with various cytoplasmic proteins such as actin and kinases11, Nef could invade B cells not only from the plasma membrane, but also from the cytoplasm. Nef could also invade B cells via endosomes, because conduit-associated Nef often co-localized with vesicules containing β-adaptin, a component of the clathrin-AP-2 complex39. The involvement of clathrin-dependent endocytosis was further indicated by the fact that Nef transfer to B cells was attenuated by inactivation of β-arrestin 2 and dynamin Ia, which facilitate formation of clathrin-coated endosomes41, or by disruption of ExxxLL(160), which mediates Nef interaction with AP-238.
Nef could also traffic to B cells via a clathrin-independent TGN-targeting endocytic pathway, because some Nef co-localized with tubular structures and TGN46 within conduits. In addition, mutations of EEEE(62), which triggers PACS-1-dependent recruitment of Nef-containing endosomes to the TGN14, impaired Nef trafficking to B cells. Consistent with its inability to interact with Nef14, ARF6, which routs HLA-I-containing endosomes to the TGN39. The involvement of clathrin-dependent endocytosis was further indicated by the fact that Nef transfer to B cells was attenuated by inactivation of β-arrestin 2 and dynamin Ia, which facilitate formation of clathrin-coated endosomes41, or by disruption of ExxxLL(160), which mediates Nef interaction with AP-238.
Nef could also traffic to B cells via a clathrin-independent TGN-targeting endocytic pathway, because some Nef co-localized with tubular structures and TGN46 within conduits. In addition, mutations of EEEE(62), which triggers PACS-1-dependent recruitment of Nef-containing endosomes to the TGN14, impaired Nef trafficking to B cells. Consistent with its inability to interact with Nef14, ARF6, which routs HLA-I-containing endosomes to the TGN14, played a marginal role in Nef trafficking to B cells. The mechanism by which organelle-associated Nef affects B cell signaling remains unknown. One possibility is that Nef inhibits NF-κB-dependent CSR-inducing signals by interacting with vacuolar ATPase11,53. In addition to endosomes, Nef may utilize exosomes to target B cells. This possibility is consistent with earlier data showing dendritic cell internalization of HIV-1 particles to multivesicular endocytic bodies, followed by extracellular release of infectious exocytic vesicles54. Nef could enhance this process by promoting proliferation of multivesicular bodies and remodeling of the actin cytoskeleton55.
Nef may also utilize contact-independent mechanisms to invade immune cells and their precursors. Indeed, B cells and hematopoietic cells become dysfunctional after capturing soluble Nef from the extracellular environment8,48. In vivo, extracellular Nef presumably originates from its passive release by dying infected cells8,48,56. The absence of significant cell death in cultures with infected macrophages possibly reflects the lack of multiple immune and non-immune death-inducing signals usually present in vivo and explains why supernatants from these cultures neither contained Nef nor had overt class switch-inhibiting activity. In light of these considerations, we propose that Nef utilizes both contact-dependent and contact-independent mechanisms to generate suppressive signals in bystander non-infected B cells.
In summary, our work indicates that HIV-1 exploits Nef-trafficking connectivity networks to evade virus-specific IgG2 and IgA production by systemic and intestinal B cells. Thus, small molecules capable of inhibiting conduit formation and/or Nef function could be useful to enhance protection against HIV-1.
Methods
Tissue, blood and serum specimens
Lymph node, spleen, gut and tonsil tissues from 20 HIV-1+ patients with chronic infection and 13 HIV-1− patients with reactive inflammation were obtained from a repository at the Department of Pathology of Weill Medical College of Cornell University (Supplementary Table S1 online). LTNPs with transfusion-acquired infection by ΔNef-HIV-1 or sexually-acquired infection by wt HIV-1 and control donors matched for age, sex and/or transfusion are from Australia and were previously described45. The Institutional Review Board of Weill Medical College of Cornell University approved studies involving both tissue and blood specimens. These specimens were drawn from healthy donors and infected patients upon obtaining informed consent.
Cells
IgD+ B cells were sorted from the peripheral blood of healthy donors as previously described34. THP-1 and U937 (American Type Tissue Collection) are human myeloid cell lines with macrophage-like properties. U1 is a chronically infected subclone of U937 cells (from AIDS Research and Reference Reagent Program at National Institutes of Health (NIH)). U1 cells expressed HIV-1 upon exposure to 30 ng/ml of TNF-α (R&D Systems) for 24 h. Activated U1 cells were washed twice with phosphate buffer solution before using them in co-cultures. Finally, 293T epithelial cells were used in experiments involving wt HIV-1 and ΔNef-HIV-1 virions. Cultures were carried out in complete RPMI 1640 medium supplemented with 10% fetal bovine serum.
Cultures and reagents
IgD+ B cells were co-cultured with THP-1 cells, U1 cells, 293 cells, or macrophages at a 2:1 ratio. After 48 h, B cells were sorted with biotinylated mouse monoclonal antibody (mAb) HIB19 to CD19 (BD Biosciences PharMingen) and anti-biotin MicroBeads (Miltenyi), washed, and seeded at 1 × 105/100 μl with or without TD stimuli, including 100 ng/ml of CD40L (Immunex) and 30 ng/ml of IL-10 (Sigma-Aldrich). In some experiments, co-cultures were performed in the presence of 2 mM azide (Sigma-Aldrich), 2.5 mM latrunculin-B, 1 mM colchicin, 10 mM nocodazole, 2.5 mM wortmannin (Calbiochem), 50 nM AD101 (Schering Plough Research Institute), or 50 nM AMD3100 (AnorMed Incorporated). To obtain macrophages, monocytes sorted with CD14 Microbeads (Miltenyi) were incubated as adherent monolayers in 15-cm tissue-culture dishes using complete medium containing 25 ng/ml of monocyte colony-stimulating factor (PeproTech) for the initial 2 d and complete medium lacking monocyte colony-stimulating factor for the following 4 d. Differentiated macrophages were treated with trypsin and seeded into 24 well plate or 3cm glass-bottom dish at 1 × 106/well one day before virus infection.
In vitro infections
To generate viral stocks, HeLa cells were transfected with 30 μg of ADA, ΔNef-ADA (from M. Stevenson, University of Massachusetts Medical School), LAI, or ΔNef-LAI (from B. Berkhout, Academic Medical Center) plasmids by calcium phosphate/DNA co-precipitation and then incubated at 37°C. TCID50 was measured after collecting virions from culture supernatants by ultra-centrifugation. 20,000 TCID50/mL of virus was used in each well or dish. Free virions were washed away 24 hours later and newly produced HIV-1 virions from infected macrophages were quantified by p24 ELISA at d 7 post-infection.
Flow cytometry
Splenic B cells and macrophages from the spleen of infected patients undergoing post-traumatic splenectomy (the Institutional Review Board of Weill Medical College of Cornell University approved studies involving splenic specimens, which were collected upon obtaining informed consent) and B cells from co-cultures with in vitro infected macrophages were stained with fluorescein-conjugated mAb SJ25-C1 to CD19 or biotin-conjugated mAb 61D3 to CD14 (SouthernBiotech), followed by staining with strepatavidin-PerCp-Cy5.5. Cells were then fixed, permeabilized with Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit (BD Biosciences PharMingen), and further stained with goat polyclonal antibody (pAb) VA-19 to Nef and mAb 24-4 to p24 (Santa Cruz). Donkey anti-goat Alexa Fluor 546 and goat anti-mouse IgG2b Alexa Fluor 647 were sequentially used as secondary reagents. At least 5 × 104 cells were acquired with a LSRII analyzer (BD Biosciences PharMingen).
Immunohistochemistry and immunofluorescence analysis
Paraformaldehyde-fixed frozen tissue sections 5-15 μm in thickness were blocked with 1% bovine serum albumin-phosphate buffer solution mixed with whole human IgG or human FcγR blocking reagent (Miltenyi) for 1 h at 25 °C and then stained with appropriate primary mAbs or polyclonal antibodies (pAbs) (Supplementary Table S3 online) and secondary reagents as previously described8. Nuclei were visualized with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) (Boehringer Mannheim). Images were acquired with an Axiovert 200M microscope (Carl Zeiss). Control stainings and quantification of GC-associated proteins were performed as described in Supplementary Methods. To visualize CD19, CD14, Nef and p24 in B cells and macrophages, cells were centrifuged, fixed in methanol and labeled with DAPI and appropriate primary and secondary antibodies (Supplementary Table S3 online).
Plasmids, nucleofections and lipofections
Nef-eGFP was obtained by ligating a NA7 Nef-coding sequence from the Nef-dsRED vector (from S.J. Burakoff, New York University) with the pEGFP-N1 vector. Nef-eGFP Nef mutants were generated through a QuickChange II Site-Directed Mutagenesis Kit (Stratagene) and primers described in Supplementary Methods. THP-1 cells were nucleofected with Nef-eGFP using Cell Line Nucleofector Kit V and a Nucleofector II device (Amaxa). Nef-eGFP was nucleofected in the presence or absence of the following DN constructs: dynamin-K44A and β-arrestin-2-V54D (from Dr. M.G. Caron, Duke University), ARF6-T27N (from J.G. Donaldson, NIH), RhoA-N19, Cdc42-N17, Rac1-N17 (from Y. Zheng, Cincinnati Children's Hospital Medical Center), and Vav2-R/S (from P. Marignani, Dalhousie University). Nucleofection efficiency was usually greater than 70%. Nucleofected cells expressing eGFP were FACSorted before using them in co-cultures with B cells. More than 80% of THP-1 cells were viable after nucleofection. In additional experiments, 293T cells were treated with lipofectamine (Invitrogen) to transfect either a wt HIV-1-expressing NL4-3/9-7 plasmid or a ΔNef-HIV-1-expressing NL4-3/9-7-dsRed plasmid in which the nef gene was replaced by a dsRed-encoding sequence (from J.P. Moore, Weill Medical College of Cornell University). Transfection efficiency was routinely greater than 90%.
Wide-field and confocal microscopy
Imaging studies on fixed cells were performed as follows. 1 × 106/ml B cells were incubated with pre-warmed serum free medium containing 10 mM CellTracker (Invitrogen) for 30 min at 37 °C. After an additional incubation at 37 °C and appropriate washes, B cells were co-cultured with eGFP-THP-1, wt eGFP-Nef-THP-1, mutant eGFP-Nef-THP-1, U1/HIV-1 or TNF-α-induced U1/HIV-1 cells on a glycine-coated cover glass. Cells were subsequently fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton-100, and stained with goat pAb vA-19 to HIV Nef (Santa Cruz), mouse mAb Kal-1 to HIV p24 (Dako), mouse mAb 100/1 to β-adaptin (AbCam), or rabbit pAb NV110-40769 to TGN46 (Novus) and appropriate secondary reagents. Alternatively, WGA Alexa Fluor® 488 conjugate (Molecular Probes, Inc.) was incubated overnight with infected primary macrophages or macrophage-B cell co-cultured on glass-bottom dish to a final concentration of 3 μg/ml. Cells were washed gently and fixed in 4% paraformaldehyde containing 4% sucrose in phosphate buffer solution for 10 min at 37°C, followed by permeabilization with 0.2% Triton-100 in phosphate buffer solution for 4 min at 25 °C. Nef, p24, TGN46 and β-adaptin were stained as described. B cells were visualized using A-11 mAb to Pax5 (Santa Cruz). Slides were analyzed by wide-field microscopy as described8 or by confocal microscopy. Imaging studies on live cells were performed as follows. 1 × 106/ml B cells pre-loaded with CellTracker or 200 nM LysoTracker (Invitrogen) were co-cultured with THP-1 cells expressing wt or mutant Nef-eGFP B cells at 37°C on MatTek collagen-coated glass-bottom microwell dishes (MatTek Corporation). In some experiments, live THP-1 cells were pre-loaded with the lipophilic tracer 1,19-dioctadecyl-3,3,39,39-tetrametylindodicarbocyanine (DiD) (Invitrogen) or with CellTracker. Cells were analyzed by confocal microscopy on a heated CO2 chamber. Differential interfering contrast (DIC) and epifluorescence images were acquired at appropriate intervals with a Zeiss LSM 510 laser scanning confocal microscope. Time-lapse and three-dimensional movies were generated as reported in Supplementary Methods.
ELISAs and QRT-PCRs
Total and virus-specific IgM, IgG, IgA, IgG1, IgG2, IgG3, IgG4, IgA1 and IgA2 were detected as described in Supplementary Methods. To detect ferritin, macrophage supernatants were incubated overnight at 4 C° on microplates coated with mouse RF-80A pAb to ferritin (Lee Solutions). Ferritin from human liver (Calbiochem) was used to generate a standard curve. A peroxidase-conjugated mouse H117 mAb to ferritin (Leinco Technologies) and the substrate 3.3′,5,5′-tetramethylbenzidine (Kirkegaard and Perry) were used in sequential steps. Readings were performed at 450 nm. Iα1-Cα1, Iα2-Cα2, AICDA, and Iα-Cμ transcripts were measured by QRT-PCR as previously described8.
Immunogold transmission electron microscopy
THP-1 cells with or without Nef-eGFP were plated with B cells onto 35 mm Petri dishes for two days. Cells were then collected and pelleted and fixed in 4% paraformaldehyde with 1% glutaraldehyde in PBS for 15 min at 25 °C. After washing, pellets were dehydrated in series of graded ethanol in the order of 50, 70, 85, 95, 100%, 15 minutes each, while progressively lowering the temperature from 25 °C to −20 °C by the time 100% ethanol was used. Pellets were then infiltrated sequentially with 3:1, 1:1 of ethanol : resin (L R White, London Resin Co., LTD), 30 min each, followed with pure resin for 24 hrs, all at -20 °C. After exchanging to fresh resin, pellets were then polymerized at 50 °C for 24 hrs, completed with UV light exposure until the block was totally hardened. For labeling, 65 nm in thickness of sections were mounted on nickel grids and incubated with blocking buffer for goat secondary antibodies for 10 mins (Aurion) while the control sections were incubated with plain blocking buffer. Sections were then incubated with mouse anti-GFP (eBioscience) at 1:100 in blocking buffer for 1 h. After five 5-min washes with blocking buffer, secondary goat pAb to mouse IgG conjugated with 6 nm gold (Aurion) was added and further incubated for 1 hour. After three 3-min washes with PBS and two 1-min washes with distilled H2O, sections were incubated with uranyl acetate for 15 min followed with Lead Citrate for 5 min. THP-1 cells lacking Nef-eGFP were consistently negative for immunogold staining.
Statistical analysis
Values were calculated as mean standard deviation for at least three separate experiments done in triplicate. The significance of differences between experimental variables was determined with the Student's t-test and a p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank M. Stevenson (University of Massachusetts Medical School), S.J. Burakoff (New York University) M.G. Caron, (Duke University), J.G. Donaldson (NIH), Y. Zheng (Cincinnati Children's Hospital Medical Center), P. Marignani (Dalhousie University), J.P. Moore (Weill Medical College of Cornell University), amd A. Pernis (Columbia University) for reagents and discussions. This work was supported by NIH research grants R01 AI057653, R01 AI057653-S1 and R01 AI074378, by an Irma T. Hirschl Career Scientist Award, and by a CLL Research Grant from the Cornell Comprehensive Cancer Center (to A.C.); by funds from NIH T32 grant AI07621 (to W.X.); and by a Cancer Research Institute fellowship (to P.S.).
Footnotes
Author Contributions: W.X. and P.A.S. designed and performed research; B.H and K.C. performed research and discussed data; J.S.S., W.B.D., A. Chadburn, D.M.K. and A. Chiu provided samples and discussed data; M.S., T.J.K. and R.W.S provided reagents and performed research; S.C.B. provided clinical data; L.C.G. performed electron microscopy; and A. Cerutti designed research, discussed data and wrote the paper.
Competing Interest Statement: The authors declare that they have no competing financial interests.
References
- 1.Davis DM. Intercellular transfer of cell-surface proteins is common and can affect many stages of an immune response. Nat Rev Immunol. 2007;7:238–243. doi: 10.1038/nri2020. [DOI] [PubMed] [Google Scholar]
- 2.Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 3.Sherer NM, Mothes W. Cytonemes and tunneling nanotubules in cell-cell communication and viral pathogenesis. Trends Cell Biol. 2008;18:414–420. doi: 10.1016/j.tcb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watkins SC, Salter RD. Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity. 2005;23:309–318. doi: 10.1016/j.immuni.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Fackler OT, Alcover A, Schwartz O. Modulation of the immunological synapse: a key to HIV-1 pathogenesis? Nat Rev Immunol. 2007;7:310–317. doi: 10.1038/nri2041. [DOI] [PubMed] [Google Scholar]
- 6.Sherer NM, et al. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol. 2007;9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sowinski S, et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol. 2008;10:211–219. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 8.Qiao X, et al. Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat Immunol. 2006;7:302–310. doi: 10.1038/ni1302. [DOI] [PubMed] [Google Scholar]
- 9.Muratori C, et al. Macrophages transmit human immunodeficiency virus type 1 products to CD4-negative cells: involvement of matrix metalloproteinase 9. J Virol. 2007;81:9078–9087. doi: 10.1128/JVI.00675-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kestler HW, 3rd, et al. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65:651–662. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- 11.Peterlin BM, Trono D. Hide, shield and strike back: how HIV-infected cells avoid immune eradication. Nat Rev Immunol. 2003;3:97–107. doi: 10.1038/nri998. [DOI] [PubMed] [Google Scholar]
- 12.Fackler OT, Luo W, Geyer M, Alberts AS, Peterlin BM. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol Cell. 1999;3:729–239. doi: 10.1016/s1097-2765(01)80005-8. [DOI] [PubMed] [Google Scholar]
- 13.Peng B, Robert-Guroff M. Deletion of N-terminal myristoylation site of HIV Nef abrogates both MHC-1 and CD4 down-regulation. Immunol Lett. 2001;78:195–200. doi: 10.1016/s0165-2478(01)00250-4. [DOI] [PubMed] [Google Scholar]
- 14.Blagoveshchenskaya AD, Thomas L, Feliciangeli SF, Hung CH, Thomas G. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell. 2002;111:853–866. doi: 10.1016/s0092-8674(02)01162-5. [DOI] [PubMed] [Google Scholar]
- 15.Thoulouze MI, et al. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity. 2006;24:547–561. doi: 10.1016/j.immuni.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Burton DR. Antibodies, viruses and vaccines. Nat Rev Immunol. 2002;2:706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- 17.Lane HC, et al. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1983;309:453–458. doi: 10.1056/NEJM198308253090803. [DOI] [PubMed] [Google Scholar]
- 18.Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol. 2009;9:235–245. doi: 10.1038/nri2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binley JM, et al. Differential regulation of the antibody responses to Gag and Env proteins of human immunodeficiency virus type 1. J Virol. 1997;71:2799–2809. doi: 10.1128/jvi.71.4.2799-2809.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regulier EG, et al. Persistent anti-gag, -Nef, and -Rev IgM levels as markers of the impaired functions of CD4+ T-helper lymphocytes during SIVmac251 infection of cynomolgus macaques. J Acquir Immune Defic Syndr. 2005;40:1–11. doi: 10.1097/01.qai.0000173702.05308.c4. [DOI] [PubMed] [Google Scholar]
- 21.Martinez V, et al. Combination of HIV-1-specific CD4 Th1 cell responses and IgG2 antibodies is the best predictor for persistence of long-term nonprogression. J Infect Dis. 2005;191:2053–2063. doi: 10.1086/430320. [DOI] [PubMed] [Google Scholar]
- 22.Schafer F, et al. Lack of simian immunodeficiency virus (SIV) specific IgA response in the intestine of SIV infected rhesus macaques. Gut. 2002;50:608–614. doi: 10.1136/gut.50.5.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mestecky J, et al. Paucity of antigen-specific IgA responses in sera and external secretions of HIV-type 1-infected individuals. AIDS Res Hum Retroviruses. 2004;20:972–988. doi: 10.1089/aid.2004.20.972. [DOI] [PubMed] [Google Scholar]
- 24.De Milito A, et al. Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood. 2004;103:2180–2186. doi: 10.1182/blood-2003-07-2375. [DOI] [PubMed] [Google Scholar]
- 25.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–239. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 26.He B, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol. 2006;176:3931–3941. doi: 10.4049/jimmunol.176.7.3931. [DOI] [PubMed] [Google Scholar]
- 27.Swingler S, et al. Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe. 2008;4:63–76. doi: 10.1016/j.chom.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moir S, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. 2008;205:1797–1805. doi: 10.1084/jem.20072683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Titanji K, et al. Loss of memory B cells impairs maintenance of long-term serological memory during HIV-1 infection. Blood. 2006;108:1580–1587. doi: 10.1182/blood-2005-11-013383. [DOI] [PubMed] [Google Scholar]
- 30.Hart M, et al. Loss of discrete memory B cell subsets is associated with impaired immunization responses in HIV-1 infection and may be a risk factor for invasive pneumococcal disease. J Immunol. 2007;178:8212–8220. doi: 10.4049/jimmunol.178.12.8212. [DOI] [PubMed] [Google Scholar]
- 31.Moir S, et al. Perturbations in B cell responsiveness to CD4+ T cell help in HIV-infected individuals. Proc Natl Acad Sci U S A. 2003;100:6057–6062. doi: 10.1073/pnas.0730819100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poudrier J, et al. The AIDS disease of CD4C/HIV transgenic mice shows impaired germinal centers and autoantibodies and develops in the absence of IFN-gamma and IL-6. Immunity. 2001;15:173–185. doi: 10.1016/s1074-7613(01)00177-7. [DOI] [PubMed] [Google Scholar]
- 33.Junt T, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, et al. Epithelial cells trigger frontline immunoglobulin class switching through a pathway regulated by the inhibitor SLPI. Nat Immunol. 2007;8:294–303. doi: 10.1038/ni1434. [DOI] [PubMed] [Google Scholar]
- 35.Klein U, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7:773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 36.Shaffer AL, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg ME, Iafrate AJ, Skowronski J. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. Embo J. 1998;17:2777–2789. doi: 10.1093/emboj/17.10.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. 1994;76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 39.Greenberg ME, et al. Co-localization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 down-regulation. Embo J. 1997;16:6964–6976. doi: 10.1093/emboj/16.23.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piguet V, et al. Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the μ chain of adaptor complexes. Embo J. 1998;17:2472–2481. doi: 10.1093/emboj/17.9.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg M, DeTulleo L, Rapoport I, Skowronski J, Kirchhausen T. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr Biol. 1998;8:1239–1242. doi: 10.1016/s0960-9822(07)00518-0. [DOI] [PubMed] [Google Scholar]
- 42.Litinskiy MB, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez B, et al. Plasma levels of B-lymphocyte stimulator increase with HIV disease progression. Aids. 2003;17:1983–1985. doi: 10.1097/00002030-200309050-00018. [DOI] [PubMed] [Google Scholar]
- 44.He B, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A2 class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 45.Dyer WB, et al. Strong human immunodeficiency virus (HIV)-specific cytotoxic T-lymphocyte activity in Sydney Blood Bank Cohort patients infected with nef-defective HIV type 1. J Virol. 1999;73:436–443. doi: 10.1128/jvi.73.1.436-443.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burton DR, et al. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 47.Popovic M, et al. Persistence of HIV-1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 2005;102:14807–14812. doi: 10.1073/pnas.0506857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prost S, et al. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARγ/STAT5 signaling pathway in macaques. J Clin Invest. 2008;118:1765–1775. doi: 10.1172/JCI33037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. 2005;79:10978–10987. doi: 10.1128/JVI.79.17.10978-10987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santa-Marta M, Aires da Silva F, Fonseca AM, Rato S, Goncalves J. HIV-1 Vif protein blocks the cytidine deaminase activity of B-cell specific AID in E. coli by a similar mechanism of action. Mol Immunol. 2006;44:583–590. doi: 10.1016/j.molimm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Begum NA, et al. Requirement of non-canonical activity of uracil DNA glycosylase for class switch recombination. J Biol Chem. 2007;282:731–742. doi: 10.1074/jbc.M607439200. [DOI] [PubMed] [Google Scholar]
- 52.Mann J, et al. Functional analysis of HIV type 1 Nef reveals a role for PAK2 as a regulator of cell phenotype and function in the murine dendritic cell line, DC2.4. J Immunol. 2005;175:6560–6569. doi: 10.4049/jimmunol.175.10.6560. [DOI] [PubMed] [Google Scholar]
- 53.Conboy IM, Manoli D, Mhaiskar V, Jones PP. Calcineurin and vacuolar-type H+-ATPase modulate macrophage effector functions. Proc Natl Acad Sci U S A. 1999;96:6324–6329. doi: 10.1073/pnas.96.11.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiley RD, Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1 trans infection. Proc Natl Acad Sci U S A. 2006;103:738–743. doi: 10.1073/pnas.0507995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa LJ, et al. Interactions between Nef and AIP1 proliferate multivesicular bodies and facilitate egress of HIV-1. Retrovirology. 2006;3:33. doi: 10.1186/1742-4690-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fujii Y, Otake K, Tashiro M, Adachi A. Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett. 1996;393:93–96. doi: 10.1016/0014-5793(96)00859-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.